

Abstract
The zymogen granule is the specialized organelle in pancreatic acinar cells for digestive enzyme storage and regulated secretion and is a classic model for studying secretory granule function. Our long term goal is to develop a comprehensive architectural model for zymogen granule membrane (ZGM) proteins that would direct new hypotheses for subsequent functional studies. Our initial proteomics analysis focused on identification of proteins from purified ZGM (Chen, X., Walker, A. K., Strahler, J. R., Simon, E. S., Tomanicek-Volk, S. L., Nelson, B. B., Hurley, M. C., Ernst, S. A., Williams, J. A., and Andrews, P. C. (2006) Organellar proteomics: analysis of pancreatic zymogen granule membranes. Mol. Cell. Proteomics 5, 306–312). In the current study, a new global topology analysis of ZGM proteins is described that applies isotope enrichment methods to a protease protection protocol. Our results showed that tryptic peptides of ZGM proteins were separated into two distinct clusters according to their isobaric tag for relative and absolute quantification (iTRAQ) ratios for proteinase K-treated versus control zymogen granules. The low iTRAQ ratio cluster included cytoplasm-orientated membrane and membrane-associated proteins including myosin V, vesicle-associated membrane proteins, syntaxins, and all the Rab proteins. The second cluster having unchanged ratios included predominantly luminal proteins. Because quantification is at the peptide level, this technique is also capable of mapping both cytoplasm- and lumen-orientated domains from the same transmembrane protein. To more accurately assign the topology, we developed a statistical mixture model to provide probabilities for identified peptides to be cytoplasmic or luminal based on their iTRAQ ratios. By implementing this approach to global topology analysis of ZGM proteins, we report here an experimentally constrained, comprehensive topology model of identified zymogen granule membrane proteins. This model contributes to a firm foundation for developing a higher order architecture model of the ZGM and for future functional studies of individual ZGM proteins.
The acinar cells of the exocrine pancreas are the functional units of digestive enzyme synthesis, storage, and secretion. Digestive enzymes are stored in large vesicles within these cells known as zymogen granules (ZGs).1 The ZG is the secretory organelle responsible for transport, storage, and secretion of digestive enzymes and has long been a model for understanding secretory granule functions (1, 2). It is believed that the ZG membrane (ZGM) carries at least part of the molecular machinery responsible for digestive enzyme sorting, granule trafficking, and exocytosis. Therefore elucidating the ZGM molecular architecture is critical for studying ZG function. The overall goal of our studies is to build a quantitative, architectural model of the ZGM that will direct new hypotheses for subsequent functional analysis of this prototypic secretory granule. This model will ultimately comprise not only the complete protein components of the ZGM but also their membrane topologies, absolute quantities, and protein complexes with which they are associated. As the first step toward this goal, we recently conducted the first comprehensive proteomics analysis of ZGM and identified over 100 proteins (3). The next question to be addressed is how these proteins are organized across the ZGM, namely the membrane topology of ZGM proteins.
The topological organization of a ZGM protein relative to the lipid bilayer dictates its accessibility to interacting partners and modifying enzymes. Therefore, an accurate topology model describing the number of transmembrane spans and the orientation of a ZGM protein is essential for understanding its correct function. This importance is highlighted in the case of syncollin that was originally suggested as a Ca2+-sensitive regulator of the SNARE complex but then identified as a luminal peripheral membrane protein likely playing a role in ZG maturation (4). Despite the importance of membrane topology, there is currently no experimental method able to derive full-topology models in a global manner. The development of a topology model still largely relies on sequence-based computational algorithms to predict transmembrane domains. However, such predictions are not always consistent among different algorithms and usually do not clarify the orientation of a protein. Therefore, experimentally determined reference points are needed to constrain the topology prediction. Traditionally a protease protection assay or glycosylation mapping is used to obtain topology information on an individual protein basis. Very recently, large scale topology mappings have been applied to Escherichia coli and yeast to determine the locations of the membrane protein carboxyl termini using fusion proteins with topology reporters (5, 6). In mammalian cells, a fluorescence-based protease protection technique was introduced to characterize the topology of a small set of green fluorescent protein fusion proteins in live cells (7). Both of the above strategies required exogenous expression of fusion proteins, which can be labor-intensive and may sometimes introduce artifacts.
