

Abstract
The objective of this study is to determine the molecular factors that lead to β-lactamase inhibitor resistance for the variant M69V in SHV-1 β-lactamase. With mechanism-based inhibitors, the β-lactamase forms an acyl-enzyme that consists of a trans-enamine derivative in the active site. The present study focuses on these intermediates by introducing the mutation E166A that greatly retards deacylation. Thus, by comparing the properties of the E166A and the M69V-E166A forms we can explore the consequences of the resistance mutation at the level of the enamine acyl-enzymes. The reactions between the β-lactamase and the inhibitors tazobactam, sulbactam and clavulanic acid are followed in single crystals of the enzymes by using a Raman microscope. The resulting Raman difference spectroscopic data provide detailed information on conformational events involving the enamine species as well as an estimate of their populations. The Raman difference spectra for each of the inhibitors in the E166A and the M69V-E166A variants are very similar. In particular, detailed analysis of the main enamine Raman vibration near 1595 cm−1 reveals that the structure and flexibility of the enamine fragments are essentially identical for each of the three inhibitors in E166A and in the M69V-E166A double mutant. This finding is in accord with the X-ray derived structures, presented herein at 1.6 to 1.75 Å resolution, of the trans– enamine intermediates formed by the three inhibitors in M69V-E166A. However, a comparison of Raman results for M69V-E166A and E166A, show that the M69V mutation results in a 40%, 25% or negligible reduction in enamine population when the β-lactamase crystals are soaked in 5mM tazobactam, clavulanic acid and sulbactam solutions, respectively. The levels of enamine from tazobactam and clavulanic acid can be raised by increasing the concentrations of inhibitor in the mother liquor. Thus, the sensitivity of population levels to concentration of inhibitor in the mother liquor focuses attention on the properties of the encounter complex preceding acylation. It is proposed that for small ligands, such as tazobactam, sulbactam and clavulanic acid, the positioning of the lactam ring in the active site in the correct orientation for acylation is only one of a number of poorly defined conformations. For tazobactam and clavulanic acid the correctly oriented encounter complex is even less likely in the M69V variant leading to a reduction in inhibition of the enzyme via formation of the acyl-enzyme and the onset of resistance. Analysis of the X-ray structures of the three intermediates in M69V-E166A demonstrates that, compared to the structures for the E166A form, the oxyanion hole becomes smaller providing one explanation as to why acylation may be less efficient following the M69V substitution.
β-lactamases (E.C.3.5.2.6) are a major mechanism of defense used by bacteria to protect themselves against the lethal action of β-lactam antibiotics (1). β-Lactamase enzymes are classified into four major classes, Ambler class A–D, based on their amino acid sequence similarity (2). Class A β-lactamases are serine hydrolases produced by both Gram positive and Gram negative bacteria (3). These serine hydrolases are able to inactivate a wide range of substrates (penicillins, cephalosporins, extended spectrum cephalosporins, and carbapenems) making their presence in enteric bacteria (e.g., Escherichia coli and Klebsiella pneumoniae) extremely problematic (4). Three β-lactamase inhibitors (clavulanate, sulbactam, and tazobactam) (Figure 1) are currently available (5, 6). These inhibitors are paired with β-lactam antibiotics to prevent their degradation by bacteria possessing class A β-lactamases. The β-lactamase inhibitors undergo complex reactions with the enzyme that are outlined in Scheme 1. Despite extensive study of the mechanism of inhibition by these compounds, questions still remain as to the nature and importance of the various intermediates (7–19).
Figure 1.
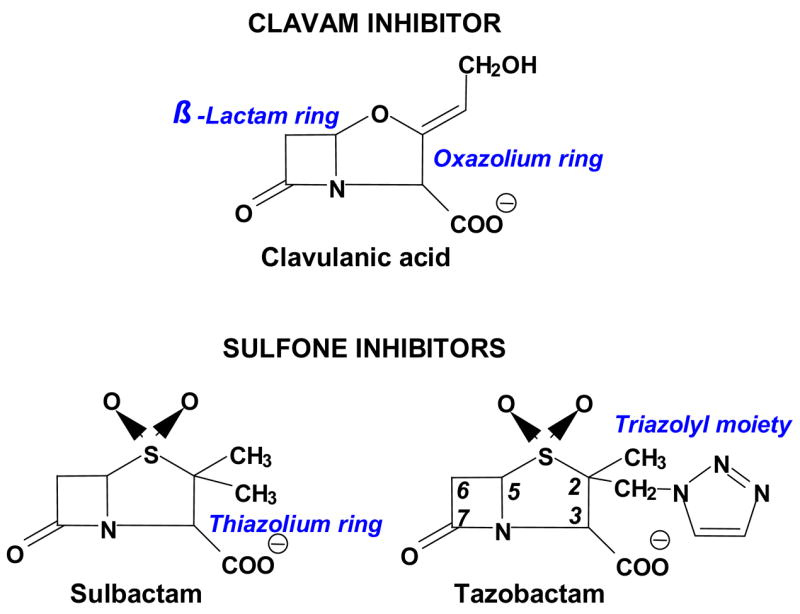
Class A β-lactamase inhibitors
Scheme 1.

Proposed reaction mechanism for Class A β-lactamase with a generalized inhibitor
SHV-1, a class A β-lactamase, is found primarily in K. pneumoniae and is of significant clinical importance since extended spectrum and inhibitor resistant variants have been described in this family (20). Moreover, structural and analytical studies have established that SHV is an excellent model for mechanistic studies of class A β-lactamase inhibition. Recently, we have shown that the tracking and trapping of intermediates in SHV β-lactamase crystals can be accomplished using an interplay of two techniques, X-ray and Raman crystallography (21–23). These efforts were aided by using a deacylation deficient variant of the SHV-1 enzyme, E166A. We have shown that in three inhibitor bound crystal structures, a trans-enamine intermediate is formed, an indication that all these inhibitors could follow a common pathway to inhibition (22, 23). Both from Raman and X-ray crystallographic data it was evident that tazobactam forms the most conformationally stable trans-enamine while sulbactam and clavulanate form less stable trans-enamine intermediates. These observations could, in part, explain their clinical efficacy. The crystallographic observations of these inhibitor reaction intermediates sets the stage to further our understanding how resistance to β-lactamase inhibitors can occur.
Alterations at M69 have been described in many TEM-type inhibitor resistant variants and recently an M69I variant in SHV (SHV-49) was observed clinically (24). This SHV variant had previously been shown to increase resistance to clavulanic acid and to a lesser degree resistance to sulbactam and tazobactam in laboratory constructs of SHV and the 94% sequence identical OHIO-1 (25–27). Helfand, et al., have also studied the role of substitution at M69 by site-saturation mutagenesis (25). The findings indicated that substitution of -Ile, -Leu or -Val significantly increased the MICs and IC50s for all three inhibitors; while -Lys, -Tyr and -Phe substitution leads to cephalosporinase activity. These results indicate that the size and nature of the residue at the M69 position impacts the substrate specificity. An overall trend for OHIO-1 M69I was an apparent slower turnover rate and a significant reduction in the affinity for inhibitors. Although the Ki and kinact of the inhibitors are affected by the M69 mutation, β-lactam substrates are still remarkably well hydrolyzed since there is no clear overall trend in either kcat or Km in SHV and TEM β-lactamases (26–28).
