

Abstract
Latent membrane protein-1 (LMP1), the major oncoprotein of Epstein-Barr virus (EBV), is likely responsible for many of the altered cellular growth properties in EBV-associated cancers including nasopharyngeal carcinoma (NPC). In this study, the effects of LMP1 on cell growth and migration were studied in the context of the EBV-positive C666-1 NPC cell line. In the soft agar transformation and transwell metastasis assays, LMP1 enhanced cell growth and migration through activation of phosphatidylinositol 3-kinase (PI3K)/Akt and NFκB signaling. Inhibitors of PI3K, Akt and NFκB signaling dramatically reduced these enhanced properties. An IκBα super-repressor also blocked these effects. However, constitutive activation of Akt alone did not alter cell growth, suggesting that both PI3K/Akt and NFκB activation are required by LMP1. These enhanced effects required the full-length LMP1 encompassing both the PI3K/Akt activating C-terminal activation region (CTAR) 1 and the non-redundant NFκB activating regions CTARs 1 and 2. LMP2A, a latent protein that is also frequently expressed in NPC, similarly activates the PI3K/Akt pathway, however its over-expression in C666-1 cells did not affect cell growth or migration. LMP1 also decreased expression of the junctional protein plakoglobin which was shown to be partially responsible for enhanced migration induced by LMP1. This study reveals that in epithelial cells the transforming properties of LMP1 require activation of both PI3K/Akt and NFκB and demonstrates that the loss of plakoglobin expression by LMP1 is a significant factor in the enhanced migration.
Keywords: Epstein-Barr virus, latent membrane protein-1, Akt, NFκB, plakoglobin
Introduction
Epstein-Barr virus (EBV) is a ubiquitous γ-herpesvirus that infects more than 90% of the adult human population, and is associated with malignancies of epithelial and lymphocyte origins (1). Cancers linked to EBV include the epithelial cell cancer nasopharyngeal carcinoma and B cell malignancies including Burkitt lymphoma, Hodgkin disease, post-transplant lymphoma and AIDS-associated lymphoma (1). These malignancies are associated with the expression of EBV latent genes which are classified into types I, II and III (2). Type I latency, typical of Burkitt lymphoma, has the most restricted expression profile such that only the EBV nuclear antigen 1 (EBNA1), BamHI-A transcripts and the untranslated nonpolyadenylated EBERs are expressed. Type II latency is associated with nasopharyngeal carcinoma and Hodgkin lymphoma, and expresses latent membrane proteins 1 and 2 (LMP1, LMP2A and LMP2B) in addition to the transcripts expressed in type I latency. In Type III latency all of the latency genes are expressed including EBNA -2, -3A, -3B, -3C and –LP and is only found in cancers linked to immunosuppression such as post-transplant lymphoma and AIDS-associated lymphoma. Of these latent proteins, LMP1, EBNA-1, -2, -3A and –3C are required for EBV-induced B cell transformation (2).
As LMP1 and LMP2A are frequently detected in EBV-associated nasopharyngeal carcinoma, the effects of these genes on growth regulation is thought to contribute to the development of cancer (1). LMP1 can transform Rat-1 and human embryonic lung fibroblasts to form foci, grow in soft agar, and form tumors in nude mice. This property requires the activation of PI3K/Akt signaling induced by the C-terminal activation region (CTAR) 1 domain (3, 4). In addition, transgenic mice expressing LMP1 in B-lymphocytes develop B cell lymphomas or develop epidermal hyperplasia when expressed in mouse epidermis (5, 6). The effects of LMP1 on cell signaling and expression include activation of transcription factors (NFκB, ERK, p38, AP1) (7, 8), cytokines (IL6, IL8, IL10), anti-apoptotic proteins (A20, bcl-2) (9, 10) and in epithelial cells, proteins that modulate adhesion and invasion (E-cadherin, MMP9, MUC1) (11, 12). These properties have been largely attributed to two C-terminal domains, CTAR1 and CTAR2. Both CTAR1 and CTAR2 can activate IκBα-dependent canonical NFκB signaling while CTAR1 can also activate the NIK-dependent non-canonical NFκB pathway (8, 9, 13). LMP1 CTAR1 also uniquely induces p50/p50 NFκB homodimers in association with the transactivating IκB member bcl-3 (14, 15). This property has been linked to the induction of the epidermal growth factor receptor (EGFR) in the etiology of NPC (14-16). The transforming properties of LMP1 in both fibroblasts and epithelial cells have been shown to require activation of PI3K/Akt and ERK-MAPK (3, 7, 17). LMP2A also affects cellular growth properties, and can transform several epithelial cell lines and inhibit differentiation. LMP2A has been shown to activate the proto-oncogenic Wnt signaling pathway in a PI3K-dependent manner (18-20). These properties of both LMP1 and LMP2A likely contribute to the development of NPC (10).
To further investigate the transforming properties of LMP1 and LMP2A, their effects on the growth and migration properties of epithelial cells were studied in the context of an EBV-positive NPC cell line, C666-1. C666-1 cells are unique in that they are the only NPC cell line that has retained the EBV episome. These cells express very low levels of LMP1 and LMP2A (21). However it has been shown that the low level of LMP1 expression and its induction of Akt signaling are still critically required for the survival of C666-1 cells (22). The data presented in this study reveal that enhanced expression of LMP1 induces growth and migration and these effects require activation of PI3K/Akt and IκBα-dependent canonical NFκB signaling. Interestingly, this study shows that LMP1-mediated down-regulation of plakoglobin is a major factor in promoting enhanced migration.
Results
LMP1 activates Akt and down-regulates IκBα and plakoglobin levels
To investigate the effects of LMP1 and LMP2A on the growth of C666-1 cells, full-length LMP1, LMP1-CTAR1 that is deleted for CTAR2 and LMP1-CTAR2 that is deleted for CTAR1, and full-length LMP2A were stably expressed by retroviral transduction. Expression of the HA-tagged LMP1, CTAR1 and CTAR2 constructs was relatively equivalent (Fig. 1A). Using an LMP2A-specific antibody, expression of LMP2A was confirmed and was increased above the basal levels detected in the pBabe vector control (Fig. 1A). LMP1 activates PI3K/Akt signaling and subsequently inactivates GSK3β (3, 23). LMP2A also activates PI3K/Akt signaling, leading to inactivation of GSK3β and activation of β-catenin signaling (18, 24). Activation of PI3K/Akt is required for the transformation by LMP1 and LMP2A (3, 19, 25). To assess the effects of LMP1, CTAR1, CTAR2 or LMP2A on activation of PI3K/Akt in C666-1 cells, activated phosphorylated Akt and inactivated phosphorylated GSK3αβ isoforms were identified by immunoblotting. The control cell line (pBabe) had detectable levels of phosphorylated Akt, which was increased by the expression of LMP1, CTAR1, CTAR2 and LMP2A (Fig. 1B). This differs from previous studies where LMP1-CTAR2 was not sufficient to induce Akt activation (3). It is possible that in the context of basal levels of LMP1 expression, over-expression of CTAR2 can complement full-length LMP1 signaling and enhance Akt activation. Phosphorylation and inactivation of GSK3αβ was also enhanced in LMP1, CTAR1, CTAR2 and LMP2A expressing cells, correlating with the enhanced activation of Akt (Fig. 1B).
