

Abstract
Lipopolysaccharide (LPS) binding protein (LBP) plays an important role in regulating leukocyte responses to LPS. Remarkably, it may either augment these responses at low LBP concentrations or inhibit them at high concentrations. We previously reported that native high-density lipoprotein (HDL) augments human monocyte responses to LPS by suppressing the inhibitory activity of high concentrations of LBP, a process that occurs before HDL can inhibit the response by subsequently binding and neutralizing LPS. We now show that this novel activity is conferred largely by an HDL component protein, apolipoprotein (apo)A-II. Purified apoA-II was highly active in our assays. We also found that HDL from apoA-II-deficient mice was almost completely inactive, whereas the activities of HDLs that lacked apoA-I, apoC-I, apoE, or apoC-III were similar to that of wild-type HDL. Decreased activity was also observed in rabbit HDL, which is naturally deficient in apoA-II. Incorporating human apoA-II into rabbit HDL increased its activity to levels found in human HDL. Our investigation of the mechanism of apoA-II activity revealed that LBP promoted the formation of large LPS aggregates with low bioactivity and that apoA-II inhibited the formation of these aggregates without binding and directly inhibiting LPS bioactivity. Our results suggest a novel pro-inflammatory activity of apoA-II that may help maintain sensitive host responses to LPS by suppressing LBP-mediated inhibition. Our findings also raise the possibility that the decline of plasma apoA-II during sepsis may help control the response to LPS.
Keywords: LBP, endotoxin, HDL, apoA-II, LPS
Introduction
Recognition of lipopolysaccharide (endotoxin or LPS) by MD-2-TLR41,2 is an important determinant of the innate immune response to Gram-negative bacteria. Sensitive host responses to LPS are orchestrated by several LPS binding proteins. LBP,3-5 a secreted protein found in the plasma and extravascular fluids, transfers LPS to CD14,6 an LPS binding receptor that exists in soluble or membrane-bound forms. CD14 greatly increases host cell sensitivity to LPS by transferring LPS to the MD-2-TLR4 receptor complex. Sentinel responses to LPS often confer resistance to infection, whereas the response to LPS during uncontrolled infection contributes to the pathophysiology of severe sepsis and septic shock. Therefore, animal hosts have evolved numerous mechanisms for regulating the response to LPS. LBP can enhance or suppress the host response to LPS by mechanisms that depend on the LBP concentration.7 Its ability to enhance the response to LPS may account for the impaired resistance to infection in LBP-deficient mice.4,8-11 Optimal CD14-mediated cell stimulation by LPS occurs at LBP concentrations that are considerably lower than those of normal human plasma.7,12 High acute-phase concentrations, such as those found in septic patients, can strongly inhibit cell responses to LPS.13 Intraperitoneal LBP administration can rescue animals from the toxic effects of LPS or Gram-negative bacteria.7 Thus, LBP may help to control the response to LPS when LBP levels rise in plasma13 and extravascular fluids9,14-16 during Gram-negative sepsis. The inhibitory activity of LBP may be explained, in part, by its ability to shuttle LPS into plasma lipoproteins,17,18 which can neutralize LPS bioactivity. Other inhibitory mechanisms of LBP are indicated by its ability to inhibit LPS in the absence of plasma. These mechanisms may involve the ability of LPS to form complexes with LBP that are internalized by membrane-bound CD14 (mCD14) but contribute little to signal transduction19 and the ability of LBP to remove LPS that has already bound to mCD14 on cell surfaces.20
We previously reported that a protein component of native HDL (nHDL) augments cell responses to LPS by suppressing the inhibitory activity of LBP.12 Although this finding appears to contradict a widely-accepted view that nHDL and other plasma lipoproteins provide a host mechanism for controlling responses to LPS,21 this apparent discrepancy can be explained by the ability of nHDL to differentially affect early and late (or prolonged) responses to LPS (discussed by Thompson and Kitchens12). We found that, in the absence of nHDL, LBP inhibits the response to LPS at LBP concentrations that are equivalent to those in normal plasma (∼3 μg/ml), whereas nHDL restores LPS bioactivity under these conditions. Therefore, we hypothesized that nHDL helps maintain sensitive host responses to LPS by suppressing LBP inhibitory activity.
In many septic patients, LBP levels increase dramatically, causing greater LPS inhibition,12,13 whereas circulating levels of HDL and its most abundant normal protein components, apolipoprotein (apo)A-I and apoA-II, decline sharply.12,22 The decline of HDL during acute inflammation and infection has been thought to impair the neutralization of circulating LPS. To the contrary, we found that LPS bound more rapidly to plasma lipoproteins in the serum of septic patients, even when HDL levels were low.23 Septic patients also have lower levels of apoC-I,12,22,24,25 a predominantly HDL-associated protein that can enhance in vivo responses to LPS.26 Thus, increased levels of LBP and decreased levels of nHDL during septic infection may help control the host response to LPS.
Our investigation of the protein components of nHDL revealed that apoA-II plays a major role in regulating the inhibitory activity of LBP in vitro. Here, we show direct evidence that LBP promotes the formation of large LPS aggregates that have little or no LPS bioactivity and that apoA-II suppresses this inhibitory activity of LBP by its unique ability to prevent LPS aggregation without directly inhibiting LPS bioactivity.
Materials and Methods
Cells
THP-1 cells (a human monocyte cell line) were cultured in 1,25-dihydroxyvitamin D3 (BioMol, Plymouth Meeting, PA, USA) for 4 days to induce mature monocyte characteristics.20 The cells (5 × 105 cells/50 μl) were stimulated with LPS (usually 20 pg/ml) in suspension at 37°C for 4 h in serum-free medium (SFM) containing RPMI-1640, 20 mM HEPES buffer pH 7.4 and 0.1 mg/ml bovine serum albumin (BSA) as previously described.12 LPS, LBP, nHDL or apolipoproteins were added to the cells simultaneously. Tumor necrosis factor (TNF) was measured in cell-free supernatants by ELISA (BD Pharmingen, San Jose, CA, USA). To compensate for day-to-day variations in TNF production among groups of more than one experiment, the data from separate experiments were normalized to the average positive control value and expressed as mean ± SD.