Crystallographic analyses of such mutations have been carried out previously in related β-lactamases, TEM-32 (M69I/M182T) and TEM-34 (M69V) (29), and found the mutations introduce subtle active site changes that result in an effect on enzyme catalysis and inhibitor recognition. This study pointed to a possible role for the local environment of S130 for the inhibitor resistance trait of M69 variants. This effect was also postulated to be mediated via S70 changes that were observed in the uncomplexed TEM variant structures (29). In the wt SHV and TEM β-lactamases, S130 is shown to act as a second nucleophile for irreversible inhibition by β-lactamase inhibitors (30), so the changes observed in the orientation of the S130 side chain in the M69V and M69I TEM structures could negatively affect irreversible inhibition (29). In a different crystallographic study involving TEM-33 (M69L) (28), structural changes were not observed, yet molecular dynamics simulations hinted at subtle changes in enzyme dynamics. In this case, it was concluded that the mechanism of inhibitor resistance for the M69L mutation in TEM is the reduced affinity for the inhibitors, which likely affects the rate of the formation of the initial acyl inhibitor complex.
In the current study, we report the Raman and X-ray crystallographic analyses of the SHV-1 variant M69V-E166A complexed with all three inhibitors. Both methods demonstrate that little or no differences occur for the conformations of the intermediate bound to E166A or M69V-E166A. However, the Raman data show that differences in the active site populations make two of the compounds less effective inactivators of the M69V variant and the X-ray data provide a rationale for this in terms of subtle changes in active site residues.
Materials and Methods
Inhibitors
Sodium clavulanate (Glaxo-Smith-Kline), sulbactam (Pfizer) and tazobactam (Wyeth Pharmaceuticals) were gifts of the respective companies. Stock solutions of these inhibitors at 20 mM concentration in 2 mM HEPES buffer, pH 7.0 were prepared for “soak in” experiments with the protein crystals.
Protein isolation and purification
The E166A SHV and M69V-E166A β-lactamase were generated by site specific mutagenesis (21, 31). The E166A/M69V-E166A variants of SHV β-lactamase protein were isolated and purified as previously described (31). An additional gel filtration HPLC purification step was performed using a Sephadex Hi Load 26/60 column (Pharmacia, Uppsala, Sweden) and elution with phosphate buffered saline, pH 7.4.
Crystallization and structure determination
E166A SHV and M69V-E166A were concentrated to 5 mg/mL in 2 mM HEPES buffer, pH 7.0 for crystallization per the protocol of Kuzin et al. (30). Crystals were prepared using the sitting drop method. Drops of 10 μL volume were prepared mixing 4 parts of protein solution with 1 part CYMAL-6 5.6 mM (Hampton Research, Laguna Niguel, CA) and 5 parts PEG-6000 (Hampton Research) 20–30% w/v in HEPES 0.1M pH 7.0. The reservoir solution consisted of PEG-6000 20–30% w/v in HEPES 0.1M pH 7.0. The wells were sealed with packing tape and stored at room temperature. Crystals, typically 300 × 300 × 300 μm in size, were transferred from the mother liquor solution into a 0.1 M HEPES pH 7.0 solution and washed four times to remove excess PEG-6000 prior to use with the Raman microscope. Then they were transferred to a siliconized glass cover slip into 3 μL 0.1 M HEPES pH 7.0 and brought into a hanging drop set up. Crystal protein concentrations were estimated to be 28 mM (James Knox, personal communication) based on unit cell dimensions of a = 49.6 Å, b = 55.6 Å and c = 87.0 Å and an occupancy of 4 molecules/unit cell. These crystals were used for soaking experiments to track inhibitor intermediates. The Raman studies carried out at 5mM concentrations indicated that all three inhibitors inhibited the enzyme in the trans-enamine intermediate form as was observed for the single E166A mutant. Maximal trans-enamine formation was achieved in roughly 10–20 minutes yet, unlike for sulbactam, the amount of this intermediate for tazobactam and clavulanic acid was substantially reduced in the double mutant compared to the single E166A mutant. Based on the Raman results, we adjusted the soaking procedure by increasing the soaking concentration from 5mM to 50mM for each of the three inhibitors to increase the amount of trans-enamine intermediate. The compounds were soaked using the procedure as reported previously (21, 23). The best data set collected is from a single crystal cut into three pieces and each soaked with one of the three inhibitors for 10 minutes. Diffraction data were collected at the ALS Berkley 4.2.2. synchrotron facility for the three inhibitor soaked crystals of M69V/E166A mutant. The data processing is done with d*TREK (32) and the reflection files are further process with CCP4 (33). Crystallographic refinement was carried out in CNS (34) using parameter and topology files for the trans-enamine reaction intermediate generated in PRODRG (35). The E166A mutant structures complexed to either tazobactam (1RCJ), sulbactam (2A3U), and clavulanic acid (2A49) were used as the starting model for refinement of their respective inhibitor complexes to the E166A/M69V double mutant. For crystallographic refinement cross-validation, 10% of the reflections where used for the Rfree calculations. Different Rfree sets where used to refine each structure and biased was removed by carrying out a simulated annealing step to 3000K in CNS. The temperature factors where restrained during refinement and the RMSD values are listed in Table 1. The occupancy of the ligand was refined as 1.0 although also refinement with ligand occupancies of 0.8 and 0.9 yielded comparable results. Iterative model building was performed in O (36). Waters were selected with |Fo|-|Fc| density above 3σ, and in good hydrogen bonding geometry with nearby protein atoms and/or water molecules. The crystallographic structures of M69V/E166A bound to tazobactam, sulbactam and clavulanic acid were refined to 1.75, 1.70, and 1.6Å resolution, respectively. The final refined R-factors and Rfree values are shown in Table 1. Each model contained a covalently bound inhibitor, a fully ordered cymal-6 molecule and one partial cymal-6 molecule. Two of the structures contained a partially ordered HEPES molecule (clavulanic acid and sulbactam bound structures).
Table 1.