Figure 1.
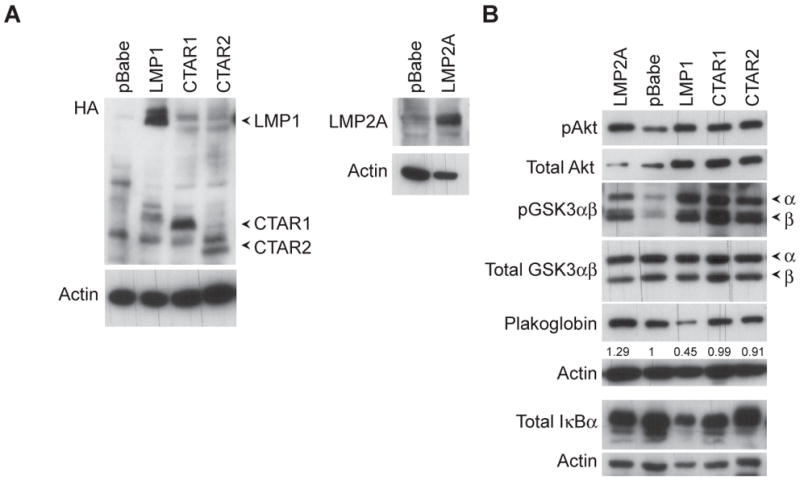
LMP1 down-regulates plakoglobin levels. (A) Stable expression of HA-tagged LMP1, CTAR1, CTAR2 or LMP2A from the pBabe vector in C666-1 cells was analyzed by immunoblot analysis. Arrows indicate the LMP1-specific band. (B) The effects of LMP1, CTAR1, CTAR2 and LMP2A expression on phosphorylated Akt (pAkt), phosphorylated GSK3αβ, IκBα and plakoglobin levels were analyzed by immunoblot analysis. Actin was used as a loading control. Densitometry with Image J software was performed to normalize plakoglobin levels to the corresponding actin levels. Fold change of the normalized plakoglobin levels relative to the pBabe control is indicated below the immunoblots.
Activation of NFκB signaling by LMP1 is required for B cell transformation but not rodent fibroblast transformation (3, 26, 27). Both CTAR1 and CTAR2 domains of LMP1 have been shown to induce NFκB signaling (13, 28), however the CTAR2 domain is considered the major activating domain that can activate the canonical IκBα-dependent activation of NFκB (29, 30). To assess the activation of the canonical pathway, total levels of IκBα were detected by immunoblot analysis. Decreased IκBα was only observed in cells over-expressing full-length LMP1 (Fig. 1B). In the background of basal LMP1 expression, over-expression of CTAR1 or CTAR2 was not sufficient to further decrease IκBα levels compared to the pBabe control. These findings indicate that over-expression of LMP1, CTAR1, CTAR2 and LMP2A were all able to further activate Akt signaling to comparable levels, however, only full-length LMP1 dramatically affected the canonical NFκB pathway. These differences in signaling properties may induce differences in cellular growth potential.
Nasopharyngeal carcinoma is a highly metastatic and invasive malignant tumor (10). It has been reported that LMP1 induces epithelial cell migration through down-regulation of proteins involved in cell adhesion including E-cadherin and up-regulation of proteins involved in opposing cell adhesion, or the degradation of the extracellular matrix including activation of the ERK-MAPK pathway and the up-regulation of MUC1 and MMP9 (11, 12, 17). To identify whether other proteins involved in cell-cell adhesion contributes to LMP1-induced migration, the levels of plakoglobin were analyzed by immunoblot analysis. Plakoglobin is found at both adherens junctions and desmosomes, and loss of plakoglobin has been associated with breast and ovarian cancers and accounts for increased keratinocyte motility (31, 32). C666-1 cells over-expressing full-length LMP1 had decreased plakoglobin levels. A representative experiment is shown and densitometry revealed that plakoglobin expression was decreased 55% compared to the pBabe control (Fig. 1B). Over-expression of CTAR1, CTAR2 or LMP2A did not decrease plakoglobin levels below the pBabe control (Fig. 1B), suggesting that both CTAR1 and CTAR2 domains of LMP1 are required in the down-regulation of plakoglobin. It appears that in NPC cells, the full-length LMP1 is required to affect multiple pathways linked to oncogenesis, including down-regulation of plakoglobin and the activation of IκBα-dependent NFκB signaling. To investigate any contributions of Akt and NFκB signaling to the regulation of plakoglobin levels, additional constructs affecting these pathways were utilized.
To discern the involvement of the canonical NFκB pathway on plakoglobin expression, the IκBα super-repressor (IκBαSS32/36AA) was expressed in the LMP1 over-expressing cells. The IκBα super-repressor contain serine-to-alanine mutations at amino acids 32 and 36, and is unable to be phosphorylated and degraded (14). Expression of the HA-tagged IκBα super-repressor and total IκBα was verified by immunoblot analysis to total IκBα levels (Fig. 2A). The IκBα super-repressor stabilized endogenous IκBα levels, possibly by competitive binding, suggesting that the IκBα super-repressor was effectively inhibiting degradation of IκBα (Fig. 2A). LMP1 expression was detected with anti-LMP1 specific antibodies, a combination of four monoclonals that detect LMP1 and multiple degradation fragments (Fig. 2A). Expression of LMP1 in IκBαSS32/36A-expressing cells was comparable to the pHSCG vector control and co-expression of IκBαSS32/36A from the pHSCG retroviral vector did not significantly affect LMP1 expression from the pBabe retroviral vector (Fig. 2A). Expression of the IκBα super-repressor did not reduce LMP1-mediated phosphorylation and activation of Akt when compared to pBabe levels, however, it did inhibit the effects of LMP1 on reduction of plakoglobin (Fig. 2A)
Figure 2.