High-density lipoproteins
nHDL was isolated from human, murine, or rabbit plasma containing EDTA by ultracentrifugal flotation in KBr (1.21 > d > 1.063) as previously described.27 Murine HDLs were concentrated on Centricon YM-100 membranes (Millipore, St Louis, MO, USA) without significant loss of protein. The protein content of the HDL preparations was measured by the Pierce BCA assay (Pierce Chemical Co., Rockford, IL, USA) using BSA as a standard. To analyze HDL proteins, samples were heated to 100°C for 5 min in SDS sample buffer with or without dithiothreitol (DTT), and the samples were run on 4-20% SDS-PAGE gradient gels and stained with Coomassie Blue. Human plasma was obtained by informed consent from healthy volunteers according to protocols approved by the Institutional Review Board (UT Southwestern Medical Center). Wild-type mice, ApoC-I knock-out mice, ApoE knock-out mice, and ApoC-I/C-III/E triple knock-out mice (all C57BL/6 background) were from the breeding facility of Leiden University Medical Center. ApoA-I knock-out mice were kindly provided by Dr Philip Shaul (UT Southwestern Medical Center). ApoA-II knock-out mice28 (stock #006258) were from The Jackson Laboratory (Bar Harbor, ME, USA). The animal protocol was approved by the Institutional Animal Care and Use Committee (UT Southwestern Medical Center) and by the Institutional Ethics Committee for Animal Care and Experimentation (Leiden University Medical Center). Freshly prepared normal rabbit plasma was obtained from Pel-Freez (Rogers, AR, USA).
Reagents
Purified human apolipoproteins A-II, C-I, and E were from BioDesign International (Saco, ME, USA) and apoA-I was a gift of Dr Peter Lerch (Swiss Red Cross, Bern, Switzerland). Synthetic apoC-I was made by total peptide synthesis of the mature (57 amino acids) protein (Protein Chemistry Technology Center, UT Southwestern Medical Center, Dallas, TX, USA). Recombinant human LBP was produced by baculovirus expression20 and ran as a single band on Coomassie Blue-stained SDS-PAGE gels. LPS from Escherichia coli O14 and biosynthetically labeled [3H]-LPS (1.5 × 106 dpm/μg)29 from E. coli LCD25 were provided by Dr Robert Munford. The [3H]-LPS is an Ra chemotype LPS with a molecular mass of ∼5000 Da. Other reagents were from Sigma-Aldrich (St Louis, MO, USA) unless otherwise specified.
Western blotting and fluorography
Samples containing purified apolipoproteins, LBP, and [3H]-LPS were mixed with Tris-glycine sample buffer without SDS or DTT and were run on 10% native PAGE gels and transferred to Immobilon-P membranes. Immunoblot detection antibodies were goat anti-human apoA-II-biotin (#12B-G1a), goat anti-human apoA-I-biotin (#11B-G2b), goat anti-human apoC-I-biotin (#31B-G1a), all from Academy Biomedical (Houston, TX, USA), and goat anti-human apoE-biotin (#K74180B) from BioDesign (Saco, ME, USA). Rabbit anti-human LBP (#K1970602) was a gift of Dr Stephen Carroll (XOMA, Berkeley, CA, USA). Detection was by streptavidin-peroxidase or donkey anti-rabbit IgG-peroxidase followed by Enhanced Chemiluminescence on Hyperfilm ECL (Amersham Biosciences, Pittsburg, PA, USA). To detect [3H]-LPS, the gels were fixed, incubated with Amplify Fluorographic Reagent (Amersham Biosciences, Piscataway, NJ, USA), dried, and exposed to film according to the manufacturer’s instructions. [3H]-LPS was also detected on Immobilon-P membranes by soaking the membrane in Amplify Reagent and exposing the wet, plastic-wrapped membrane to film. To measure the bioactivity of LPS in different regions of the native gel, the corresponding gel piece was cut out of an unfixed gel and crushed with a Teflon pestle in a micro-centrifuge tube containing gel running buffer. The crushed gel was centrifuged, and the eluted [3H]-LPS was measured in the supernatant by liquid scintillation counting. The LPS concentration was determined by the known specific radioactivity of the [3H]-LPS preparation. The LPS was then diluted in SFM, and its bioactivity was measured by the THP-1 cell response assay.
Statistical analysis
The data were analyzed using Prism v.5.0 software from GraphPad (San Diego, CA, USA). Significant differences (P < 0.05) from controls were determined by two-tailed P-values of the unpaired t-test to compare means.
Results
ApoA-II prevents LBP-mediated inhibition of LPS bioactivity
We previously showed12 that LBP augments the response to LPS maximally at ∼10-3 μg LBP/ml in serum-free conditions in the absence of HDL in both THP-1 cells and normal human monocytes. Higher concentrations of LBP increasingly inhibit the response to LPS. As shown in a representative experiment (Fig. 1A), HDL augments the response to LPS only when LBP is inhibitory (see Thompson and Kitchens12 for detailed data and statistics). Similar results were obtained when IL-8 and IL-1β were used as response readouts.12 In the experiments shown in Figure 1B, we tested the abilities of purified apolipoproteins to augment responses that were inhibited by LBP. The proteins tested are normally associated with HDL. ApoA-I and apoA-II constitute 65% and 20%, respectively, of the total HDL protein, and apoA-I,30-32 apoE,33 and apoC-I26 are known to interact with LPS. As shown in Figure 1B, apoA-II was the most potent protein tested. The activity of 100 μg/ml of apoA-II exceeded that of 200 μg/ml of nHDL, whereas apoA-I was relatively weak, and little or no activity was observed using apoE and apoC-I. Human apoA-II normally exists as a homodimer;34 in experiments not shown, we found that the activity of monomeric apoA-II was similar to that of the dimeric protein. The protein preparations had no stimulatory activity in the absence of LPS (data not shown).
Fig. 1.
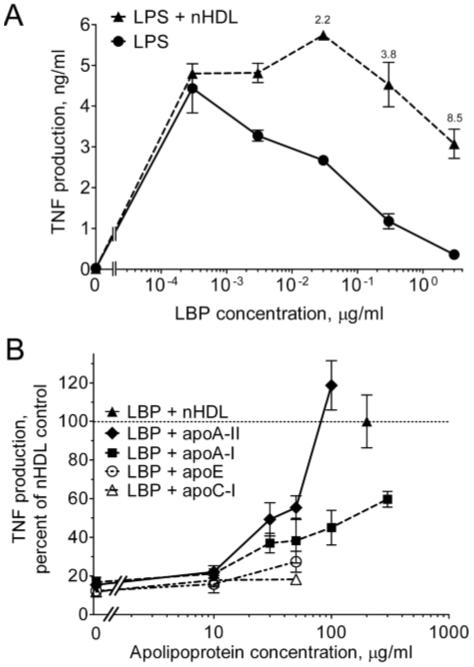
The apoA-II component of nHDL prevents the inhibition of LPS bioactivity by LBP. (A) THP-1 cells were incubated for 4 h at 37°C in the presence or absence of nHDL (200 μg protein/ml) and the indicated concentrations of LBP in SFM containing 20 pg/ml of LPS. TNF was measured in the culture supernatants by ELISA. Error bars denote mean ± range of duplicate determinations in a single experiment. TNF was undetectable in the absence of LPS and in the presence of nHDL alone (not shown). Annotations above the symbols denote the fold increase induced by nHDL. The experiment was repeated with similar results. (B) THP-1 cells were incubated with LPS in the presence of increasing concentrations of the indicated apolipoproteins or nHDL (200 μg/ml) in SFM containing an inhibitory concentration of LBP (1 μg/ml). The data are expressed as the percentage of the fold augmentation of TNF obtained with nHDL. Error bars are mean ± SEM of 3-5 experiments performed in duplicate. The dashed line (denoting 100%) indicates the activity of nHDL.