Because of its ability to analyze endogenous proteins, a mass spectrometry-based proteomics approach provides a promising alternative to the above strategies. In a pioneering study, Wu et al. (8) reported the topology analysis of a number of Golgi membrane proteins and demonstrated the application of shotgun proteomics to high throughput topology analysis. However, the method did not provide a means for relative quantification between samples (9); therefore the potential of quantitative proteomics for global topology mapping of organellar membrane proteins has not been fully exploited. The value of a quantitative approach to topology is that it allows development of a more powerful statistical model to discriminate between states. In the current study, as a second step toward a comprehensive architectural model of the ZGM, we combined a global protease protection analysis with iTRAQ-based quantification and developed a statistical model to assign topologies to membrane protein domains. By applying this method to systematic topology analysis of endogenous ZGM proteins, we significantly extended the proteins identified on ZGM and more importantly provided experimentally constrained topology information for all identified proteins. This comprehensive topological map of ZGM proteins bridges between cataloging individual ZGM proteins and building protein-protein interaction networks of ZGM. It will aid development of interaction models and provides a firm foundation for future functional studies of individual ZGM proteins.
EXPERIMENTAL PROCEDURES
Reagents and Materials—
Percoll was purchased from Amersham Biosciences, sequencing grade modified trypsin was from Promega (Madison, WI), and proteinase K was from Invitrogen. iTRAQ™ reagents were from Applied Biosystems (Foster City, CA). α-Cyano-4-hydroxycinamic acid and other reagents were obtained from Sigma. Anti-Rab27B, anti-Rab3D, and anti-secretory carrier membrane protein (SCAMP) 1 antibodies were gifts from Drs. T. Izumi, M. McNiven, and D. Castle, respectively. Anti-synaptotagmin-like protein 1 (Slp1), anti-myosin Vc, and anti-polymeric immunoglobulin receptor (pIgR) antibodies were gifts from Drs. S. Catz, R. Cheney, and C. Okamoto, respectively. Anti-syntaxin 7 antibody was from Synaptic Systems (Goettingen, Germany), and anti-amylase antibody was from Sigma. Anti-ectonucleoside triphosphate diphosphohydrolase 1 (ENTP1) and anti-Rap1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Strong cation exchange (SCX) MicroSpin™ columns were from The Nest Group, Inc. (Southborough, MA). The Zorbax C18 reversed-phase cartridge and Zorbax 300 SB C18 reversed-phase analytical column were purchased from Agilent (Palo Alto, CA). HPLC grade water and acetonitrile (Optima) were purchased from Fisher. All chemicals were of analytical grade and used as received.
Isolation of Zymogen Granules and Purification of Zymogen Granule Membranes—
ZGs and ZGM were purified as described in our early proteomics study (3), and more details are available in a more recent publication (10). After the procedure, purified ZGMs were pelleted and stored at −80 °C until use.
Proteinase K Digestion of Intact Zymogen Granules—
For protease protection studies, ZGs purified in the Percoll gradient were collected and directly diluted in homogenization buffer without any additional centrifugation step to minimize mechanical shearing. No protease inhibitors were included in Percoll gradient and the dilution buffer to avoid interference with subsequent protease digestion. Proteinase K was chosen in the study because of its nonspecificity and high activity at low temperature and neutral pH. The ZG suspension was divided evenly in four groups. Proteinase K (∼150 μg each) was added to two groups of ZGs at an estimated enzyme to substrate ratio of 1:50 (mass to mass). An equal amount of buffer was added to the other two groups of ZGs as controls. The ZGs were incubated at 4 °C for 15 or 30 min. After digestion, ZG suspensions were diluted with homogenization buffer containing 1 mm fresh PMSF to terminate digestion. ZGs were collected by centrifuging at 1700 × g for 10 min at 4 °C. The supernatants were removed, and ZGs were resuspended in ZG lysis buffer. ZG membranes were purified as described above under “Isolation of Zymogen Granules and Purification of Zymogen Granule Membranes,” and the ZG content from each group was saved.