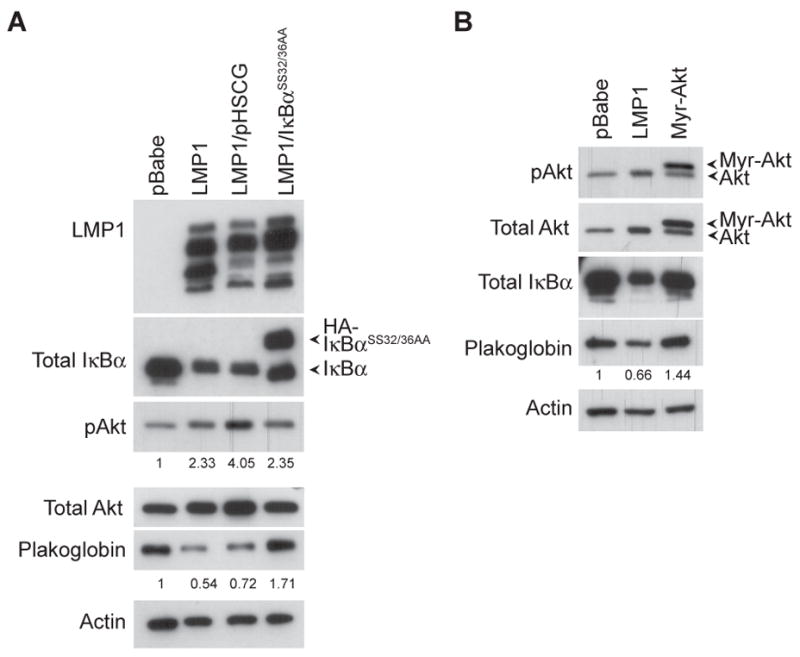
Down-regulation of plakoglobin by LMP1 is dependent on the IκBα regulated canonical NFκB pathway. (A) Expression and the effects of the HA-tagged IκBα super-repressor (IκBαSS32/36AA) from the pHSCG vector, on LMP1’s down-regulation of plakoglobin was analyzed by immunoblot analysis. Activation of Akt was detected by the phosphorylation of Akt (pAkt). (B) Expression and the effects of the constitutively active myristylated-Akt (myr-Akt) and LMP1 on plakoglobin levels was compared by immunoblot analysis. Actin was used as a loading control. Densitometry with Image J software was performed to normalize pAkt and plakoglobin levels to the corresponding actin levels. Fold change of the normalized pAkt and plakoglobin levels relative to the pBabe control is indicated below the immunoblots.
To determine the effects of Akt activation on plakoglobin expression, a constitutively activated myristylated-Akt was stably expressed in C666-1 cells. Antibodies for total Akt detected the myristylated-Akt which migrated slightly slower than endogenous Akt (Fig. 2B). The myristylated-Akt was activated as detected by the phosphorylated Akt levels (Fig. 2B). Expression of the myristylated-Akt did not affect total IκBα or plakoglobin levels when compared to the pBabe control (Fig. 2B). These data indicate that the effects of LMP1 expression on plakoglobin requires activation of NFκB and that activation of Akt alone does not affect plakoglobin expression.
Full-length LMP1 enhances cell growth through activation of PI3K and IκBα-dependent canonical NFκB signaling
To analyze the LMP1-mediated effects on C666-1 growth, MTS and soft agar colony assays were performed. The MTS assay measures the metabolic activity of mitochondrial dehydrogenase as an indicator of cell cycle induction and growth. Full-length LMP1 dramatically induced cell growth and high levels of MTS activity (Fig. 3A). The rate of growth was not altered by CTAR1, CTAR2 or LMP2A expression compared to the pBabe control.
Figure 3.
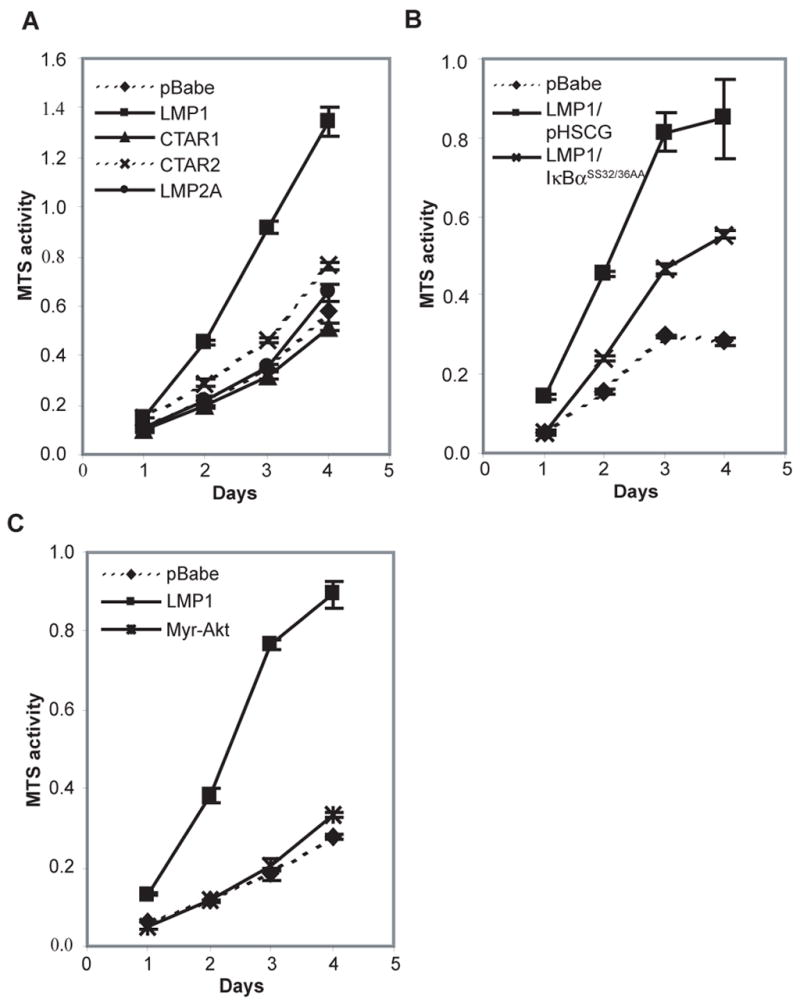
LMP1 induced growth requires both CTAR1 and 2 domains, and is dependent on NFκB activation. (A-C) MTS assays were performed to measure the metabolic activity of C666-1 cells stably expressing LMP1, CTAR1, CTAR2, LMP2A, the constitutively activated myristylated-Akt (myr-Akt), the pBabe vector control and the effects of the IκBα supper-repressor (IκBαSS32/36AA) on the growth of LMP1-expressing cells.
To investigate the contributions of PI3K/Akt and NFκB signaling to LMP1-induced growth, inhibitors and constructs activating or suppressing these pathways were employed. Expression of the IκBα super-repressor reduced the enhanced growth of LMP1 cells in the MTS assay while expression of myristylated-Akt did not enhance cell growth. (Fig. 3B and 3C). These findings suggest that the canonical NFκB pathway is critically involved in LMP1-induced growth but that activation of Akt alone does not alter cell growth.
The soft agar colony formation assay measures anchorage-independent cell growth as an indicator of transformation. Although pBabe control cells were able to form colonies and were visible by two weeks post-seeding (data not shown), cells over-expressing LMP1 were able to form large colonies by only four days post-seeding (Fig. 4A). In agreement with the effects on activation of cell signaling pathways, expression of CTAR1, CTAR2, or LMP2A did not enhance growth in soft agar.
Figure 4.