We next tested effects of nHDLs that lacked specific apolipoproteins. HDL was prepared from the plasma of gene knock-out mice that lacked apoA-I, apoA-II, apoC-I, or apoE, and from triple knock-out mice that lacked apoC-I, apoC-III, and apoE; the HDL preparations were tested for their ability to augment the cell response to LPS in the presence of an inhibitory concentration of LBP. As shown in Figure 2, the activity of apoA-II-deficient HDL was not significantly different from that of the control without HDL, whereas all of the other HDL preparations significantly increased the response to LPS. The activities of the HDLs from apoA-I, apoE, and apoC-I knock-out mice were not significantly different from that of wild-type HDL. HDL from apoC-I/C-III/E triple knock-out mice and human HDL were somewhat more active than the wild-type HDL. Similar results were obtained when the data were expressed as the fraction of the activity of wild-type HDL within each experiment (data not shown). The HDL preparations had no stimulatory activity in the absence of LPS (data not shown). The results suggest that the ability of nHDL to suppress LBP inhibitory activity is conferred principally by apoA-II and that apoA-I, apoC-I, apoE, and apoC-III do not make singularly important contributions.
Fig. 2.
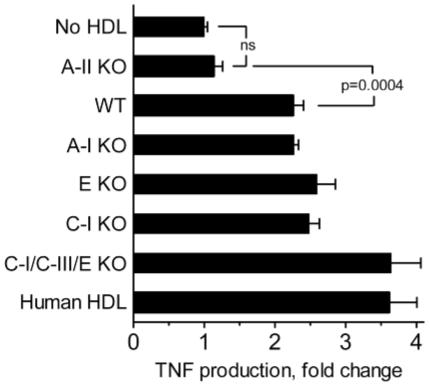
The activity of nHDL requires apoA-II but not apoA-I, apoC-I, apoC-III, or apoE. THP-1 cells were stimulated with LPS as described in the caption to Figure 1 in the presence of an inhibitory concentration of LBP and 100 μg/ml of nHDL isolated from humans, wild-type mice, or apolipoprotein gene knock-out mice as indicated. TNF production is expressed as the fold change from the control (no HDL). Error bars denote mean ± SEM of duplicate determinations in 2-9 experiments (n = 4-18). All HDLs were significantly different from the control (P < 0.0001) except for apoA-II knock-out HDL (P = 0.2). Wild-type HDL was significantly different from apoA-II knock-out HDL (P = 0.0004), apoC-I/C-III/E knock-out HDL (P = 0.0013), and human HDL (P = 0.0019).
To obtain a more abundant source of apoA-II-deficient HDL for apoA-II incorporation experiments, we turned to rabbit HDL, which contains little or no apoA-II due to low expression of the apoA2 gene.35 Although rabbit HDL showed significant activity in our assay compared to the control without HDL (Fig. 3), the activity of rabbit HDL was significantly lower than that of human HDL (Fig. 3). Analysis of HDL proteins on SDS-PAGE gels showed that apoA-II was undetectable in the rabbit HDL (Fig. 3B). The incorporations were performed by incubating human apoA-II with rabbit plasma followed by HDL isolation from the plasma mixtures. The apoA-II content of the resulting HDL (Fig. 3, lanes 3 and 4) were somewhat higher and lower, respectively, than that of human HDL (Fig. 3, lane 1). Incorporation of purified human apoA-II into the rabbit HDL restored the activity to the same level as that of human HDL. In contrast, the incorporation of apoE (Fig. 3, lane 5) had no effect on HDL activity in these assays. The results support the hypothesis that apoA-II plays a key role in augmenting the cell response to LPS by suppressing the inhibitory activity of LBP.
Fig. 3.
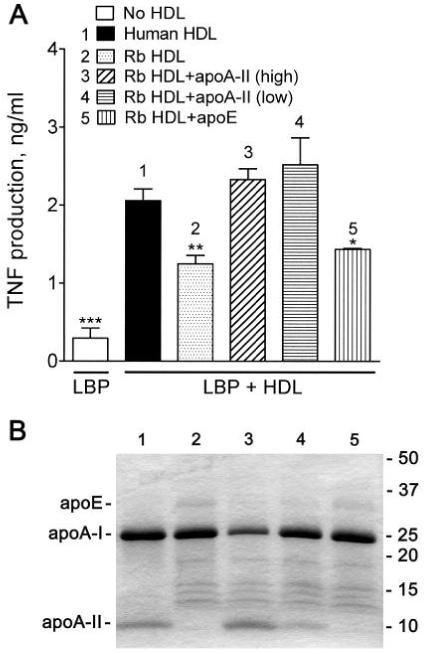
Incorporation of apoA-II restores normal activity to apoA-II-poor nHDL. (A) Rabbit plasma, which contains very low concentrations of apoA-II, was incubated at 37°C for 30 min with 400 μg/ml (3) of purified human apoA-II, the normal apoA-II concentration of human plasma, or with 133 μg apoA-II/ml (4) to allow apoA-II incorporation into HDL; human apoE (5) was used as a control at 50 μg/ml, the normal concentration of apoE in human plasma. The HDL was then isolated by ultracentrifugal flotation. THP-1 cells were stimulated with LPS as described in the caption to Figure 1 in the presence of an inhibitory concentration of LBP (1 μg/ml) and HDL (200 μg protein/ml) isolated from control human (1) or rabbit (2) plasma or HDL from rabbit plasma (3-5) containing the indicated apolipoproteins. Error bars are mean ± SEM of four determinations in two incorporation experiments. Significant differences from the activity of human HDL are denoted * P = 0.0163, ** P = 0.0043, *** P = 0.0001. (B) The above HDL preparations (5 μg protein) were separated on 4-20% SDS-PAGE gradient gels (reducing conditions), and the proteins were stained with Coomassie Blue. The lane numbers refer to the samples described in (A).