In-solution Digestion and iTRAQ Labeling—
For in-solution digestion, ZGM pellets were solubilized on ice in 50 μl of buffer containing 500 mm tetraethylammonium bicarbonate, 8 m urea, and 0.4% SDS. Protein concentrations were determined with the Bio-Rad Bradford assay kit. ZGM proteins (30 μg) from each group were reduced with 5 mm tris(2-carboxyethyl)phosphine for 1 h at 37 °C, and then cysteines were blocked with 10 mm methyl methanethiosulfonate for 20 min at room temperature. The protein solution was diluted four times with 0.5 m tetraethylammonium bicarbonate containing 3 μg of trypsin (Promega sequencing grade, 1:10, w/w) and incubated at 37 °C overnight. To quantitatively distinguish protease-sensitive and -protected peptides, multiplexed isobaric tags (iTRAQ reagents) were used to label tryptic peptides from control and proteinase K-treated ZGM samples, respectively (114 and 115 iTRAQ reporters for controls and 116 and 117 iTRAQ reporters for proteinase K-treated ZGMs). The labeling procedure used was according to the protocol provided by the manufacturer. The reaction was stopped by diluting the mixture with 10 volumes of SCX buffer containing 10 mm KH2PO4 and 15% acetonitrile with pH adjusted to 3.
2D LC-MALDI-MS/MS—
As described in our early proteomics study (3), the combined iTRAQ-labeled peptide mixture was first fractionated on a SCX MicroSpin column. The eluate from each salt step was first desalted and concentrated on a reversed-phase cartridge (Zorbax C18; 5 mm × 0.3-mm inner diameter; 5-μm particles; Agilent) and then separated by a reversed-phase column (Zorbax 300 SB C18 column, 75 μm × 150 mm, 3.5-μm particles) on an Agilent 1100 HPLC system. The column effluent was mixed with MALDI matrix (2 mg/ml α-cyano-4-hydroxycinnamic acid) and spotted on 1536-well OptiTOF™ MALDI target plates that were later analyzed by tandem mass spectrometry.
The MS and MS/MS spectra were acquired on an Applied Biosystems 4800 Proteomics Analyzer (TOF/TOF) (Applied Biosystems/MDX Sciex, Foster City, CA) in positive ion reflection mode with a 200-Hz neodymium-doped yttrium aluminium garnet (Nd:YAG) laser operating at 355 nm. Accelerating voltage was 20 kV with a 400-ns delay. For MS/MS spectra, the collision energy was 2 keV, and the collision gas was air. Each MALDI plate was calibrated on nine calibration wells using standards from Applied Biosystems with a 20-ppm mass accuracy in the MS mode. Both MS and MS/MS data were then acquired in the sample wells using the instrument default calibration. Typical MS spectra were obtained with the minimum possible laser energy to maintain the best resolution. Single stage MS spectra for the entire samples were collected first, and in each sample well MS/MS spectra were acquired from the 12 most intense peaks above the signal to noise ratio threshold of 30.
Database Search and Statistic Analysis of iTRAQ Results—
Database searching was performed using Applied Biosystems GPS Explorer™ v3.6. This software interacts directly with the Oracle database in which the mass spectrometer stores its data and submits monoisotopic peak lists in batch to a local version of the Mascot search engine (v2.1) for protein identities (11). No additional peak list filtering was specified. Peak lists were generated by the mass spectrometer during data acquisition based on a specified signal to noise threshold (30 in this case). To estimate the false positive rate (FPR) of peptide identifications in each data set, a target-decoy database was generated by manually concatenating the forward and reverse sequences in the International Protein Index (IPI) rat database (version 3.18 with 38,873 of Rattus norvegicus proteins and 77,746 total sequences). The decoy database method is being increasingly used as an independent estimate of FPR in database searching (12, 13). At any given probability threshold, the number of matches to reversed sequences can be counted and compared with the total number of peptide assignments above that threshold to derive an estimate of the FPR.
The database searches were performed with the following parameters: up to one missed cleavage, amino-terminal and lysine modification by iTRAQ reagent and methyl methanethiosulfonate modification on cysteine as fixed modifications, and methionine oxidation as a variable modification. For all the searches, precursor ion mass tolerance was set as 150 ppm, and fragment ion mass tolerance was defined as 0.6 Da. A stringent threshold of 1% FPR (13, 14), which corresponds to a Mascot ion score ≥29 and GPS Explorer ion score confidence interval greater than 90%, was used to include peptides for protein identification and peptide quantification. All the unique peptides were manually examined to generate a minimal list of protein identifications according to the strategy proposed by Nesvizhskii et al. (15, 16). To conclude the identification of a specific isoform in a protein family (e.g. Rabs), at least one unique peptide has to be detected that is not shared by other isoforms in the same protein family. In a few incidences where different isoforms (e.g. Rap1A and -1B) could not be distinguished based on the detected peptides, the common protein name (e.g. Rap1) was reported without specifying its isoform. Spectra with de novo sequence annotations were interpreted and documented using a locally developed software program, MSExpedite available through ProteomeCommons.