Data Collection and Refinement Statistics for M69V E166A mutant
| Data Collection | |||
|---|---|---|---|
| Clavulanic acid | Sulbactam | Tazobactam | |
| Space Group | P212121 | P212121 | P212121 |
| Unit cell dimensions (Å) | a=49.68 b=55.20 c=83.49 | a=49.59 b=55.31 c= 83.63 | a=49.67 b=55.18 c=83.53 |
| Wavelength (Å) | 1.46 | 1.46 | 1.46 |
| Resolution (Å) | 19.91–1.60 | 9.99–1.70 | 19.91–1.75 |
| Redundancy | 4.83 | 1.91 | 3.00 |
| Total number of Reflections | 142212 | 86698 | 134196 |
| Number of unique Reflections | 29464* (26486) | 25354* (25308) | 23825* (23773) |
| <I>/<σI> | 9.8 (2.6) | 9.9 (2.2) | 11.2 (3.0) |
| Rmerge (%) | 9.4 (50.6) | 5.4 (32.7) | 5.9 (29.9) |
| Completeness (%) | 95.1 (90.9) | 93.4 (92.2) | 99.9 (99.7) |
| Refinement | |||
| R-factor % | 16.00 | 17.51 | 15.92 |
| R-free % | 19.96 | 21.09 | 19.05 |
| RMSD from ideality | |||
| Bond lengths (Å) | 0.009 | 0.008 | 0.009 |
| Angles (°) | 1.59 | 1.49 | 1.58 |
| RMSD B-factors atoms (Å2) | |||
| Bonded main chain | 2.1 | 2.1 | 2.2 |
| Bonded side chain | 2.8 | 3.6 | 3.8 |
| Angle main chain | 4.5 | 2.7 | 2.8 |
| Angle side chain | 5.6 | 4.9 | 5.1 |
| Average B-factor | |||
| Protein (Å2) | 12.47 | 17.55 | 14.95 |
| Inhibitor (Å2) | 34.30 | 23.13 | 20.93 |
| Water (Å2) | 29.00 | 32.51 | 31.99 |
Data sets were processed with the Friedel mates kept separate. The numbers in parenthesis are unique reflections used for refinement
Raman crystallography
The Raman microscope system has been described previously (37, 38). A 647 nm Kr+ laser beam with 120 mW laser power (Innova 70 C, Coherent, Palo Alto CA) was focused onto the protein crystals, suspended on the underside of a siliconized quartz cover slip in a 3μL drop. A 20 × objective gave an approximately 20 μm spot size on the crystal. The crystals and the laser spot were visualized with real time color video display to assure proper alignment and that no light-induced damage was occurring in the crystal. During data collection, spectra were acquired over 10 second intervals and 10 spectra were averaged for each acquisition time point. Spectra of the apo-β-lactamase protein crystals were obtained and subsequently we infused the inhibitor into the protein crystal by adding 1 μL of the inhibitor solution to the drop to achieve a final drop volume of 4 μL and a final inhibitor concentration of 5 mM. Spectra of the β-lactamase-inhibitor complex were then acquired serially every 2–3 minutes following addition of the inhibitor. An apo-β-lactamase spectrum was subtracted from the inhibited protein spectra at varying time intervals following addition of inhibitor, according to Equation (1).
| (1) |
Here f is a subtraction scale factor selected in order to minimize the protein amide I band from apo-protein in the difference spectra. Typically f has a value of 0.95 to 1.0. Data collection and subtractions were performed using HoloGRAMS and GRAMS/AI 7 software (ThermoGalactic, Inc., Salem, N.H.). Raman spectra of the inhibitors in solution were obtained using the Raman microscope. Spectra were obtained of 4 μL drops of inhibitor solutions prepared at varying inhibitor concentrations in HEPES pH 7.0. The peak heights of various Raman bands in the inhibitor spectra were examined as a function of concentration to prepare concentration calibration curves.
The kinetic plots (Figures 5–7) were obtained by measuring the integrated intensity of the enamine feature in the difference spectrum near 1595 cm−1 and dividing this value by the integrated amide I band at 1655 cm−1 in the parent spectrum. The initial data points in Figures 5–7 are above zero since these data are collected in the time interval from 60 to 160 secs after the inhibitor is introduced into the hanging drop.
Figure 5.

Time dependence of the enamine peak area near 1593 cm−1 (normalized to the amide I band) for the E166A (upper trace) and M69V-E166A (lower trace) SHV β-lactamase variant crystals and tazobactam (5mM in the mother liquor)
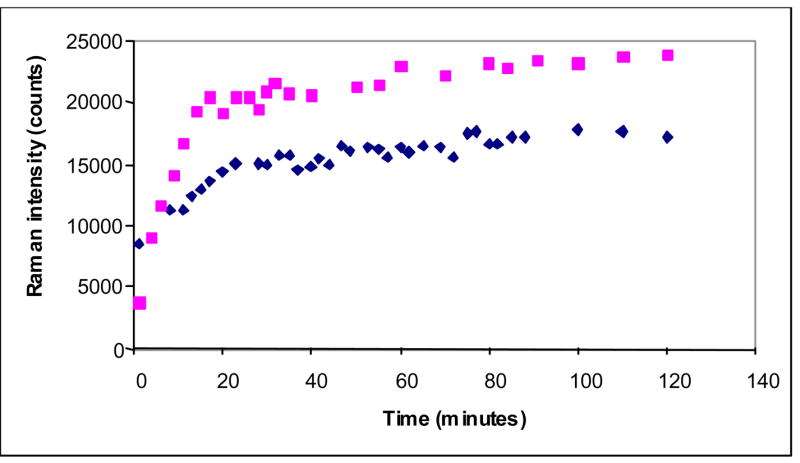
Figure 7.

Time dependence of the enamine peak area near 1599 cm−1 (normalized to the amide I band) for the E166A (cyan squares) and M69V-E166A (blue diamonds) SHV β-lactamase variant crystals and sulbactam 5mM in the mother liquor
Calculations
Ab initio quantum mechanical calculations were performed to predict the Raman spectra of model intermediate compounds using Gaussian 03 ™ (39) software. Calculations were performed at the Hartree-Fock level using the 6-31+G (d) basis set.
Results and Discussion
Inhibitors form very similar trans-enamine conformations in E166A and E166A-M69V variants
a) Raman spectroscopic evidence
Figure 2, compares the Raman difference spectra of the enamine species in single crystals of E166A and M69V-E166A for tazobactam. The two spectra are remarkably similar throughout, suggesting that the trans-enamine assumes a very similar conformation in M69V-E166A compared to E166A SHV-1 β-lactamase. In particular, the positions and band profiles of the O=C-C=C-NH- stretching frequency near 1595 cm−1 are identical, to within experimental error of ± 2 cm−1 (Table 2). In the same way the Raman difference spectra for the intermediate formed by sulbactam and clavulanic acid are essentially identical in the E166A and M69V-E166A variants (Supplementary Figures 1S and 2S). The exact position of the intense enamine mode near 1595 cm−1 is a function of the geometry of the trans-enamine skeleton. Thus, the most planar trans-enamine formed by clavulanic acid has νO=C-C=C-NH- at 1612 cm-1, whereas the intermediate from tazobactam has a C-C=C-N dihedral angle of 168 ° and this distortion can account in part, for the symmetric stretch occurring at the lower frequency of 1593 cm−1(22). Taken together, these findings suggest that the conformations of the trans-enamine heavy atom skeleton are essentially identical for each inhibitor in E166A and M69V-E166A SHV-1 β-lactamase. Moreover, as discussed in Padayatti et al. (22), the shape of the band profile near 1595 cm−1 reflects the degree of static and/or dynamic disorder in the “tails” of the enamine extending beyond the N4 atom. Thus, the identity in band profiles for each of the E166A and M69V-E166A pairs means that the degree of conformational flexibility in each tail must be very similar, too.
Figure 2.

Raman difference spectra of the E166A and M69V-E166A β-lactamase crystal with tazobactam 5mM in the mother liquor (Hepes 0.1M, pH 7). The vertical bar represents the intensity of a 20 000 photon event.
Table 2.