ApoA-II does not bind to LBP
To begin to understand the mechanism of apoA-II activity in our assays, we first looked for evidence of a protein-protein interaction between apoA-II and LBP. Mobility shift analysis of the proteins on native polyacrylamide gels did not reveal significant evidence of association between apoA-II and LBP. As shown in Figure 4, LBP migrated near the top of the native gels, whereas apoA-II migrated faster through the gel. Pre-incubation of equimolar mixtures of LBP and apoA-II in the presence or absence of LPS resulted in a shift of only a trace amount of the total apoA-II into the LBP region of the gel to a location that did not precisely correspond to the location of LBP. In other experiments (not shown), we were unable to demonstrate LBP binding to apoA-II on microtiter plates. The results suggest that apoA-II does not form complexes with LBP in the presence or absence of LPS; however, the possibility of a transient or low-affinity interaction cannot be excluded.
Fig. 4.
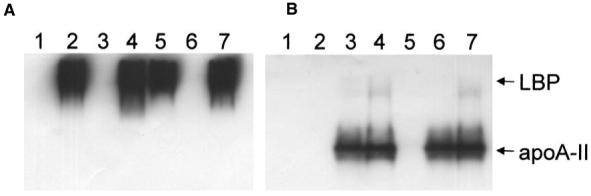
Apo-II does not co-migrate with LBP on native gels. Equimolar mixtures of LBP (1.2 μg), apoA-II (0.34 μg), and [3H]-LPS (0.1 μg) were incubated in 7.5 μl of SFM without BSA for 30 min at 37°C in the following combinations: (1) LPS, (2) LPS + LBP, (3) LPS + apoA-II, (4) LPS + LBP + apoA-II, (5) LBP, (6) apoA-II, and (7) LBP + apoA-II. The mixtures were then run on 10% native gels and subjected to Western blotting to detect LBP (A) and apoA-II (B).
ApoA-II prevents the formation of large inhibitory LPS-LBP complexes
We also explored the possibility that the activity of apoA-II involves its ability to alter the physical state of LPS. Mixtures of LPS and LBP can form LPS-LBP complexes of undetermined size that are internalized by mCD14 but have little ability to signal.19 To test the hypothesis that apoA-II prevents the formation of such LPS-LBP complexes, we analyzed LPS-LBP interactions using fluorographic detection of [3H]-LPS on native PAGE gels. LPS exists principally in aggregates, particularly in the presence of physiological culture medium (SFM), which contains divalent calcium ions. As shown in Figure 5A, [3H]-LPS ran at the top of the gel (lane 2), whereas the detergent effects of sodium deoxycholate (DOC) disaggregated the LPS36 and greatly increased its mobility through the gel (lane 1). Increasing concentrations of LBP further decreased the mobility of [3H]-LPS reflecting increased LPS-LBP complex sizes that ultimately became too large to enter the gel (Fig. 5A, lanes 2-5). ApoA-II inhibited the ability of LBP concentrations below 30 μg/ml to increase the LPS aggregate size (Fig. 5A, lanes 6-9). To confirm that LBP promoted the formation of complexes that were too large to enter the gel, we measured the recovery of [3H]-LPS from the stacking gel wells (W). As shown in Figure 5B, very little [3H]-LPS could be recovered from the gel well after applying LPS alone, whereas exposure to 3 μg/ml LBP increased the amount of recoverable [3H]-LPS by approximately 13-fold. The recovery of [3H]-LPS in the gel well was greatly decreased by adding apoA-II to the LPS and LBP mixture.
Fig. 5.
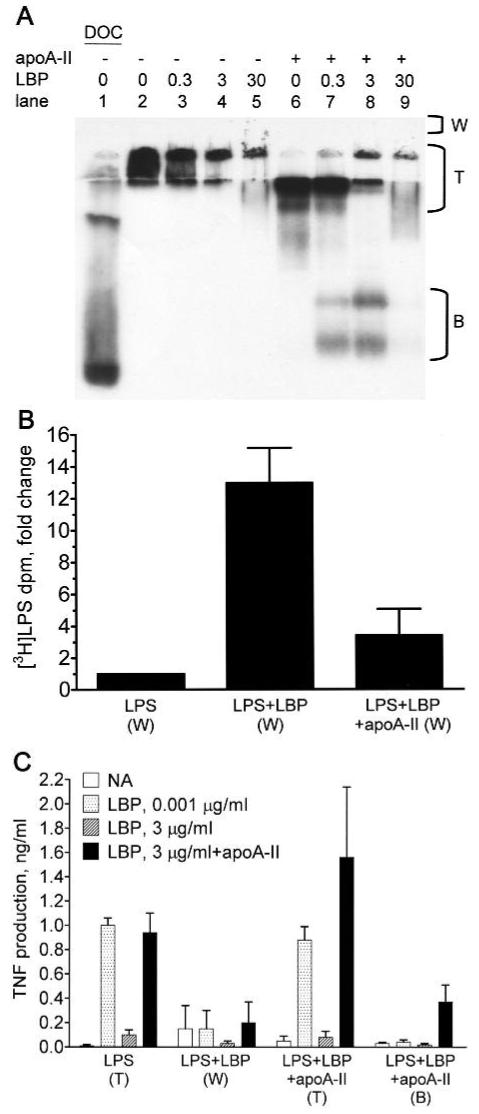
ApoA-II prevents the formation of large LPS-LBP complexes that inhibit LPS bioactivity. [3H]-LPS (0.1 μg) was incubated for 30 min at 37°C in 7.5 μl of SFM without BSA containing the indicated concentrations (μg/ml) of LBP in the presence or absence of 1.4 μg of apoA-II (apoA-II:LPS molar ratio = 4). (A) The mixtures were run on a 10% native gel and subjected to fluorography to detect the [3H]-LPS. Region (T) includes material near the top of the running gel and the stacking gel. (B) The indicated mixtures of [3H]-LPS alone (LPS), [3H]-LPS and LBP (3 μg/ml; LPS+LBP), and [3H]-LPS, LBP, and apoA-II (LPS+LBP+apoA-II) were run on separate gels as described above. Material that did not enter the gel was rinsed out of the wells above the stacking gel (region ‘W’ in panel A) and counted for recovery of [3H]-LPS. The recovered tritium dpm were 895 ± 236 (LPS), 11,951 ± 5296 (LPS+LBP), and 2980 ± 1287 (LPS+LBP+apoA-II; mean ± SD; n = 3 experiments). The data were normalized and expressed as the fold-change from LPS alone. Error bars denote mean ± SD. (C) To determine bioactivity of the LPS recovered from the indicated regions of gels, the LPS concentration was determined by counting tritium dpm, and 100 pg/ml of LPS was incubated with THP-1 cells in the presence of the indicated concentrations of LBP and apoA-II (100 μg/ml) as described in the caption to Figure 1. Error bars denote mean ± SD (n = 5-8) of duplicate determinations in 3 or 4 experiments.