For quantification, the peptide identifications and their iTRAQ ratios were exported to Excel from the GPS Explorer™ MS/MS summary tables. The software exports ratios based on peak areas for each iTRAQ label. The table was sorted by ion scores in a descending order, and only peptides within 1% FPR, again corresponding to Mascot ions score ≥29, were selected for further analysis. The identified peptides were grouped according to their protein names and accession numbers. The peptides identified as the same sequence from multiple sample wells were counted as one unique peptide, and an average iTRAQ ratio and its corresponding S.D. was obtained for each unique peptide. For double duplex experiments, a ratio between the average of two proteinase K-treated samples and that of two control samples, referred to as “PK/CT,” was used for each unique peptide. Abundance ratio calculations included corrections for overlapping isotopic contributions (both natural and enriched 13C components).
Statistical Modeling of the iTRAQ Ratio Distributions in the Topology Studies—
A statistical model was developed to estimate the probability that any observed peptide is derived from either the luminal or the cytoplasmic fraction based on the observed iTRAQ ratio. A two-component mixture model is used (17, 18) that functions by fitting two separate curves to the observed bimodal distribution of all peptide iTRAQ ratios in each data set (Fig. 4). For these data, a normal curve is used to model the lower ratio cytosolic fraction, and a γ distribution is used for the luminal fraction. The distributions are fitted to the data in a semisupervised manner: fundamentally an expectation-maximization algorithm is used to learn an optimal fit for the distributions to the data set in an automated way starting from training distributions of peptides known to be cytosolic and luminal, but the fit is assisted by setting guiding parameters for intensity and shape based on both the training data and manual curation. The expectation-maximization algorithm is an iterative, two-step optimization approach in which the parameters of each distribution are used to calculate, at each iteration, the probability of each peptide belonging to both the cytosolic and luminal fractions. These probabilities are then used to adjust the distribution shapes by weighting the contribution of each peptide ratio to each of the two distributions in a manner proportional to its probability of being in that distribution. The algorithm proceeds in this manner, by successively recalculating probabilities after each adjustment of the curve and then adjusting the curve based on these weighted probabilities, until a convergence is achieved. The probabilities of each peptide are then reported as a Bayesian probability. For the cytosolic fraction the probability of the peptide being cytosolic given a peptide ratio r is calculated as follows.
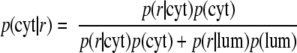 |
(Eq. 1) |
The numerator of this formula may be interpreted as the probability of having the iTRAQ ratio of r and being cytosolic, and the denominator may be interpreted as the overall probability of having the iTRAQ ratio r. The p(r|cyt) and p(r|lum) are the values calculated for the cytosolic and luminal distributions, respectively, at a given value of r; finally the p(cyt) and p(lum) terms are the “prior” proportion of cytosolic to luminal peptides in the data set. The luminal probability at any value of r is calculated in an analogous manner. For these data, the probabilities of peptides known with high confidence to be either cytosolic or luminal were fixed to “1” for the corresponding fraction (and “0” for the other) and modeled in combination with the unknown peptides. This had the effect of guiding the model learning, a method utilized in Marelli et al. (17) as well.
Fig. 4.

Statistical model for estimating peptide topology. Shown are histograms for iTRAQ ratios for two independent experiments. The distribution of all peptides in each data set is shown as green. The two final curves learned by the model for both luminal (red) and cytosolic (blue) distributions are plotted on the figure. The dashed distributions indicate the histograms of the training data sets used as a starting point for the model. The training distributions have been scaled by a factor of 10.