Characteristics of the main enamine feature given by the three clinical inhibitors used with two variants (E166A and M69V-E166A) of SHV-1 β-lactamase
| Tazobactam derived enamine ±2 cm−1 | Sulbactam derived enamine± 2 cm−1 | Clavulanic acid derived enamine± 2 cm−1 | |
|---|---|---|---|
| Enamine peak Raman shift for various inhibitors with E166A/M69V-E166A β-lactamase | 1593/1593 cm−1 | 1599/1601 cm−1 | 1612/1610 cm−1 |
| Enamine peak width of various inhibitors with E166A SHV-1 β-lactamase | 18 cm−1 | 25 cm−1 | 32 cm−1 |
| Enamine peak width of various inhibitors with M69V-E166A SHV-1 β-lactamase | 18 cm−1 | 28 cm−1 | 34 cm−1 |
The one reproducible difference in all three data sets (e.g. Figure 2) concerns the broad feature near 1520 cm−1 in the E166A case. For M69V-E166A SHV-1 β-lactamase the broad band moves to near 1495 cm−1.
Using the simplest trans-enamine, formed by clavulanic acid, we undertook ab initio quantum mechanical calculations with the Gaussian 03 software (39). These are detailed in the Supplementary Material and confirm the identity of the relatively intense mode in the 1600 cm−1 region, as a O=C-C=C-NH- symmetric stretch. Further progress in the use of calculations requires the use of isotopically labeled inhibitors (see Supplementary Material).
b) X-ray crystallographic evidence
All three inhibitors are observed as trans-enamine intermediates with tazobactam being well ordered compared to the more flexible sulbactam and clavulanic acid tails (Figure 3). This is reminiscent of our previous analysis of each inhibitor in the E166A structure (22). The clavulanic acid intermediate is refined as a decarboxylated trans-enamine intermediate due to the absence of electron density for this moiety, as in the single mutant E166A structure. This is confirmed by the absence of a –COO− symmetric stretch near 1400 cm−1 in the Raman spectrum seen in Supplementary Material, Figure 2S (22). B-factor refinement and occupancy refinement for each of the three inhibitors indicated that the occupancies are ≥80%. The validity of the models was evaluated using PROCHECK (40). In all three structures, over 90% of the phi-psi angles are in the most favored region.
Figure 3.
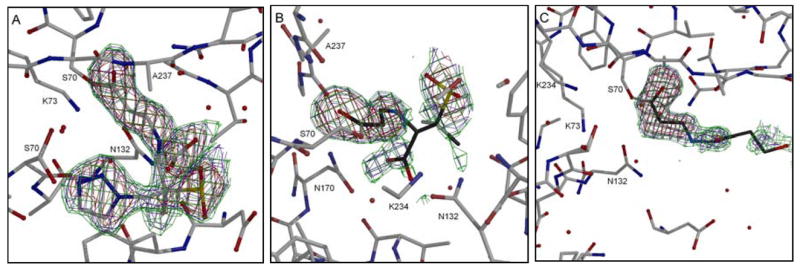
Electron density of the inhibitors in the active site of M69V/E166A SHV-1 β-lactamase. The omit Fobs− Fcalc difference electron density of the active site of the double mutant protein shows the density of covalently bound trans-enamine intermediate for tazobactam (A), sulbactam (B), and clavulanic acid (C). The omit density is contoured at 3.0 σ (red), 2.0 σ (blue), and 1.5σ (green) and is calculated at the end of refinement after omitting the ligands from the map calculation. The omit map suggested clavulanic acid is in a decarboxylated state similar to that observed when complexed in the E166A single mutant structure.
The M69V/E166A structures of the bound inhibitors are quite similar to that of the single E166A mutant structures yielding r.m.s.d’s for Cα atoms of 0.129 Å for the tazobactam bound structures, 0.140 Å for the sulbactam bound structures, and 0.149 Å for clavulanic acid bound structures. Despite the overall similarity, the M69V mutation has resulted in noteworthy conformational shifts in flanking β-strands, in active site residues, and in the bound inhibitors, interactions (Figure 4). Albeit slightly shifted in concert with active site shifts, the Serine70-O-C(=O)-C=C-NH- fragment is in a similar conformation for the single and double mutant variants. This is in complete agreement with the Raman data that indicate essential identity in the trans-enamine fragments. These structural similarities and differences will be discussed below. The ability to compare not one, but three V69 mutant structures with three M69 structures enhances the significance of subtle conformational differences if the changes are consistent between both sets of three structures.
Figure 4.
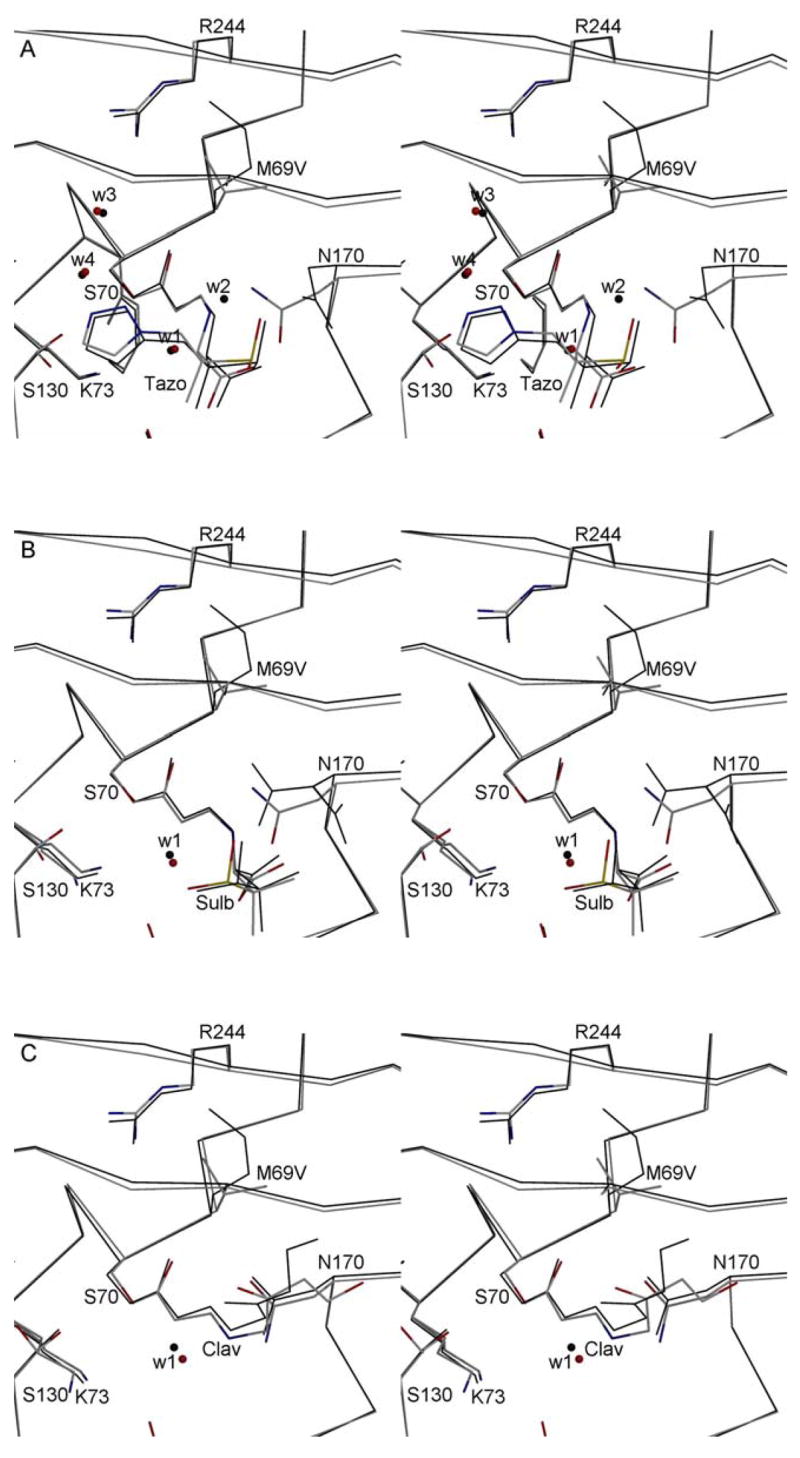
Stereo diagrams depicting the active sites of the inhibitor bound structures of the E166A and inhibitors resistant M69V/E166A variants. For each of the inhibitors, the M69V/E166A double mutant (grey with colored N, O, and S atoms) and the single E166A mutant (black lines) are superimposed. Tazobactam (tazo, 3A), sulbactam (sulb, 3B), and clavulanic acid (clav, 3C) are shown covalently attached to S70. The E166A SHV-1 β-lactamase structures used for the super positions have PDB identifiers 1RCJ (complexed with tazobactam), 2A49 (complexed with sulbactam), and 2A3U (complexed with clavulanic acid) with E166A) are used for comparison of the respective inhibitor bound double mutant structures. Water molecules present near the inhibitor are shown as small spheres. The catalytic water involved in deacylation (W1) is highlighted. For tazobactam in the single mutant structure, a ligand induced shift causes N170 to reorient thereby shifting the position of catalytic water to a new position (W2). Two additional waters are depicted in the tazobactam bound structures (W3 & W4) which are key for interacting with either tazobactam and K234 and/or R244.