To measure the bioactivity of the LPS applied to native gels, we measured the abilities of equal concentrations of [3H]-LPS recovered from the well and from various gel fractions to stimulate TNF production by THP-1 cells. In each experiment, the reference standard for bioactive LPS was material recovered from the region designated ‘T’ in Figure 5A, derived from samples containing LPS alone. As shown in Figure 5C, the bioactivity of LPS-LBP complexes that were too large to enter the gel (LPS+LBP [W]) was greatly reduced compared to that of the LPS that entered the gel (LPS [T]). The LPS+LBP mixtures that entered the gel were fully active when assayed in the presence of the low concentration of LBP (1.54 ± 0.28 ng/ml TNF [n = 4], not shown in panel C). These data support the hypothesis that LBP reduces LPS bioactivity by promoting the formation of large complexes that have reduced ability to trigger TLR4. The LPS from mixtures of LPS, LBP, and apoA-II that migrated near the top of the gel (LPS+LBP+apoA-II [T]) had essentially full bioactivity in the presence of 0.001 μg/ml LBP. Some of the LPS from these mixtures ran as smaller, high-mobility aggregates, which had reduced LPS bioactivity (LPS+LBP+apoA-II [B]). However, the bioactivity was partially restored by incubating these smaller LPS aggregates with apoA-II and LBP. Whereas a high concentration of LBP (3 μg/ml) in the activity assay inhibited the bioactivity of all LPS fractions, adding apoA-II to the LBP restored the activity in the LPS (T) fractions and enhanced the activity of the (LPS+LBP+apoA-II [T]) fractions by up to 2-fold. Attempts to restore the activity of the large LPS-LBP complexes (LPS+LBP [W]) with apoA-II and LBP resulted in low and variable activity. This may be explained by the results of experiments shown in Figure 6. When LPS was pre-incubated for 30 min with high concentrations of LBP, the low bioactivity of the LPS could not be restored by subsequently adding apoA-II. However, apoA-II could restore approximately half of the activity if added 5 min after LBP. Taken together, these results suggest that LBP inhibits LPS bioactivity by promoting the formation of large LPS aggregates; this process is inhibited by apoA-II, and LBP inhibition rapidly becomes irreversible unless apoA-II is present.
Fig. 6.
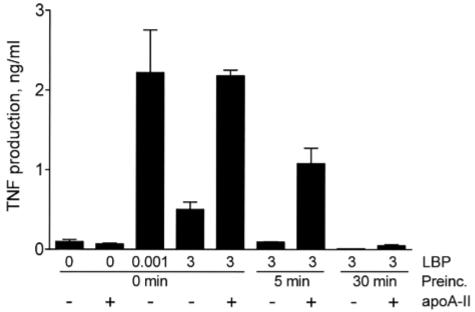
Rescue of LPS bioactivity by apoA-II is prevented by prolonged pre-incubation of LPS with LBP. LPS (20 pg/ml) was pre-incubated with PBS or 3 μg/ml of LBP at 37°C for the indicated times. The mixtures were then incubated with THP-1 cells in the presence or absence of apoA-II (100 μg/ml) to measure LPS bioactivity as described in the caption to Figure 1. Error bars denote mean ± SD (n = 4) of duplicate determinations in two experiments.
HDL apolipoproteins disaggregate LPS, whereas apoA-II fails to bind and inhibit LPS bioactivity
A common property of apolipoproteins is their ability to bind lipids, and the ability to interact with LPS is a known property of several apolipoproteins; apoA-I can bind and inactivate LPS, and apoC-I and apoE can disaggregate LPS and modulate its bioactivity.26,30-33,37 To investigate further the interactions of apoA-II with LPS, we compared its ability to bind and disaggregate LPS with those of apoA-I, apoC-I, and apoE using native gel assays. To obtain a clear view of [3H]-LPS-apolipoprotein co-migration, we reduced the ability of the medium to maintain a high state of LPS aggregation by eliminating calcium from the mixtures. As shown in Figure 7B, [3H]-LPS ran near the top of the gel in the absence of apolipoproteins, whereas all of the apolipoproteins partially disaggregated the LPS. Similar results were obtained with monomeric apoA-II (data not shown). To determine whether the LPS remained bound to the apolipoproteins during migration through the gel, we transferred the material in the gels to membranes for Western blot detection of the proteins and fluorographic detection of the transferred [3H]-LPS. As shown in Figure 7C, the LPS did not significantly alter the mobility of apoA-II, and it showed little or no co-migration with apoA-II. In contrast, LPS altered the migration of the other apolipoproteins and co-migrated with a large fraction of each protein. The results suggest that apoA-II interacts transiently with LPS, whereas prolonged complex formation occurs between LPS and apoA-I, apoC-I, or apoE. The experiments shown in Figure 7 were also performed under conditions in which LPS and apolipoproteins were pre-incubated in SFM. Although the presence of the calcium component of SFM promoted more LPS aggregation and resulted in slower mobility of the LPS on the native gels, LPS altered the mobilities of apoA-I, apoC-I, and apoE, whereas the mobility of apoA-II was unaffected (data not shown). Taken together, these results suggest that apoA-II does not form complexes with LPS.
Fig. 7.
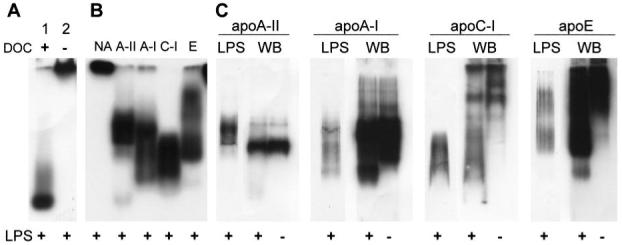
ApoA-II disaggregates LPS without forming LPS-apoA-II complexes. [3H]-LPS was incubated in 7.5 μl of HEPES-buffered saline containing 0.1 mM EDTA and the indicated proteins for 30 min at 37°C. The mixtures were run on 10% native PAGE gels and subjected to fluorography to detect [3H]-LPS or Western blotting to detect the proteins. (A) To show LPS disaggregation, [3H]-LPS (0.05 μg) was incubated in the presence (lane 1) or absence (lane 2) of sodium deoxycholate (1.5 mg/ml); the mixtures were run on a separate gel. (B,C) [3H]-LPS and the indicated apolipoproteins were incubated alone or in equimolar mixtures (1.7 μg apoA-II, 2.8 μg apoA-I, 0.66 μg apoC-I, or 3.4 μg apoE with 0.5 μg LPS), and the gels were run concurrently. (B) [3H]-LPS was detected by fluorography. (C) The gel was transferred to Immobilon-P membranes for Western blot (WB) detection of the indicated proteins. The same membranes were then fluorographed to detect transferred [3H]-LPS (LPS).