LMP1 enhanced soft agar colony formation requires both CTAR1 and 2 domains and is dependent on PI3K and NFκB signaling. (A) C666-1 cells stably expressing LMP1, CTAR1, CTAR2, LMP2A or the pBabe vector control was assessed for anchorage-independent growth using the soft agar colony assay. (B) The signals required by LMP1 for enhanced soft agar colony formation was assessed by using inhibitors of PI3K (LY294002), Akt (Akt inhibitor I) and NFκB (BAY11-7085) signaling, compared to the DMSO control. (C) The requirement of canonical NFκB signaling for LMP1-induced soft agar colony formation was assessed using the IκBα super-repressor (IκBαSS32/36AA), compared to the pHSCG vector control. (D) Expression of the constitutively active myristylated Akt (myr-Akt) was used to assess whether activation of Akt signaling is sufficient to enhance soft agar colony growth.
To assess the contribution of activated NFκB and Akt on LMP1-induced growth effects, inhibitors of PI3K (LY294002), Akt (Akt inhibitor I) and NFκB (BAY11-7085) signaling were tested in the soft agar assay. The inhibitors LY294002 and BAY11-7085 have been previously shown to have an inhibitory effect on the growth of LMP1-induced lymphomas (27). The NFκB inhibitor, BAY11-7085, inhibits the phosphorylation of IκBα and thus specifically targets the canonical NFκB pathway. All of these inhibitors blocked the enhanced soft agar colony growth induced by LMP1 (Fig. 4B). In agreement with previous assays, expression of the IκBα super-repressor inhibited the LMP1-induced growth in soft agar while myristylated-Akt did not induce growth (Fig. 4C and 4D). These data indicate that activation of NFκB and PI3K/Akt are required for LMP1-enhanced growth, however, activation of Akt alone is not sufficient to affect the growth of C666-1 cells. The requirement of both of these pathways is also supported by the observation that only full-length LMP1 is able to enhance cell growth and induce both PI3K/Akt signaling and the degradation of IκBα (Fig. 1B).
Full-length LMP1 enhances migration through activation of PI3K/Akt and IκBα-dependent canonical NFκB signaling
To investigate the effects of LMP1 on cell motility, LMP1, CTAR1, CTAR2 and LMP2A expressing cells were analyzed using a transwell migration assay. This assay measures the number of cells that migrate through a porous membrane in the direction of a chemoattractant. Since C666-1 cells are grown on fibronectin-coated culture dishes, fibronectin was used as the chemoattractant. Full-length LMP1 induced a 5-fold enhancement in migration that was not induced by over-expression of CTAR1 or CTAR2 alone (Fig. 5A). Although LMP2A has been shown to enhance migration in some epithelial cells (33-35), LMP2A did not induce migration in C666-1 cells.
Figure 5.

LMP1 induced migration requires both CTAR1 and 2 domains and is dependent on PI3K and NFκB signaling. (A) C666-1 cells stably expressing LMP1, CTAR1, CTAR2, LMP2A or the pBabe vector control was assessed for metastasis in the transwell migration assay. (B) The signals required by LMP1 for enhanced migration was assessed by using inhibitors of PI3K (LY294002), Akt (Akt inhibitor I) and NFκB (BAY11-7085) signaling at the indicated concentrations, compared to the DMSO control. (C) The ability of the PI3K inhibitor (LY294002), NFκB inhibitor (BAY11-7085) and Akt inhibitor (Akt inhibitor I) to block PI3K/Akt and IκBα-dependent NFκB signaling was assessed by immunoblot analysis after an overnight treatment, and blotted for activated phosphorylated Akt (pAkt), inactivated phosphorylated GSK3 (pGSK3αβ) and total IκBα levels. Actin was used as a loading control. Densitometry with Image J software was performed to normalize pAkt and IκBα levels to the corresponding actin levels. Fold change of the normalized pAkt and IκBα levels relative to the pBabe control is indicated below the immunoblots. White line indicates that intervening lanes have been spliced out. (D) LMP1-induced migration is dependent on IκBα-dependent NFκB signaling and activation of Akt is not sufficient to enhance migration. Expression of the constitutively active myristylated Akt (myr-Akt) was used to assess whether activation of Akt signaling is sufficient to enhance migration. The requirement of canonical NFκB signaling for LMP1-induced migration was assessed using the IκBα super-repressor (IκBαSS32/36AA), compared to the pHSCG vector control.
Similar to the growth studies, inhibitors of PI3K (LY294002), Akt (Akt inhibitor I) and NFκB (BAY11-7085) signaling inhibited LMP1-induced migration (Fig. 5B). The effect of the inhibitors at blocking Akt activation and IκBα degradation was determined by immunoblot analysis. LY294002 efficiently blocked activation of Akt as detected by the decreased phosphorylated Akt levels (Fig. 5C). Akt has been shown to phosphorylate IκBα and induce its degradation, however LY294002 treatment only further enhanced the degradation of IκBα induced by LMP1 (Fig. 5C). This suggests that the decreased migration observed with LY294002 is due to blocking PI3K/Akt signaling without blocking LMP1-induced degradation of IκBα. Although Akt inhibitor I was not as potent as LY294002 at inhibiting Akt phosphorylation, Akt inhibitor I was able to block phosphorylation and the activation of Akt at 20μM, a dose where the strongest effect was observed at inhibiting migration (Fig. 5C). To confirm the inhibition of Akt at 20μM, further immunoblot analysis was performed to determine the block in phosphorylation of the Akt target, GSK3β. A dose dependent inhibition of GSK3β phosphorylation was apparent from 10μM and was very evident at 20μM (Fig. 5C), indicating effective inhibition of Akt activation. Similarly to LY294002 treatment, Akt inhibitor I did not block, and even further enhanced the down-regulation of IκBα by LMP1 (Fig. 5C). This suggests that the inhibition of LMP1-induced migration by Akt inhibitor I was due to inhibition of Akt activation without blocking LMP1-induced degradation of IκBα.
The BAY11-7085 NFκB inhibitor effectively inhibited LMP1-induced enhanced migration and blocked activation of NFκB as evidenced by stabilized and increased IκBα levels. It appears that BAY11-7085 does not affect the activation of Akt by LMP1, such that increasing doses of BAY11-7085 did not affect pAkt levels (Fig. 5C). The requirement of NFκB signaling in LMP1-mediated migration was further evaluated in cells expressing the IκBα super-repressor, where the super-repressor also did not affect LMP1-induced Akt activation (Fig. 2A). The requirement for NFκB signaling was confirmed by the inhibition of LMP1-induced migration by the IκBα super-repressor (Fig. 5D). This supported the BAY11-7085 effects and the requirement for NFκB signaling in LMP1-induced migration, in the absence of any perturbation of Akt activation.
Although studies with LY294002 demonstrated a requirement for PI3K/Akt signaling, expression of the myristylated-Akt only enhanced migration slightly above pBabe control levels (Fig. 5D). This suggests that similar to the effects of LMP1 on growth, activation of PI3K/Akt and IκBα-dependent NFκB signaling are required for LMP1-enhanced motility, and that activation of Akt alone aids in migration but is insufficient at inducing similar levels of migration as that observed for LMP1.