RESULTS
Applying iTRAQ-based Quantitative Proteomics to Protease Protection Analysis—
For global topology analysis of ZGM proteins, we combined a conventional protease protection assay with iTRAQ-based quantitative proteomics. Initially pilot experiments were conducted to examine the digestion conditions. In these experiments, purified ZGs were incubated with or without proteinase K for different periods of time and at different temperatures. ZGs were then pelleted by centrifugation and solubilized in lysis buffer for Western blotting analysis. The completeness of the digestion was tested using a representative cytoplasm-orientated protein, Rab3D. As shown in Fig. 1 (right), there was little or no detectable Rab3D remaining after proteinase K digestion either at room temperature for 10 min or at 4 °C for 15 or 30 min. In contrast to Rab3D, the amount of amylase, a representative ZG luminal protein, remained unchanged for up to 30 min indicating that the ZGs remained largely intact during digestion.
Fig. 1.

Work flow of iTRAQ-based topology analysis of pancreatic ZG membrane proteins. Left, isolated ZGs were treated with or without proteinase K and then lysed. ZGM proteins were digested with trypsin, and resulting peptides were labeled with separate iTRAQ reagents. The peptides were mixed and analyzed by 2D LC-MALDI-MS/MS. Right, Western blot validation of proteinase K digestion for a representative ZG luminal protein, amylase, and a representative cytoplasm-orientated ZGM protein, Rab3D. 5 μg of proteins from ZGs treated with proteinase K for 0, 10 (room temperature), 15, and 30 min (4 °C) were analyzed. ′, min.
Based on the pilot experiments, we determined the digestion conditions and scaled up the experiments 10-fold to conduct a global protease protection study on ZGs. The work flow of the iTRAQ-based topology analysis of pancreatic ZGM proteins is illustrated in Fig. 1 (left). The purified ZGs were incubated with (proteinase K-treated groups) or without (control groups) proteinase K for 15 or 30 min at 4 °C. The ZGs were lysed, and membranes were washed with KBr and Na2CO3. ZGM pellets were solubilized and then digested with trypsin. To quantitatively distinguish protease-sensitive and protease-protected peptides, the tryptic peptides were labeled with distinct iTRAQ reagents to compare the relative abundance of each peptide in control and proteinase K-treated samples. Because proteinase K treatment should remove most cytoplasmic ZGM protein domains, we expected the peptides from these protein domains to show a significant reduction and have low iTRAQ ratios (proteinase K-treated versus control), whereas those from luminal ZGM proteins would show little change.
Proteinase K Treatment Generated Two Populations of Tryptic Peptides with Distinct iTRAQ Ratio (Proteinase K-treated Versus Control) Distributions—
In a typical global protease protection experiment, over 3200 MS/MS spectra were acquired leading to 1079 peptide identifications with a 1% or less estimated FPR by target-decoy database search. To evaluate the overall quality of the data, histograms of the iTRAQ ratio distributions of these peptides were analyzed. As shown in Fig. 2 (upper left), the iTRAQ ratios between two control groups appeared to have a normal distribution centered around 1.0 (mean ± S.D. = 1.02 ± 0.19). Similarly the iTRAQ ratios between two proteinase K-treated (15- and 30-min) groups were also distributed around 1.0. with a larger standard deviation (mean ± S.D. = 1.04 ± 0.44) compared with the control distribution because of different digestion durations (Fig. 2, upper right). By contrast, the iTRAQ ratio distributions between proteinase K-treated groups and control groups dramatically differed from a normal distribution and appeared as a bimodal distribution indicating the presence of two populations of peptides, protease-sensitive and -protected, upon proteinase K treatment (Fig. 2, bottom left and right). Upon 15-min proteinase K digestion, a relatively small population of peptides appeared to have significantly reduced iTRAQ ratios (distributed between 0 and 0.55), whereas the majority of the peptides remained largely unchanged (Fig. 2, bottom left). To further differentiate these two populations of peptides, a longer period of digestion, 30 min, was examined. Indeed more peptides had further reduced iTRAQ ratios; however, the boundary distinguishing the two populations of peptides also became less clear (Fig. 2, bottom right). Based on this observation, we hypothesized that the population with the low iTRAQ ratios contained peptides from the cytoplasm-orientated proteins and protein domains, whereas the second population with unchanged ratios included peptides from the luminal proteins and protein domains.
Fig. 2.