To determine the relative abilities of the apolipoproteins to inhibit LPS bioactivity, we incubated LPS with apoA-II and the other HDL-derived apolipoproteins and measured the abilities of the mixtures to stimulate THP-1 cells in the presence of a low stimulatory concentration of LBP that did not inhibit the cell response. As shown in Figure 8, apoA-II caused little or no decrease in LPS bioactivity, whereas the other apolipoproteins were highly inhibitory. The results suggest that, unlike the other apolipoproteins tested, apoA-II has little or no capacity to bind LPS in a way that inhibits its bioactivity.
Fig. 8.
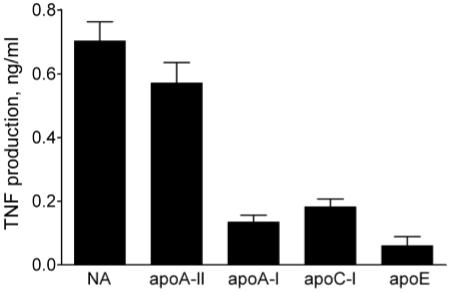
ApoA-II does not inhibit LPS bioactivity in the presence of low concentrations of LBP. LPS (20 pg/ml) was pre-incubated with PBS or indicated apolipoproteins (1 μg/ml) for 30 min at 37°C. The mixtures were incubated with THP-1 cells in the presence of 0.001 μg/ml LBP to measure LPS bioactivity as described in the caption to Figure 1. Error bars denote mean ± SD (n = 4-8) of duplicate determinations in 2-4 experiments.
Discussion
These results reveal a novel pro-inflammatory role for apoA-II in its ability to augment the bioactivity of bacterial LPS by preventing LBP-mediated inhibition. Although several other HDL-associated apolipoproteins have been reported to bind LPS and alter its bioactivity,26,30-33,37 to our knowledge, there are no previous reports of LPS interactions with apoA-II.
The findings suggest that the mechanism of the effects of apoA-II on LBP inhibitory activity involve its ability to prevent the formation of large LPS-LBP complexes in which LPS bioactivity is inhibited (Fig. 5) combined with the relative inability of apoA-II to bind and inhibit LPS activity (Figs 7 and 8). However, we also found surprisingly low bioactivity in LPS that had been extensively disaggregated by the combined action of LBP and apoA-II (Fig. 5, gel region ‘B’) or by deoxycholate (data not shown). These observations may be consistent with a previous report that highly disaggregated or monomeric LPS has very little bioactivity in the presence of LBP.38 Although monomeric LPS that is associated with soluble CD1439 or MD-240 possesses a high degree of bioactiv ity, the ability of LBP to augment the bioactivity of LPS that is not already associated with such LPS receptors appears to depend on the interaction of LBP with aggregated LPS. The data thus show that LPS bioactivity is diminished both by excessive aggregation in the presence of high concentrations of LBP or by excessive disaggregation (Fig. 5). It should be noted, however, that the loss of LPS bioactivity by disaggregation resulting from the combined action of apoA-II and LBP may occur only when the disaggregated LPS is separated from the original mixture of proteins (Fig. 5, gel region ‘B’). These mixtures show no significant loss of LPS activity if left undisturbed (Fig. 5C [T] and Fig. 6).
Although we attribute the apoA-II mechanism to its direct effects on the physical state of LPS, the data do not exclude the possibility that protein-protein interactions between apoA-II and LBP could inhibit the formation of large LPS-LBP complexes. Others have shown that apoA-II can bind to phospholipid transfer protein (PLTP),34 a member of the LBP family of lipid transfer proteins,5 and that apoA-II can inhibit the activity of hepatic lipase.34 However, we did not observe significant co-migration of apoA-II with LBP on native PAGE gels, suggesting that there may be little or no direct interaction between these two proteins. Others have shown evidence of apoA-I interactions with LBP,41 but we found that it has relatively little ability to prevent LBP from inhibiting LPS activity (Fig. 1B). The poor activity of apoA-I and the other apolipoproteins tested may be attributed, at least in part, to their ability to bind (Fig. 7) and inhibit (Fig. 8) LPS directly. Although apoA-II can result in a slight inhibition of LPS activity in the presence of very low concentrations of LBP (Fig. 8), apoA-II appears to have no inhibitory effects even at high concentrations in the presence of high concentrations of LBP (Fig. 5C [T] and Fig. 6).
The ability of apoA-II to augment the response to LPS appears to be mechanistically different from that of apoC-I, an HDL component that has in vivo pro-inflammatory activity during LPS administration and Gram-negative bacterial infection.26 These studies showed that apoC-I inhibits the clearance of circulating LPS, which may prolong its stimulatory activity, and that apoC-I also has LBP-like activity in macrophage cultures. The extent to which apoC-I can substitute for LBP in vivo is still unclear, since LBP-deficient mice have a pronounced defect in their ability to respond to low concentrations of LPS in whole blood ex vivo42 and in one LBP knock-out model in vivo.4 However, in vivo responses to LPS were not impaired in another LBP knock-out model.42 Whereas low concentrations of apoC-I augment macrophage responses in the absence of LBP,26 high apoC-I concentrations, such as those found in whole blood, are inhibitory (data not shown). In our monocyte cultures, apoC-I only slightly augmented the response in the presence of an inhibitory concentration of LBP (Fig. 1B), and it inhibited LPS bioactivity when allowed time to bind to the LPS (Fig. 8). Thus, in contrast to apoC-I, we found that apoA-II did not inhibit LPS bioactivity and that its ability to augment the response to LPS occurred only in the presence of inhibitory concentrations of LBP. Experiments are in progress to determine whether apoA-II has pro-inflammatory effects in vivo.