Restoration of plakoglobin blocks LMP1-induced migration
The loss of E-cadherin has been linked to the increased invasiveness of LMP1 expressing cells (36). Recently a mutational analysis of LMP1 in Rat1 cells indicated that LMP1 also down-regulated another cell-adhesion protein, plakoglobin, through the TRAF binding domain in CTAR1 and a region between CTAR1 and CTAR2 (amino acids 220–378) (7). In this study, the loss of plakoglobin only occurred in C666-1 cells expressing full-length LMP1 (Fig. 1A) and this loss correlated with enhanced migration (Fig. 5). In addition, plakoglobin levels were restored in IκBα super-repressor expressing cells that had reduced migration (Fig. 2A and 5D). Therefore, the role of plakoglobin in LMP1-induced migration was evaluated by restoring plakoglobin levels in LMP1 expressing cells using a wild-type plakoglobin construct. Expression of the myc-tagged wild-type plakoglobin was detected using antibodies against c-myc (Fig. 6A). Expression of the myc-tagged plakoglobin was much higher in LMP1-expressing cells than in the pBabe/DsRed control cells. This may be due to mechanisms that control plakoglobin stability induced by endogenous levels of plakoglobin. Densitometry revealed that plakoglobin levels had been restored, albeit above pBabe/DsRed control levels, in LMP1 expressing cells (6.78-fold above pBabe/DsRed control cells compared to the reduction to 0.6-fold in the parental LMP1 expressing cells), (Fig. 6A). Restoration of plakoglobin levels in LMP1-expressing cells did not affect the expression levels of LMP1, however, in the transwell migration assay, restoration of plakoglobin in LMP1-expressing cells decreased migration approximately 50% compared to LMP1 alone (Fig. 6B). A slight decrease in migration was also observed when plakoglobin was over-expressed in pBabe control cells (Fig. 6B). These data indicate that plakoglobin levels affect LMP1-mediated migration.
Figure 6.
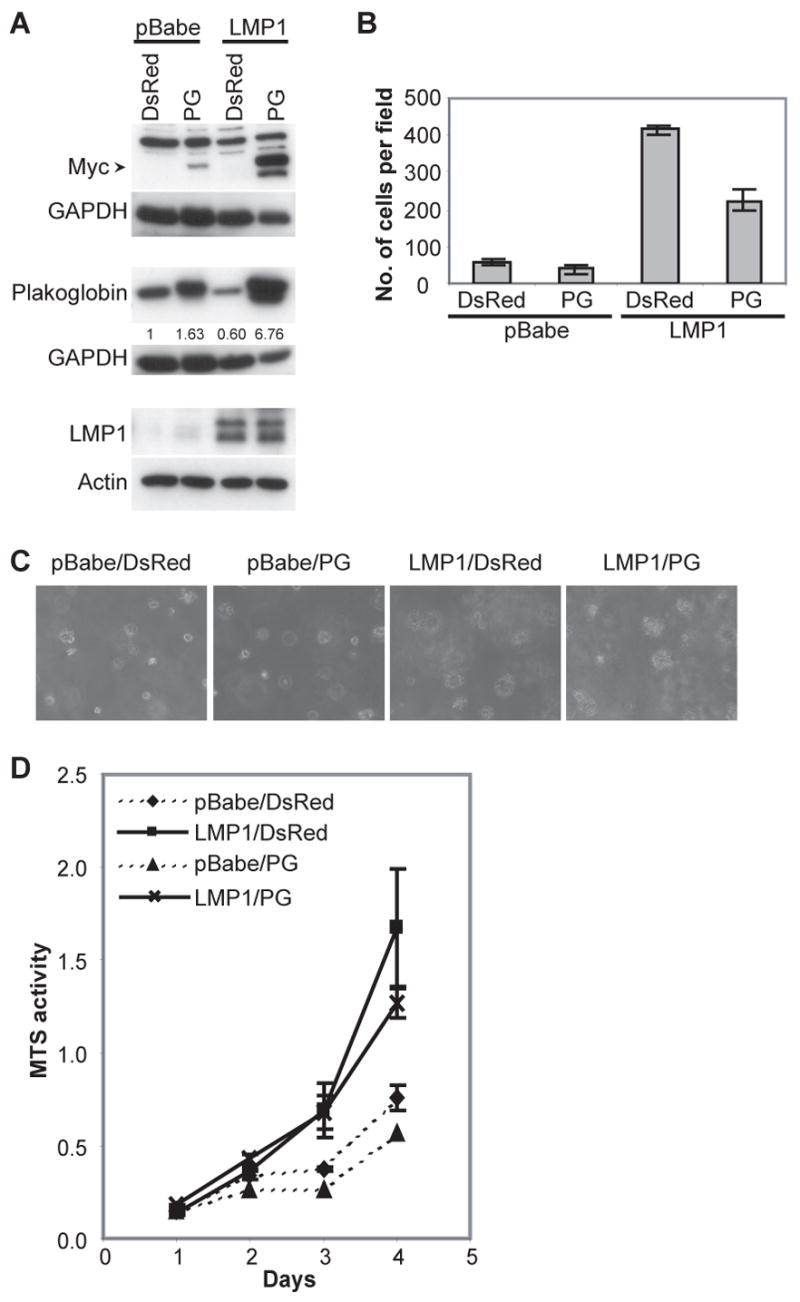
LMP1 induced migration is blocked by restoring plakoglobin levels. (A) Stable expression of LMP1 in the pBabe vector and a myc-tagged plakoglobin (PG) or the DsRed vector control from C666-1 cells was analyzed by immunoblot analysis. Actin and GAPDH were used as loading controls. Densitometry with Image J software was performed to normalize plakoglobin levels to the corresponding GAPDH levels. Fold change of the normalized plakoglobin levels relative to the pBabe/DsRed control is indicated below the immunoblots. (B) The effects of restoring PG levels on the migration of LMP1-expressing C666-1 cells were assessed by the transwell migration assay. (C) Soft agar transformation and (D) MTS assays were performed to assess the effect of expressing a PG construct on the growth of pBabe and LMP1-expressing C666-1 cells, compared to the DsRed vector control.
In addition to cell migration, plakoglobin has also been linked with growth altering effects (31, 32). To determine if the restoration of plakoglobin in LMP1-expressing cells affects other growth properties, soft agar transformation and MTS assays were performed. Changes in growth were not detected when plakoglobin was restored in LMP1-expressing cells (Fig. 6C and 6D), in that the number and size of colonies in soft agar and the rate of metabolism were comparable to LMP1-expressing cells that have lost plakoglobin expression. This data indicate that in C666-1 cells, LMP1-induced loss of plakoglobin contributes to migration but is not required for the growth promoting properties associated with LMP1 function.
Discussion
This study reveals that LMP1 contributes to the transformation of NPC through activation of PI3K/Akt and IκBα-dependent canonical NFκB signaling. Both signaling pathways were required for transformation and in agreement, both CTAR1 and CTAR2 domains were required for enhanced growth and migration through CTAR1-mediated activation of PI3K/Akt and the activation of canonical NFκB signaling primarily from CTAR2. The activation of these pathways have also been previously shown to be required for LMP1-mediated transformation in rodent and human fibroblasts and for transformation of B lymphocytes by EBV (3, 27, 37). The data presented here also indicates that the loss of plakoglobin is an additional target of LMP1 that contributes to LMP1-induced migration.