Differences in trans-enamine populations in E166A and M69V-E166A variants
Figures 5, 6, and 7 compare the intensity of the main trans-enamine mode and hence the enamine population, for the E166A and M69V-E166A reacting with tazobactam, clavulanic acid and sulbactam, respectively. In each experiment the crystal was soaked in mother liquor containing 5 mM inhibitor. For tazobactam the enamine population in the double variant reaches only 60% of the one found for E166A, where in the latter we know that the active sites are fully occupied (23). The intermediate formed from clavulanic acid in M69V-E166A has about 75% of the population found for E166A, whereas the populations formed in both mutant enzymes by sulbactam are very similar.
Figure 6.
Time dependence of the enamine peak area near 1610 cm−1 (normalized to the amide I band) for the E166A (upper trace) and M69V-E166A (lower trace) SHV β-lactamase variant crystals and clavulanic acid (5mM in the mother liquor)
Thus, while the X-ray data and the Raman data in Figure 2 (and Supplementary Data Figures 1S, 2S), demonstrate that at the structural level the enamines in E166A and M69V-E166A are very similar or identical, for two out of three of the inhibitors, we have evidence for lower intermediate populations in the M69V-E166A mutant that carries the resistance conferring alteration. This shows that in the crystal complexes, tazobactam and clavulanic acid are less effective at blocking the active site in M69V-E166A at the trans-enamine level. Since we cannot detect structural differences in the enamine populations in E166A and M69V-E166A, it is apparent that differences involving other steps in the enzyme reaction must account for the change in acyl-enzyme population by modifying the balance between acylation and deacylation kinetics.
There are several lines of evidence suggesting that changes in the acylation step, including the orientation of the β-lactam ring in the active site, are an important factor in causing low populations of acyl-enzyme intermediates. Meroueh et al. (28) solved the structures of WT TEM-1 β-lactamase and the M69L mutant and undertook molecular dynamics calculations on these protein forms. Their conclusion was that “the inhibitor-resistant trait is a relatively modest elevation of the dissociation constant for the formation of the pre-acylation complex”. The lack of favorable active site inhibitor contacts is likely to render the small β-lactams sensitive to retarded acylation when the M69L mutation occurs near the active site. Part of this retarded acylation may be due to M69V perturbing the stereochemistry associated with the oxyanion hole as is found in the X-ray structures of inhibitors bound to M69V-E166A. This is discussed in the following section, along with changes in other key active site residues caused by the M69V substitution.
In our experiments involving SHV crystals, acylation by the inhibitors is slow and a lag phase of 10–30 minutes is seen before the maximum enamine population is observed (21). Acylation is undoubtedly retarded by the presence of the E166A mutation, since E166A plays a role in the acylation step, probably acting as a general base. Of course, this factor is the same for both the E166A and M69V-E166A species. Our present observations pertain to the crystal phase and given the subtlety of the interactions in the active site, direct comparison to the solution state cannot be made. Considerable insight comes from observing the equilibrium enamine population set up in the crystals. For E166A SHV, soaking with 5 mM tazobactam in the mother liquor leads to full active site occupancy by the trans-enamine intermediate, whereas sulbactam and clavulanic acid reach only 45% and 70% occupancy, respectively. However, by raising the concentration of sulbactam and clavulanic acid to more than 10 mM, ≥80% active site occupancy of the respective enamine is achieved (22). This indicates immediately that this is an equilibrium involving productive (for acylation) complexes in the active sites and that raising the concentration of ligand increases the rate of formation of the imine-to-cis enamine-to-trans enamine populations.
Disappearance of the trans-enamine likely occurs via equilibriation with the imine complex and which subsequently hydrolyzes or undergoes irreversible inactivation (Scheme 1). However, the X-ray structures for E166A (22, 23) show the presence of only trans enamine for all three inhibitors. Moreover, hydrolysis is likely to be slow since the key base for deacylation E166 has been removed. Thus, we posit that the greater part of the differences we see for the steady state enamine population levels in E166A SHV crystals is caused by differential acylation with tazobactam being the most efficient acylating substrate and sulbactam the least.
These arguments can be extended to the double mutant M69V-E166A. The M69V replacement has its greatest effect on the enamine population from tazobactam in the M69V-E166A crystal. At 5 mM soaking conditions the steady state population is about 60% that seen for E166A alone. Raising the tazobactam to 10mM in the mother liquor does not increase the level (unpublished results), but we were able to reach ≥80% occupancy with the 50 mM soaks utilized for the X-ray analysis. This indicates a higher threshold of tazobactam is needed to increase productive encounter complexes. Assuming that deacylation rates are approximately constant for all three compounds, the active site differences between E166A and M69V-E166A β-lactamase that reduce the level of enamine from tazobactam (Figure 5) have a lesser effect on clavulanic acid (Figure 6) and no detectable effects on the enamine population from sulbactam (Figure 7).
The idea that tazobactam, sulbactam and clavulanic acid are poor acylating agents due to the lack of suitable interactions and steric complementarity with the active site also provides a rationale as to why the inhibitor-resistant β-lactamase is still able to hydrolyze penicillin-based substrates. For the inhibitors, acylation is a critical event and mutations in the enzyme bring the rate of acylation below the level to achieve complete inhibition. However, penicillin-like substrates are larger molecules that have more opportunities for hydrophobic or specific interactions with active site groups. Thus, penicillins likely reside in the active site for longer compared to the inhibitors and this increases the chances of a successful acylation event. The more efficient acylating properties of the penicillins make them less vulnerable to critical reductions in enzyme efficiency when point mutations occur in or near the active site.