The amino acid sequences of mouse and human apoA-II differ by approximately 40%, and they produce contrasting effects on lipoprotein metabolism when expressed in transgenic mice.34 Despite these differences, we found that the activity of wild-type mouse HDL was only slightly lower than that of human HDL in our assays (Fig. 2). The more robust activity of human HDL may be attributed, in part, to its higher apoA-II content. The existence of homodimers due to the presence of a cysteine residue at position 6 is a unique feature of human apoA-II, in contrast to the apoA-II monomers that are found in other species. We tested both dimeric and monomeric human forms of apoA-II and found that they had identical effects on LBP activity. Although apoA-II is an abundant component of the plasma of humans, simians, rodents, and fish, it is either absent or expressed at low levels in rabbits, dogs, pigs, and chickens. Our experiments with rabbit HDL revealed a reduced, but partial, ability to suppress LBP inhibitory activity (Fig. 3). These results raise the possibility that rabbit HDL contains unique proteins that may have apoA-II-like activity.
ApoA-II is the second most abundant protein constituent of HDL; however, despite many studies, its role in HDL metabolism is still unclear.34 Studies in transgenic mice suggest that apoA-II can confer other pro-inflammatory properties to HDL, characterized by the inability of apoA-II-enriched HDL to protect against LDL oxidation and by its ability to stimulate lipid hydroperoxide formation and monocyte transmigration.43 These pro-inflammatory activities of apoA-II have been linked to its pro-atherogenic properties in mouse models of atherosclerosis. Our findings expand the potential pro-inflammatory activities of apoA-II to the control of host responses to bacterial LPS, and they raise the possibility that apoA-II may play a role in antimicrobial host defense.
Acknowledgements
The authors thank Dr Robert Munford for critically reading the manuscript. This work was supported by National Institutes of Health Grant AI45896 from the National Institute of Allergy and Infectious Diseases and by the Netherlands Heart Foundation (NHS grant 2005B226 to PCNR).
References
- 1.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 2.Nagai Y, Akashi S, Nagafuku M, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 3.Schumann RR, Leong SR, Flaggs GW, et al. Structure and function of lipopolysaccharide-binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 4.Jack RS, Fan X, Bernhelden M, et al. Lipopolysaccharide-binding protein is required to combat a murine Gram-negative bacterial infection. Nature. 1997;389:742–744. doi: 10.1038/39622. [DOI] [PubMed] [Google Scholar]
- 5.Zweigner J, Schumann RR, Weber JR. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microb Infect. 2006;8:946–952. doi: 10.1016/j.micinf.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Haziot A, Ferrero E, Köntgen F, et al. Resistance to endotoxin shock and reduced dissemination of Gram-negative bacteria in CD14-deficient mice. Immunity. 1997;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 7.Lamping N, Dettmer R, Schröder NW, et al. LPS-binding protein protects mice from septic shock caused by LPS or Gram-negative bacteria. J Clin Invest. 1998;101:2065–2071. doi: 10.1172/JCI2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang KK, Dorner BG, Merkel U, et al. Neutrophil influx in response to a peritoneal infection with Salmonella is delayed in lipopolysaccharide-binding protein or CD14-deficient mice. J Immunol. 2002;169:4475–4480. doi: 10.4049/jimmunol.169.8.4475. [DOI] [PubMed] [Google Scholar]
- 9.Knapp S, de Vos AF, Florquin S, Golenbock DT, van der Poll T. Lipopolysaccharide binding protein is an essential component of the innate immune response to Escherichia coli peritonitis in mice. Infect Immun. 2003;71:6747–6753. doi: 10.1128/IAI.71.12.6747-6753.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan MH, Klein RD, Steinstraesser L, et al. An essential role for lipopolysaccharide-binding protein in pulmonary innate immune responses. Shock. 2002;18:248–254. doi: 10.1097/00024382-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Heumann D, Roger T. Initial responses to endotoxins and Gram-negative bacteria. Clin Chim Acta. 2002;323:59–72. doi: 10.1016/s0009-8981(02)00180-8. [DOI] [PubMed] [Google Scholar]
- 12.Thompson PA, Kitchens RL. Native high-density lipoprotein augments monocyte responses to lipopolysaccharide (LPS) by suppressing the inhibitory activity of LPS-binding protein. J Immunol. 2006;177:4880–4887. doi: 10.4049/jimmunol.177.7.4880. [DOI] [PubMed] [Google Scholar]
- 13.Zweigner J, Gramm HJ, Singer OC, Wegscheider K, Schumann RR. High concentrations of lipopolysaccharide-binding protein serum of patients with severe sepsis or septic shock inhibit the lipopolysaccharide response in human monocytes. Blood. 2001;98:3800–3808. doi: 10.1182/blood.v98.13.3800. [DOI] [PubMed] [Google Scholar]
- 14.Martin TR, Mathison JC, Tobias PS, et al. Lipopolysaccharide binding protein enhances the responsiveness of alveolar macrophages to bacterial lipopolysaccharide. Implications for cytokine production in normal and injured lungs. J Clin Invest. 1992;90:2209–2219. doi: 10.1172/JCI116106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vreugdenhil ACE, Snoek AMP, Greve JW, Buurman WA. Lipopolysaccharide-binding protein is vectorially secreted and transported by cultured intestinal epithelial cells and is present in the intestinal mucus of mice. J Immunol. 2000;165:4561–4566. doi: 10.4049/jimmunol.165.8.4561. [DOI] [PubMed] [Google Scholar]
- 16.Bannerman DD, Paape MJ, Hare WR, Sohn EJ. Increased levels of LPS-binding protein in bovine blood and milk following bacterial lipopolysaccharide challenge. J Dairy Sci. 2003;86:3128–3137. doi: 10.3168/jds.S0022-0302(03)73914-9. [DOI] [PubMed] [Google Scholar]
- 17.Wurfel MM, Kunitake ST, Lichenstein H, Kane JP, Wright SD. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J Exp Med. 1994;180:1025–1035. doi: 10.1084/jem.180.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vesy CJ, Kitchens RL, Wolfbauer G, Albers JJ, Munford RS. LPS binding protein and phospholipid transfer protein release lipopolysaccharides from Gram-negative bacterial membranes. Infect Immun. 1999;68:2410–2417. doi: 10.1128/iai.68.5.2410-2417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gegner JA, Ulevitch RJ, Tobias PS. Lipopolysaccharide (LPS) signal transduction and clearance. Dual roles for LPS binding protein and membrane CD14. J Biol Chem. 1995;270:5320–5326. doi: 10.1074/jbc.270.10.5320. [DOI] [PubMed] [Google Scholar]