PI3K activation by LMP1 was required for the induction of growth in soft agar and enhanced migration of C666-1 cells. PI3K is involved in the activation of multiple pathways, including Akt and the MAPK (ERK). It has been recently shown that ERK activation is required for LMP1-mediated transformation of rodent fibroblasts and was specifically activated by LMP1-CTAR1 (7, 17). The activation of PI3K by LMP1 also requires the TRAF binding site in CTAR1 (3, 7), suggesting that the requirement for PI3K activation in transformation may be due to Akt activation and/or ERK activation. However, a specific inhibitor of Akt blocked LMP1-induced growth of C666 cells in soft agar and migration while a MEK1/2 inhibitor (U0126) did not affect LMP1-induced growth and migration (data not shown). In addition, increased levels of activated phospho-ERK above pBabe basal levels were not detected in cells expressing full-length LMP1 (data not shown). These findings suggest that the requirement of PI3K signaling in LMP1-mediated transformation in C666-1 cells is due to Akt activation and not ERK induction. Although ERK has been previously shown to be important in LMP1-mediated transformation, it is possible that in the C666-1 carcinoma cells, the stable expression of EBV latent genes including low levels of LMP1, induces sufficient amounts of ERK activation such that only additional activation of Akt is required for the enhanced growth properties mediated by LMP1.
LMP2A also activates PI3K/Akt signaling and this activation is required for its ability to transform certain epithelial cell lines and inhibit keratinocyte differentiation (19, 24, 25). In C666-1 cells, LMP2A induced activation of Akt but did not induce any changes in growth or migration (Fig. 3A and 4A). Constitutively active myristylated-Akt was also insufficient to enhance growth or migration (Fig. 3C, 4D and 5D). LMP2A did not affect IκBα levels indicating that in C666-1 cells, LMP2A over-expression does not activate canonical NFκB signaling at the level of IκBα degradation. These findings further support the requirement for activation of both NFκB and Akt for enhanced growth and migration in the already transformed C666-1 cells. LMP2A-induced migration has been attributed to the activation of ERK and Syk tyrosine kinase signaling, and in primary epithelial cells via the induction of integrin-α6 (33–35, 38). The lack of an effect in migration by over-expressing LMP2A in C666-1 cells may be due to differences in cell lines or indicate that over-expression of LMP2A in C666-1 cells does not further activate signals involved in migration above those induced by basal levels of LMP2A expression.
A critical property to malignant transformation, in addition to growth induction, is the enhancement of migration leading to metastasis, a characteristic often associated with NPC (10). In epithelial cells, LMP1 disrupts cellular adhesion through down-regulation of the intercellular adhesion protein E-cadherin and up-regulation of proteins involved in the disruption of the extracellular matrix, including MMP9 and MUC1 (11, 12). Recently mapping studies have identified the TRAF-binding domain of LMP1 in the down-regulation of plakoglobin, but the functional consequences of this down-regulation were not determined (7). The data presented here indicate that loss of plakoglobin contributes to LMP1-induced migration but is not required for LMP1-enhanced growth. It is noteworthy, that restoration of plakoglobin in LMP1 over-expressing cells was not able to completely inhibit the enhanced migration to pBabe control levels. This finding suggests that other pathways affected by LMP1 contribute to enhanced migration. The inhibition of migration in response to LY294002, Akt inhibitor I and BAY11-7085 would suggest that these additional factors are controlled by PI3K/Akt and NFκB signaling.
Plakoglobin belongs to the armadillo family of proteins and is highly homologous to β-catenin (39–41). Both plakoglobin and β-catenin serve dual roles, as adhesive proteins that mediate intercellular junctions and as transcriptional regulators of T-cell factor (Tcf)/lymphoid enhancer factor (Lef)-responsive genes such as c-myc and cyclin D1 (31). Despite their similarities, they are functionally distinct such that plakoglobin and β-catenin knockout mice are both embryonic lethal and thus cannot compensate for the loss of the other (39). It is believed that both the adhesive and signaling properties of plakoglobin are important in the development of cancer (31, 41), although whether plakoglobin plays a tumor suppressive or promoting role appears to be cell type dependent. Inhibition of growth in lung cancer cells results from plakoglobin blocking β-catenin mediated Tcf/Lef activity, possibly by sequestering TCF away from the formation of a functional β-catenin/Tcf complex (42). It is currently unknown whether LMP1-induced loss of plakoglobin enhances migration through disruption of cellular junctions, or through regulation of Tcf/Lef responsive genes. Interestingly, LMP1 also stabilizes β-catenin (43) and in C666-1 cells, β-catenin is increased (data not shown) while plakoglobin is decreased. It is possible that the combined effects of plakoglobin and β-catenin may result in enhanced Tcf/Lef regulated transcription. Although loss of plakoglobin has not been documented in EBV-related carcinomas, an accumulation of nuclear β-catenin is found in NPC and this is thought to be partly mediated through the inactivation of GSK3β by LMP2A (44).
Plakoglobin is post-translationally modified by a variety of mechanisms, including phosphorylation by EGFR, Src, Fer, Fyn, GSK3β and is subject to stabilization by O-glycosylation (41). Tyrosine phosphorylation of plakoglobin by EGFR, Fer, Fyn and GSK3β are destabilizing while phosphorylation by Src stabilizes complexes with desmosomal proteins. In C666-1 cells over-expressing LMP1, AG1478 an inhibitor of EGFR did not affect transwell migration (data not shown), suggesting that the down-regulation of plakoglobin by LMP1 is not mediated through EGFR-induced degradation. Since LMP1 induces phosphorylation and inactivation of GSK3β, it is also unlikely that GSK3β is responsible for the effects of LMP1 on plakoglobin. It is presently unknown whether the effects of LMP1 on plakoglobin are due to transcriptional or post-translational mechanisms.
In summary, this study shows that PI3K/Akt and IκBα-dependent NFκB signaling are both required for LMP1-induced growth and migration in an EBV-positive NPC cell line. Interestingly, the data also indicate that the effect of LMP1 on plakoglobin levels contributes to LMP1-induced migration. These findings indicate that promotion of oncogenesis in cells that express LMP1 not only require activation of signaling pathways but also result from effects on the expression levels of specific cellular genes. The effects of LMP1 on these adhesion proteins may provide novel targets to inhibit metastasis of NPC.