We now analyze the structural differences in the single and double mutation enamine complexes to look for clues as to why the M69V substitution leads to a reduction in acylation efficiency.
Changes in the oxyanion hole may compromise acylation in the M69V-E166A variant
Insight into the acylation step would be facilitated by X-ray crystallographic analysis of Michaelis-type complexes. However for tazobactam, sulbactam and clavulanic acid with Ser70 mutated to glycine or alanine, these complexes have eluded characterization by X-ray or Raman crystallography. One likely reason for this has been discussed - the lack of sufficient ligand-active site contacts may prevent formation of “tight” Michaelis complexes with the ligand firmly bound in a unique orientation and productive in the active site. Given this situation we will discuss structural comparisons for the trans-enamine complexes between E166A and M69V-E166A variants. The findings are that there are small, but reproducible, differences involving key residues, most notably in the oxyanion hole, that can affect the initial acylation step.
When the three M69V/E166A inhibitor bound protein structures are superimposed onto the respective E166A structures, a consistent shift is observed in two adjacent β-strands that flank one side of the active site (Figure 4) and the largest shift is in β-strand residues 243–246 with a maximal shift of about 0.6–0.7 Å. The second largest β-strand shift is observed for residues 235 to 240 with the largest shift around 0.3Å with respect to the single mutant structures as observed for each of the three complexes (Figures 4 and 8). The 235–240 region of the b3 β-strand forms part of the oxyanion hole of the active site. Herein, the transition state interactions with the backbone nitrogen of A237 (together with the backbone nitrogen of S70) are stabilized.
Figure 8.
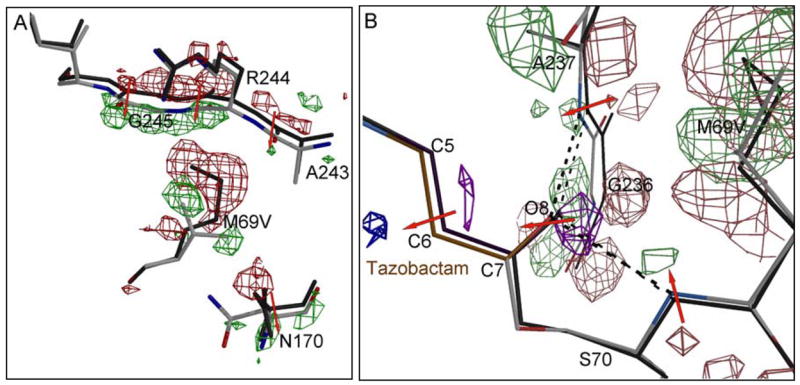
Crystallographically observed shifts in the vicinity of the M69V mutation. A) the |Fobs, M69V/E166A|-|Fobs, E166A | electron density map is contoured at 3.5 σ (green) and −3.5 σ (red). The direction of atomic shifts is indicated by a red arrow. In addition to the M69V mutation, the shifts in the strand 243–246 is readily observed in the |Fo|-|Fo| map via the positive and negative shifts peaks (phases obtained from the M69V model). The latter strand movement is likely a result of the energetic need to partially fill up the void generated by the M69V mutation. In addition, residue N170 shifted as well to accommodate the branced valine side chain. B) the |Fobs, M69V/E166A|-|Fobs, E166A | electron density map near the oxy-anion hole and acyl-bond is contoured at 3.0 σ and −3.0 σ. Positive and negative shift peaks in the protein are colored green and red, whereas positive and negative shift peaks in tazobactam are colored purple and blue, respectively.
The V69 substitution has resulted in small, but significant shifts in the neighboring residues N170 and A237, thus widening the space for the valine side chain (Supplementary Material, Figure 3S). The V69 main chain displacement has an effect in the same direction on the position of the main chain of S70 since it is the next residue in the peptide chain. Due to the shift in the main chain of residues 69 and 70, the side chain of F72 moves also in the same general direction to keep the van der Waals interactions intact (Supplementary Material, Figure 3S).
These shifts described herein are all slight (in the 0.2–0.4 Å range) yet are consistent within the two sets of three structures and are significantly higher than the r.m.s.d. values of 0.13–0.15 Å for all Cα-atoms between each single and double mutant set. In addition, these shifts are readily detected in an unbiased Fo-Fo map (Figure 8). An interesting and likely important net effect of these changes is that the oxyanion hole is 0.20 Å shorter in all 3 double mutant structures compared to their single mutant counterparts: the backbone nitrogens of S70 and A237 forming the oxyanion hole move towards each other in the three double mutant structures. This shortening of the oxy-anion hole results in a small but detectable shift in the Fo-Fo map of the O8 atom, which occupies the oxy-anion hole, for all three inhibitors. This is illustrated in Figure 8 for the tazobactam complex and is virtually identical for the other two complexes.
Although the position of the active site residue side chains in the M69V/E166A structures are identical to that observed in the respective E166A complexed and wt structures, notable differences are observed for the conserved residues K73, K234, and S130. Interestingly, these three residues adopt alternate conformations in the M69V-E166A compared to the single E166A mutant structures. In the clavulanic acid bound M69V-E166A structure, all three side chains move, whereas in the sulbactam bound structure only residues K73 and S130 adopt alternate conformations (Figure 4, main text & Figure 4S, Supplementary Material). In contrast, only residue S130 assumes an alternate conformation in the tazobactam bound double mutant structure. These three conserved residues are important for structural integrity and catalysis, although in contrast to the changes in the oxyanion hole discussed above it is difficult to immediately relate the appearance of alternate conformers to changes in acylation.
Conclusion
Using Raman spectroscopy and X-ray crystallography we have compared the trans-enamine intermediates formed by tazobactam, sulbactam and clavulanic acid in the deacylation deficient SHV-1 β-lactamase E166A with those in the enzyme that carries the M69V mutation (M69V-E166A) confering inhibitor resistance. Both the Raman and crystallographic data show that the conformations of the trans-enamines are very similar for the E166A and M69V-E1666A enzymes. However, a population analysis based on the Raman data shows that at least two of the inhibitors, tazobactam and clavulanic acid, acylate the M69V-E166A variant less efficiently. The X-ray results show that this may, in part, be due a subtle change in the oxyanion hole geometry that effects stabilization of the transition state en route to the acyl enzyme.
Supplementary Material
Figure 1S: Raman difference spectra of the E166A and M69V-E166A β-lactamase crystal with sulbactam 5mM in the mother liquor (Hepes 0.1M, pH 7). The vertical bar represents the intensity of a 5 000 photon event.
Figure 2S: Raman difference spectra of the E166A and M69V-E166A β-lactamase crystal with clavulanic acid 5mM in the mother liquor (Hepes 0.1M, pH 7). The vertical bar represents the intensity of a 5 000 photon event
Figure 3S: Conformational changes in the vicinity of the M69V mutation. Panel A: Electron density of the M69V mutation site within the tazobactam bound structure. 2Fo-Fc electron density contoured at 1.0 σ is shown for residues 67–70. Part of the covalently bound tazobactam inhibitor (tazo) is depicted in thick ball-and-stick representation. Panel B: stereo figure of a close-up superposition of the active site of the M69V/E166A and E166A single mutant SHV-1 structures. Shown are the three inhibitor bound E166A inhibitor bound structures (red lines) and the M69V/E166A variant structures (black thicker lines). The site of the inhibitors is highlighted as well as the acyl bond between the S70 and inhibitors. The oxyanion hole interactions between the O8 atom of the inhibitor and the backbone nitrogen atoms of S70 and A237 are shown as dashed lines. Atomic shifts consistently observed in all three double mutant structures with respect to the single mutant structures are highlight by black arrows indicating the direction of the shift.