- 20.Thompson PA, Tobias PS, Viriyakosol S, Kirkland TN, Kitchens RL. Lipopolysaccharide (LPS)-binding protein inhibits responses to cell-bound LPS. J Biol Chem. 2003;278:28367–28371. doi: 10.1074/jbc.M302921200. [DOI] [PubMed] [Google Scholar]
- 21.Feingold KR, Funk JL, Moser AH, Shigenaga JK, Rapp JH, Grunfeld C. Role for circulating lipoproteins in protection from endotoxin toxicity. Infect Immun. 1995;63:2041–2046. doi: 10.1128/iai.63.5.2041-2046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barlage S, Fröhlich D, Böttcher A, et al. ApoE-containing high density lipoproteins and phospholipid transfer protein activity increase in patients with a systemic inflammatory response. J Lipid Res. 2001;42:281–290. [PubMed] [Google Scholar]
- 23.Kitchens RL, Thompson PA, Munford RS, O’Keefe GE. Acute inflammation and infection maintain circulating phospholipid levels and enhance lipopolysaccharide binding to plasma lipoproteins. J Lipid Res. 2003;44:2339–2348. doi: 10.1194/jlr.M300228-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Berbée JFP, van der Hoogt CC, de Haas CJC, et al. Plasma apolipoprotein CI correlates with increased survival in patients with severe sepsis. Intensive Care Med. 2008;34:907–911. doi: 10.1007/s00134-008-1006-y. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Thompson PA, Kitchens RL. Infection induces a positive acute phase apolipoprotein E response from a negative acute phase gene: role of hepatic LDL receptors. J Lipid Res. 2008;49:1782–1793. doi: 10.1194/jlr.M800172-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berbée JFP, van der Hoogt CC, Kleemann R, et al. Apolipoprotein CI stimulates the response to lipopolysaccharide and reduces mortality in Gram-negative sepsis. FASEB J. 2006;20:2162–2164. doi: 10.1096/fj.05-5639fje. [DOI] [PubMed] [Google Scholar]
- 27.Kitchens RL, Wolfbauer G, Albers JJ, Munford RS. Plasma lipoproteins promote the release of bacterial lipopolysaccharide from the monocyte cell surface. J Biol Chem. 1999;274:34116–34122. doi: 10.1074/jbc.274.48.34116. [DOI] [PubMed] [Google Scholar]
- 28.Weng W, Breslow J. Dramatically decreased high density lipoprotein cholesterol, increased remnant clearance, and insulin hypersensitivity in apolipoprotein A-II knockout mice suggest a complex role for apolipoprotein A-II in atherosclerosis susceptibility. Proc Natl Acad Sci USA. 1996;93:14788–14794. doi: 10.1073/pnas.93.25.14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munford RS, DeVeaux LC, Cronan JE, Jr, Rick PD. Biosynthetic radiolabeling of bacterial lipopolysaccharide to high specific activity. J Immunol Methods. 1992;148:115–120. doi: 10.1016/0022-1759(92)90164-o. [DOI] [PubMed] [Google Scholar]
- 30.Emancipator K, Csako G, Elin RJ. In vitro inactivation of bacterial endotoxin by human lipoproteins and apolipoproteins. Infect Immun. 1992;60:596–601. doi: 10.1128/iai.60.2.596-601.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flegel WA, Baumstark MW, Weinstock C, Berg A, Northoff H. Prevention of endotoxin-induced monokine release by human low- and high-density lipoproteins and by apolipoprotein A-I. Infect Immun. 1993;61:5140–5146. doi: 10.1128/iai.61.12.5140-5146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma J, Liao X-L, Lou B, Wu M-P. Role of apolipoprotein A-I in protecting against endotoxin toxicity. Acta Biochim Biophys Sin. 2004;36:419–424. doi: 10.1093/abbs/36.6.419. [DOI] [PubMed] [Google Scholar]
- 33.Rensen PCN, Van Oosten M, Van de Bilt E, Van Eck M, Kuiper J, Van Berkel TJC. Human recombinant apolipoprotein E redirects lipopolysaccharide from Kupffer cells to liver parenchymal cells in rats in vivo. J Clin Invest. 1997;99:2438–2445. doi: 10.1172/JCI119427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanco-Vaca F, Escola-Gil JC, Martin-Campos JM, Julve J. Role of apoA-II in lipid metabolism and atherosclerosis: advances in the study of an enigmatic protein. J Lipid Res. 2001;42:1727–1739. [PubMed] [Google Scholar]
- 35.Lenich C, Brecher P, Makrides S, Chobanian A, Zannis VI. Apolipoprotein gene expression in the rabbit: abundance, size, and distribution of apolipoprotein mRNA species in different tissues. J Lipid Res. 1988;29:755–764. [PubMed] [Google Scholar]
- 36.Munford RS, Hall CL, Dietschy JM. Binding of Salmonella typhimurium lipopolysaccharides to rat high-density lipoproteins. Infect Immun. 1981;34:835–843. doi: 10.1128/iai.34.3.835-843.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Recalde D, Ostos MA, Badell E, et al. Human apolipoprotein A-IV reduces secretion of proinflammatory cytokines and atherosclerotic effects of a chronic infection mimicked by lipopolysaccharide. Arterioscler Thromb Vasc Biol. 2004;24:756–761. doi: 10.1161/01.ATV.0000119353.03690.22. [DOI] [PubMed] [Google Scholar]
- 38.Mueller M, Lindner B, Kusumoto S, Fukase K, Schromm AB, Seydel U. Aggregates are the biologically active units of endotoxin. J Biol Chem. 2004;279:26307–26313. doi: 10.1074/jbc.M401231200. [DOI] [PubMed] [Google Scholar]
- 39.Hailman E, Lichenstein HS, Wurfel MM, et al. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179:269–277. doi: 10.1084/jem.179.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gioannini TL, Teghanemt A, Zhang D, et al. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci USA. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massamiri T, Tobias PS, Curtiss LK. Structural determinants for the interaction of lipopolysaccharide binding protein with purified high density lipoproteins: role of apolipoprotein A-I. J Lipid Res. 1997;38:516–525. [PubMed] [Google Scholar]
- 42.Wurfel MM, Monks BG, Ingalls RR, et al. Targeted deletion of the lipopolysaccharide (LPS)-binding protein leads to profound suppression of LPS responses ex vivo, whereas in vivo responses remain intact. J Exp Med. 1997;186:2051–2056. doi: 10.1084/jem.186.12.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castellani LW, Navab M, Van Lenten BJ, et al. Overexpression of apolipoprotein AII in transgenic mice converts high density lipoproteins to proinflammatory particles. J Clin Invest. 1997;100:464–474. doi: 10.1172/JCI119554. [DOI] [PMC free article] [PubMed] [Google Scholar]