Materials and Methods
Cell culture and constructs
C666-1 cells were cultured on fibronectin-coated plates in RPMI-1640 media supplemented with heat-inactivated 10% fetal bovine serum and antibiotic/antimycotic (GIBCO). Plates were pre-coated with 10 μg/ml fibronectin (Sigma) overnight at 4°C. Stable cell lines expressing LMP1, CTAR1, CTAR2, LMP2A, myr-Akt from the retroviral vector pBabe were established by transduction and selection with 5 μg/ml puromycin (Sigma). LMP1, CTAR1, CTAR2 and LMP2A constructs in the pBabe vector have been described previously (3, 18). The myr-Akt was sub-cloned from pCMV6-myrAkt (a kind gift from CJ Der) into pBabe. The IκBα super-repressor (IκBαSS32/36AA) has been previously described (14) and was sub-cloned from pcDNA3 into pHSCG. Cells expressing the IκBα super-repressor or the pHSCG retroviral vector control were established by transduction and selected for GFP fluorescence by flow cytometry. The myc-tagged full-length plakoglobin construct (p330) expressed from the LK444 mammalian expression vector was a kind gift from Kathleen Green. Stable cell lines expressing plakoglobin were established by transfection with Lipofectamine 2000 (Invitrogen) and selection with 0.5 mg/ml G418 (GIBCO). The neomycin resistant plasmid DsRed (Clontech) was used as a control for G418 selection.
Retrovirus transduction
Retrovirus was produced in the 293T packaging cell line by co-transfecting plasmids expressing the protein of interest from a retroviral vector (pBabe or pHSCG), the VSVG envelope protein and Gag/Pol, using FuGENE6 transfection reagent (Roche). Retrovirus was harvested from clarified supernatant 48hrs post-transfection and C666-1 cells were transduced overnight in the presence of 4 μg/ml polybrene.
Immunoblot analysis
Preparation of whole cell lysates have been described previously (3). Protein concentrations were determined with the Bio-Rad DC protein assay system (Bio-Rad). Lysates were separated by denaturing SDS-PAGE and transferred to 0.45μm Optitran nitrocellulose membrane (Schleicher & Schuell). Membranes were immunoblotted with the appropriate primary antibody followed by horseradish peroxidase-tagged secondary antibodies (Amersham Biosciences and Dako) and detected with the SuperSignal West Pico System (Pierce).
Antibodies
Rabbit anti-pAkt (Ser473); anti-pGSK3α/β (Ser21/9) and anti-Akt were purchased from Cell Signaling. Rabbit anti- IκBα (clone C-21), rabbit anti-myc (clone A14), goat anti-β actin (clone I-19) and rabbit anti-GAPDH (clone FL-335) were purchased from Santa Cruz. Mouse IgG1κ anti-GSK3 was purchased from Upstate Biotechnology. Goat anti-plakoglobin was purchased from Abcam. Mouse anti-HA was purchased from Covance. Mouse anti-LMP1 (clones CS1-4) was purchased from DakoCytomation. Rat anti-LMP2 (clone 14B7) was purchased from Ascenion.
MTS assay
MTS cytotoxicity/proliferation assays were performed using the CellTiter 96 aqueous one-solution cell proliferation assay (Promega), according to manufacturer’s instructions. Cells were seeded in triplicate in a 96 well plate at 2.5 × 104 cells/ml, 100μl per well. MTS reagent was added on days 1–4 for 4hrs and absorbance was read at 490 nm, values plotted were subtracted from blanks.
Soft agar colony assay
Soft agar assays were carried out as previously described (19). Briefly, 2 × 105 cells suspended in a semi-solid Bacto agar media (0.5% Bacto agar in culture medium) were seeded on top of a Bacto agar media underlay to prevent attachment of cells to the culture plate. Suspended cells were overlaid with liquid media and where appropriate, grown in the presence of LY294002 (25 μM), Akt inhibitor I (20μM), BAY 11-7085 (10 μM) (Calbiochem) or DMSO. Cells were imaged by phase-contrast microscopy at days 4–6.
Transwell migration assay
BD Biocoat 8 μm pore size control cell culture inserts (BD Biosciences) were pre-coated with 20 μg/ml fibronectin, and 2 × 105 cells were seeded in the upper chamber in starvation medium (1% FBS). The bottom chamber consisted of starvation medium and 50 μg/ml fibronectin as chemoattractant. Where indicated, chemical inhibitors were added to both the top and bottom chambers. Following an overnight incubation, cells that had migrated to the lower side of the membrane were fixed in methanol and stained with DAPI (Molecular Probes). DAPI stained cells were visualized with fluorescence microscopy and the average number of migrated cells was calculated from eight representative fields in duplicate inserts using Image J software.
Acknowledgments
We thank Shannon Kenney for the C666-1 cells, Kathleen Green for the plakoglobin construct and Channing Der for the pCMV6-myrAkt construct. We thank Cydnie Bedford for the cloning of the pHSCG-IκBαSS32/36AA construct. We also thank Pat Kung, and Cathy Siler for critical reading of this manuscript. This work was supported by National Institutes of Health Grant CA 32979 and CA103634 (to NR-T).
References
- 1.Raab-Traub N. Pathogenesis of Epstein-Barr virus and its associated malignancies. Semin in virology. 1996;7:315–23. [Google Scholar]
- 2.Rickinson A, Kieff E. Epstein-Barr Virus and Its Replication. In: Knipe ID, editor. Field's Virology. 4. Philadelphia, PA: Lippincott Williams & Wilkins Publishers; 2001. pp. 2511–73. [Google Scholar]
- 3.Mainou BA, Everly DN, Jr, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene. 2005;24:6917–24. doi: 10.1038/sj.onc.1208846. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–40. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 5.Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V, Raab-Traub N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11963–8. doi: 10.1073/pnas.95.20.11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson JB, Weinberg W, Johnson R, Yuspa S, Levine AJ. Expression of the BNLF-1 oncogene of Epstein-Barr virus in the skin of transgenic mice induces hyperplasia and aberrant expression of keratin 6. Cell. 1990;61:1315–27. doi: 10.1016/0092-8674(90)90695-b. [DOI] [PubMed] [Google Scholar]
- 7.Mainou BA, Everly DN, Jr, Raab-Traub N. Unique signaling properties of CTAR1 in LMP1-mediated transformation. J Virol. 2007;81:9680–92. doi: 10.1128/JVI.01001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paine E, Scheinman RI, Baldwin AS, Jr, Raab-Traub N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-kappa B/Rel family proteins. J Virol. 1995;69:4572–6. doi: 10.1128/jvi.69.7.4572-4576.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Seminars in cancer biology. 2001;11:435–44. doi: 10.1006/scbi.2001.0410. [DOI] [PubMed] [Google Scholar]
- 10.Raab-Traub N. Epstein-Barr virus in the pathogenesis of NPC. Seminars in cancer biology. 2002;12:431–41. doi: 10.1016/s1044579x0200086x. [DOI] [PubMed] [Google Scholar]
- 11.Kondo S, Yoshizaki T, Wakisaka N, et al. MUC1 induced by Epstein-Barr virus latent membrane protein 1 causes dissociation of the cell-matrix interaction and cellular invasiveness via STAT signaling. Journal of virology. 2007;81:1554–62. doi: 10.1128/JVI.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsao SW, Tramoutanis G, Dawson CW, Lo AK, Huang DP. The significance of LMP1 expression in nasopharyngeal carcinoma. Seminars in cancer biology. 2002;12:473–87. doi: 10.1016/s1044579x02000901. [DOI] [PubMed] [Google Scholar]