Figure 4S: Alternate conformations of active site residues observed in the E166A/M69V double mutant structures. Depicted are the 2Fo-Fc electron density (green contoured at 1 σ) and positive omit electron density prior to including the alternate conformations (blue lines contoured at 3σ) are depicted for tazobactam (A), sulbactam (B), and clavulanic acid (C).
Figure 5S: Trans enamine intermediate of clavulanic acid (decarboxylated) linked to a derivatized serine residue. Torsional angles α, β, γ, δ are given in Table 2S.
Table 1S: Theoretical Raman shift values for the trans-enamine intermediate from clavulanic acid inhibitor linked to a serine residue, determined by Gaussian calculations
Table 2S: Torsional angles of a trans enamine intermediate of an inhibitor linked to a serine residue used for Gaussian calculations
Acknowledgments
MSH is supported by a Department of Veterans Affairs Advanced Career Development Award. RAB is supported by the Merit Review Program and NIH grant R01AI063517-01. PRC is supported by NIH GM54072. FvdA is supported by NIH grant AI062968.
Abbreviations
- S
serine
- K
lysine
- N
asparagines
- M
methionine
- R.m.s.d
Root-mean-square-deviation
- wt
wild-type
- TEM
Temoneira
- SHV
sulfhydryl reagent variable
- and MIC
minimal inhibitory concentration
References
- 1.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chemical Reviews. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 2.Helfand MS, Bonomo RA. β-lactamase: a survey of protein diversity. Curr Drug Targets Infect Disord. 2003;3:9–23. doi: 10.2174/1568005033342181. [DOI] [PubMed] [Google Scholar]
- 3.Bush K. The impact of β-lactamases on the development of novel antimicrobial agents. Curr Opin Investig Drugs. 2002;3:1284–1290. [PubMed] [Google Scholar]
- 4.Georgopapadakou NH. β-Lactamase inhibitors: evolving compounds for evolving resistance targets. Expert Opin Investig Drugs. 2004;13:1307–1318. doi: 10.1517/13543784.13.10.1307. [DOI] [PubMed] [Google Scholar]
- 5.Lee N, Yuen K, Kumana C. Clinical Role of β-Lactam/β-Lactamase Inhibitor Combinations. Drugs. 2003;63:1511–1524. doi: 10.2165/00003495-200363140-00006. [DOI] [PubMed] [Google Scholar]
- 6.Therrien C, Levesque RC. Molecular basis of antibiotic resistance and β-lactamase inhibition by mechanism-based inactivators: perspectives and future directions. FEMS Microbiol Rev. 2000;24:251–262. doi: 10.1111/j.1574-6976.2000.tb00541.x. [DOI] [PubMed] [Google Scholar]
- 7.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys 73 in a concerted base promotion of Ser 70. J Am Chem Soc. 2005;127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 8.Bonomo RA, Liu J, Chen Y, Ng L, Hujer AM, Anderson VE. Inactivation of CMY-2 β-lactamase by tazobactam: initial mass spectroscopic characterization. Biochem Biophys Acta. 2001;1547:196–205. doi: 10.1016/s0167-4838(01)00175-3. [DOI] [PubMed] [Google Scholar]
- 9.Brown RP, Aplin RT, Schofield CJ. Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry. 1996;35:12421–12432. doi: 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- 10.Fisher J, Belasco JG, Charnas RL, Khosla S, Knowles JR. β-lactamase inactivation by mechanism-based reagents. Philos Trans R Soc Lond B Biol Sci. 1980;289:309–319. doi: 10.1098/rstb.1980.0048. [DOI] [PubMed] [Google Scholar]
- 11.Fisher J, Belasco JG, Khosla S, Knowles JR. β-Lactamase proceeds via an acyl-enzyme intermediate. Interaction of the Escherichia coli RTEM enzyme with cefoxitin. Biochemistry. 1980;19:2895–2901. doi: 10.1021/bi00554a012. [DOI] [PubMed] [Google Scholar]
- 12.Fisher J, Charnas RL, Bradley SM, Knowles JR. Inactivation of the RTEM β-lactamase from Escherichia coli. Interaction of penam sulfones with enzyme. Biochemistry. 1981;20:2726–2731. doi: 10.1021/bi00513a004. [DOI] [PubMed] [Google Scholar]
- 13.Fisher J, Charnas RL, Knowles JR. Kinetic studies on the inactivation of Escherichia coli RTEM β-lactamase by clavulanic acid. Biochemistry. 1978;17:2180–2184. doi: 10.1021/bi00604a024. [DOI] [PubMed] [Google Scholar]
- 14.Charnas RL, Knowles JR. Inactivation of RTEM β-lactamase from Escherichia coli by clavulanic acid and 9-deoxyclavulanic acid. Biochemistry. 1981;20:3214–3219. doi: 10.1021/bi00514a035. [DOI] [PubMed] [Google Scholar]
- 15.Brenner DG, Knowles JR. Penicillanic acid sulfone: an unexpected isotope effect in the interaction of 6 alpha- and 6β-monodueterio and of 6, 6 dideuterio derivatives with RTEM β-lactamase from Escherichia coli. Biochemistry. 1981;20:3680–3687. doi: 10.1021/bi00516a003. [DOI] [PubMed] [Google Scholar]
- 16.Brenner DG, Knowles JR. 6-(Methoxymethylene) penicillanic acid: inactivator of RTEM β-lactamase from Escherichia coli. Biochemistry. 1984;23:5839–5846. doi: 10.1021/bi00319a025. [DOI] [PubMed] [Google Scholar]
- 17.Brenner DG, Knowles JR. Penicillanic acid sulfone: nature of irreversible inactivation of RTEM β-lactamase from Escherichia coli. Biochemistry. 1984:5833–5839. doi: 10.1021/bi00319a024. [DOI] [PubMed] [Google Scholar]
- 18.Charnas RL, Fisher J, Knowles JR. Chemical studies on the inactivation of Escherichia coli RTEM β-lactamase by clavulanic acid. Biochemistry. 1978;17:2185–2189. doi: 10.1021/bi00604a025. [DOI] [PubMed] [Google Scholar]
- 19.Hermann JC, Hensen C, Ridder L, Mulholland AJ, Holtje HD. Mechanisms of Antibiotic Resistance:QM/MM Modeling of the Acylation Reaction of Class A β-Lactamase with Benzylpenicillin. J Am Chem Soc. 2005;127:4454–4465. doi: 10.1021/ja044210d. [DOI] [PubMed] [Google Scholar]
- 20.Thomson JM, Bonomo RA. The threat of antibiotic resistance in Gram-negative pathogenic bacteria: β-lactams in peril! Curr Opin Microbiol. 2005;8:518–524. doi: 10.1016/j.mib.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 21.Helfand MS, Totir MA, Carey MP, Hujer AM, Bonomo RA, Carey PR. Following the Reactions of Mechanism-Based Inhibitors with β-Lactamase by Raman Crystallography. Biochemistry. 2003;42:13386–13392. doi: 10.1021/bi035716w. [DOI] [PubMed] [Google Scholar]
- 22.Padayatti PS, Helfand MS, Totir MA, Carey MP, Carey PR, Bonomo RA, van den Akker F. High Resolution Crystal Structures of the trans-Enamine Intermediates Formed by Sulbactam and Clavulanic Acid and E166A SHV-1 {beta}-Lactamase. J Biol Chem. 2005;280:34900–34907. doi: 10.1074/jbc.M505333200. [DOI] [PubMed] [Google Scholar]
- 23.Padayatti PS, Helfand MS, Totir MA, Carey MP, Hujer AM, Carey PR, Bonomo RA, Akker Fvd. Tazobactam Forms a Stoichiometric trans-Enamine Intermediate in the E166A Variant of SHV-1 β-Lactamase: 1.63 Å Crystal Structure. Biochemistry. 2004;43:843–848. doi: 10.1021/bi035985m. [DOI] [PubMed] [Google Scholar]
- 24.Dubois V, Poirel L, Arpin C, Coulange L, Bebear C, Nordmann P, Quentin C. SHV-49, a Novel Inhibitor-Resistant {beta}-Lactamase in a Clinical Isolate of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2004;48:4466–4469. doi: 10.1128/AAC.48.11.4466-4469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helfand MS, Hujer AM, Sonnichsen FD, Bonomo RA. Unexpected Advanced Generation Cephalosporinase Activity of the Met69Phe Variant of SHV β-Lactamase. Journal of Biological Chemistry. 2002;277:47719–47723. doi: 10.1074/jbc.M207271200. [DOI] [PubMed] [Google Scholar]
- 26.Bonomo RA, Knox JR, Rudin SD, Shlaes DM. Construction and characterization of an OHIO-1 beta-lactamase bearing Met69Ile and Gly238Ser mutations. Antimicrobial Agents and Chemotherapy. 1997;41:1940–1943. doi: 10.1128/aac.41.9.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin S, Thomas M, Shlaes DM, Rudin SD, Knox JR, Anderson VE, Bonomo RA. Kinetic analysis of an inhibitor-resistant variant of the OHIO-1 β-lactamase, an SHV-family class A enzyme. Biochem J. 1998;333(Pt 2):395–400. doi: 10.1042/bj3330395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meroueh SO, Roblin P, Golemi D, Maveyraud L, Vakulenko SB, Zhang Y, Samama JP, Mobashery S. Molecular Dynamics at the Root of Expansion of Function in the M69L Inhibitor-Resistant TEM β-Lactamase from Escherichia coli. Journal of the American Chemical Society. 2002;124:9422–9430. doi: 10.1021/ja026547q. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Minasov G, Shoichet BK. The Structural Bases of Antibiotic Resistance in the Clinically Derived Mutant beta -Lactamases TEM-30, TEM-32, and TEM-34. J Biol Chem. 2002;277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 30.Kuzin AP, Nukaga M, Nukaga Y, Hujer AM, Bonomo RA, Knox JR. Inhibition of the SHV-1 β-lactamase by sulfones: crystallographic observation of two reaction intermediates with tazobactam. Biochemistry. 2001;40:1861–1866. doi: 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]
- 31.Hujer AM, Hujer KM, Bonomo RA. Mutagenesis of amino acid residues in the SHV-1 β-lactamase: the premier role of Gly238Ser in penicillin and cephalosporin resistance. Biochimica et Biophysica Acta (BBA)/Protein Structure and Molecular Enzymology. 2001;1547:37–50. doi: 10.1016/s0167-4838(01)00164-9. [DOI] [PubMed] [Google Scholar]
- 32.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt10):1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 33.CCP4. The CCP4 suite: Programs for computational crystallography. Acta Crystallogr D. 1994;55:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.van Aalten DM, Bywater R, Findlay JB, Hendlich M, Hooft RW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput Aided Mol Des. 1996;10:255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 36.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A 47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 37.Altose MD, Zheng Y, Dong J, Palfey BA, Carey PR. Comparing protein-ligand interactions in solution and single crystals by Raman spectroscopy. Proceedings of the National Academy of Sciences. 2001;98:3006–3011. doi: 10.1073/pnas.061029598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong J, Swift K, Matayoshi E, Nienaber VL, Weitzberg M, Rockway T, Carey PR. Probing Inhibitors Binding to Human Urokinase Crystals by Raman Microscopy: Implications for Compound Screening. Biochemistry. 2001;40:9751–9757. doi: 10.1021/bi010955+. [DOI] [PubMed] [Google Scholar]
- 39.Frisch A, Frisch MJ, Trucks GW. Gaussian 03. Gaussian Inc.; Carnegie, PA: 2003. [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK- a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- 41.Foresman J, Frisch A. Exploring Chemistry with Electronic Methods. 2. Gaussian Inc.; Pittsburgh, PA.: 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S: Raman difference spectra of the E166A and M69V-E166A β-lactamase crystal with sulbactam 5mM in the mother liquor (Hepes 0.1M, pH 7). The vertical bar represents the intensity of a 5 000 photon event.
Figure 2S: Raman difference spectra of the E166A and M69V-E166A β-lactamase crystal with clavulanic acid 5mM in the mother liquor (Hepes 0.1M, pH 7). The vertical bar represents the intensity of a 5 000 photon event
Figure 3S: Conformational changes in the vicinity of the M69V mutation. Panel A: Electron density of the M69V mutation site within the tazobactam bound structure. 2Fo-Fc electron density contoured at 1.0 σ is shown for residues 67–70. Part of the covalently bound tazobactam inhibitor (tazo) is depicted in thick ball-and-stick representation. Panel B: stereo figure of a close-up superposition of the active site of the M69V/E166A and E166A single mutant SHV-1 structures. Shown are the three inhibitor bound E166A inhibitor bound structures (red lines) and the M69V/E166A variant structures (black thicker lines). The site of the inhibitors is highlighted as well as the acyl bond between the S70 and inhibitors. The oxyanion hole interactions between the O8 atom of the inhibitor and the backbone nitrogen atoms of S70 and A237 are shown as dashed lines. Atomic shifts consistently observed in all three double mutant structures with respect to the single mutant structures are highlight by black arrows indicating the direction of the shift.
Figure 4S: Alternate conformations of active site residues observed in the E166A/M69V double mutant structures. Depicted are the 2Fo-Fc electron density (green contoured at 1 σ) and positive omit electron density prior to including the alternate conformations (blue lines contoured at 3σ) are depicted for tazobactam (A), sulbactam (B), and clavulanic acid (C).
Figure 5S: Trans enamine intermediate of clavulanic acid (decarboxylated) linked to a derivatized serine residue. Torsional angles α, β, γ, δ are given in Table 2S.
Table 1S: Theoretical Raman shift values for the trans-enamine intermediate from clavulanic acid inhibitor linked to a serine residue, determined by Gaussian calculations
Table 2S: Torsional angles of a trans enamine intermediate of an inhibitor linked to a serine residue used for Gaussian calculations