- 13.Huen DS, Henderson SA, Croom-Carter D, Rowe M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;10:549–60. [PubMed] [Google Scholar]
- 14.Miller WE, Cheshire JL, Raab-Traub N. Interaction of tumor necrosis factor receptor-associated factor signaling proteins with the latent membrane protein 1 PXQXT motif is essential for induction of epidermal growth factor receptor expression. Mol Cell Biol. 1998;18:2835–44. doi: 10.1128/mcb.18.5.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thornburg NJ, Pathmanathan R, Raab-Traub N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer research. 2003;63:8293–301. [PubMed] [Google Scholar]
- 16.Thornburg NJ, Raab-Traub N. Induction of epidermal growth factor receptor expression by Epstein-Barr virus latent membrane protein 1 C-terminal-activating region 1 is mediated by NF-kappaB p50 homodimer/Bcl-3 complexes. Journal of virology. 2007;81:12954–61. doi: 10.1128/JVI.01601-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson CW, Laverick L, Morris MA, Tramoutanis G, Young LS. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. Journal of virology. 2008 doi: 10.1128/JVI.01888-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison JA, Klingelhutz AJ, Raab-Traub N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J Virol. 2003;77:12276–84. doi: 10.1128/JVI.77.22.12276-12284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholle F, Bendt KM, Raab-Traub N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol. 2000;74:10681–9. doi: 10.1128/jvi.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swart R, Ruf IK, Sample J, Longnecker R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. Journal of virology. 2000;74:10838–45. doi: 10.1128/jvi.74.22.10838-10845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung ST, Huang DP, Hui AB, et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int J Cancer. 1999;83:121–6. doi: 10.1002/(sici)1097-0215(19990924)83:1<121::aid-ijc21>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 22.Mei YP, Zhou JM, Wang Y, et al. Silencing of LMP1 induces cell cycle arrest and enhances chemosensitivity through inhibition of AKT signaling pathway in EBV-positive nasopharyngeal carcinoma cells. Cell Cycle. 2007;6:1379–85. doi: 10.4161/cc.6.11.4274. [DOI] [PubMed] [Google Scholar]
- 23.Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem. 2003;278:3694–704. doi: 10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- 24.Morrison JA, Raab-Traub N. Roles of the ITAM and PY motifs of Epstein-Barr virus latent membrane protein 2A in the inhibition of epithelial cell differentiation and activation of {beta}-catenin signaling. J Virol. 2005;79:2375–82. doi: 10.1128/JVI.79.4.2375-2382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. Journal of virology. 2007;81:9299–306. doi: 10.1128/JVI.00537-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6055–60. doi: 10.1073/pnas.100119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shair KH, Bendt KM, Edwards RH, Bedford EC, Nielsen JN, Raab-Traub N. EBV latent membrane protein 1 activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS pathogens. 2007;3:e166. doi: 10.1371/journal.ppat.0030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller WE, Mosialos G, Kieff E, Raab-Traub N. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. Journal of virology. 1997;71:586–94. doi: 10.1128/jvi.71.1.586-594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkinson PG, Coope HJ, Rowe M, Ley SC. Latent membrane protein 1 of Epstein-Barr virus stimulates processing of NF-kappa B2 p100 to p52. The Journal of biological chemistry. 2003;278:51134–42. doi: 10.1074/jbc.M304771200. [DOI] [PubMed] [Google Scholar]
- 30.Saito N, Courtois G, Chiba A, et al. Two carboxyl-terminal activation regions of Epstein-Barr virus latent membrane protein 1 activate NF-kappaB through distinct signaling pathways in fibroblast cell lines. The Journal of biological chemistry. 2003;278:46565–75. doi: 10.1074/jbc.M302549200. [DOI] [PubMed] [Google Scholar]
- 31.Chidgey M, Dawson C. Desmosomes: a role in cancer? British journal of cancer. 2007;96:1783–7. doi: 10.1038/sj.bjc.6603808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin T, Getsios S, Caldelari R, et al. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5420–5. doi: 10.1073/pnas.0501676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen MD, Young LS, Dawson CW. The Epstein-Barr virus-encoded LMP2A and LMP2B proteins promote epithelial cell spreading and motility. Journal of virology. 2005;79:1789–802. doi: 10.1128/JVI.79.3.1789-1802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu J, Lin WH, Chen SY, et al. Syk tyrosine kinase mediates Epstein-Barr virus latent membrane protein 2A-induced cell migration in epithelial cells. The Journal of biological chemistry. 2006;281:8806–14. doi: 10.1074/jbc.M507305200. [DOI] [PubMed] [Google Scholar]
- 35.Pegtel DM, Subramanian A, Sheen TS, Tsai CH, Golub TR, Thorley-Lawson DA. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: possible role in nasopharyngeal carcinoma metastasis. Journal of virology. 2005;79:15430–42. doi: 10.1128/JVI.79.24.15430-15442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fahraeus R, Chen W, Trivedi P, Klein G, Obrink B. Decreased expression of E-cadherin and increased invasive capacity in EBV-LMP-transfected human epithelial and murine adenocarcinoma cells. International journal of cancer. 1992;52:834–8. doi: 10.1002/ijc.2910520527. [DOI] [PubMed] [Google Scholar]
- 37.Cahir McFarland ED, Izumi KM, Mosialos G. Epstein-barr virus transformation: involvement of latent membrane protein 1-mediated activation of NF-kappaB. Oncogene. 1999;18:6959–64. doi: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 38.Chen SY, Lu J, Shih YC, Tsai CH. Epstein-Barr virus latent membrane protein 2A regulates c-Jun protein through extracellular signal-regulated kinase. Journal of virology. 2002;76:9556–61. doi: 10.1128/JVI.76.18.9556-9561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bierkamp C, Schwarz H, Huber O, Kemler R. Desmosomal localization of beta-catenin in the skin of plakoglobin null-mutant mice. Development (Cambridge, England) 1999;126:371–81. doi: 10.1242/dev.126.2.371. [DOI] [PubMed] [Google Scholar]
- 40.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development (Cambridge, England) 1995;121:3529–37. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 41.Yin T, Green KJ. Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol. 2004;15:665–77. doi: 10.1016/j.semcdb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Miravet S, Piedra J, Miro F, Itarte E, Garcia de Herreros A, Dunach M. The transcriptional factor Tcf-4 contains different binding sites for beta-catenin and plakoglobin. The Journal of biological chemistry. 2002;277:1884–91. doi: 10.1074/jbc.M110248200. [DOI] [PubMed] [Google Scholar]
- 43.Everly DN, Jr, Kusano S, Raab-Traub N. Accumulation of cytoplasmic beta-catenin and nuclear glycogen synthase kinase 3beta in Epstein-Barr virus-infected cells. Journal of virology. 2004;78:11648–55. doi: 10.1128/JVI.78.21.11648-11655.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morrison JA, Gulley ML, Pathmanathan R, Raab-Traub N. Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer research. 2004;64:5251–60. doi: 10.1158/0008-5472.CAN-04-0538. [DOI] [PubMed] [Google Scholar]