Histograms of tryptic peptide distributions based on different iTRAQ ratios. Tryptic peptides from four different groups of ZGMs, 15-min control, 30-min control, 15-min proteinase K treatment, and 30-min proteinase K treatment, were labeled with four different iTRAQ reagents and mixed. Histograms of peptide distributions based on different iTRAQ ratios were plotted: top left, 15-min control versus 30-min control (CT/CT); top right, 15-min proteinase K treatment versus 30-min proteinase K treatment (PK/PK); bottom left, 15-min proteinase K treatment versus 15-min control (PK/CT-15 min); bottom right, 30-min proteinase K treatment versus 30-min control (PK/CT-30 min). A total of 1079 peptides were plotted between iTRAQ ratio 0 and 1.9 with the bin size at 0.05.
To test this hypothesis, we first examined peptides from a set of ZGM proteins with known topology (Fig. 3 and supplemental Table 1). In this list, the cytoplasm-orientated proteins included molecular motor proteins dynein and myosin I, small G-proteins (five Rabs and Rap 1), and SNARE proteins VAMPs 2 and 8. The luminal proteins included seven digestive enzymes as well as colipase, GP2, GP3, Itmap1, ZG16, and syncollin whose topologies have been examined previously using conventional protease protection analyses (19–21). As shown in Fig. 3, the iTRAQ ratios (proteinase K-treated versus control for 15-min incubation) of the peptides from these proteins appeared in two distinct clusters. The peptides from cytoplasm-orientated proteins or protein domains all had low iTRAQ ratios and clustered together with a mean ± S.D. of 0.30 ± 0.15, whereas the peptides from lumen-orientated proteins or domains showed unchanged iTRAQ ratios with the mean very close to 1.0 (mean ± S.D. = 0.96 ± 0.16). The detailed information of these peptides including protein names, peptide sequence, iTRAQ ratios of each peptide, and topology annotation is provided in supplemental Table 1. Although the 15-min proteinase K treatment clearly separated the cytoplasmic peptides from luminal peptides based on their iTRAQ ratios, the 30-min treatment appeared to result in slight overdigestion. A small number of peptides from luminal proteins such as GP2 and Itmap1 showed lower iTRAQ ratios and crossed into the cytoplasmic peptides (supplemental Fig. 1 and supplemental Table 1). This suggested that prolonged exposure to proteinase K might have increased the instability of isolated ZGs and therefore the accessibility of the proteinase K to the luminal ZG proteins. For this reason, we chose a 15-min proteinase K treatment as our standard procedure and used iTRAQ ratios (PK/CT-15 min) for the global topology analysis of ZGM proteins.
Fig. 3.

iTRAQ ratio distributions of tryptic peptides from ZGM proteins with known topology. A histogram of iTRAQ ratios (proteinase K versus control-15 min) from identified peptides illustrates the presence of two distinct clusters of tryptic peptides corresponding to the peptides from cytoplasm-orientated and lumen-orientated ZGM proteins, respectively. The ZG proteins contributing to each cluster are listed on each side of the histogram, and the cytoplasmic and luminal peptides are distinguished with two separate labels in the histogram. In the three bins with overlaps between two clusters, the luminal peptides are plotted on top of the cytoplasmic peptides.
We repeated the iTRAQ-based global protease protection study multiple times under these experimental conditions, and the results were very reproducible. The iTRAQ ratio distributions (green curves) of unique peptides from two independent experiments are shown in Fig. 4. The left panel displays the same experiment as shown in Fig. 2, and the right panel displays a repeated experiment. In both experiments, over 3000 MS/MS spectra (3243 in experiment 1 and 3066 in experiment 2) were acquired in which 1079 (experiment 1) and 895 (experiment 2) peptides were identified within a 1% FPR. Similar to the first experiment, the iTRAQ ratios in the second experiment also exhibited a bimodal distribution, and peptides from the same set of ZGM proteins used in Fig. 3 also separated into two distinct clusters based on their iTRAQ ratios (supplemental Table 2).
Statistical Modeling to Classify Peptides as Cytoplasmic or Luminal with a Calculated Probability Based on Their Observed iTRAQ Ratios—
The results from ZG proteins of known topology highlighted the potential of the quantitative protease protection analysis to distinguish cytoplasmic and luminal peptides and therefore derive topologies of the corresponding proteins. For these data, the bimodal distribution observed suggests that a simple thresholding heuristic could have been utilized to classify the majority of peptides as either cytoplasmic or luminal (e.g. a ratio ≤0.5 is cytoplasmic). However, we sought to develop a more precise measure because of the presence of a number of borderline peptides as well as the fact that the most discriminative threshold tended to vary somewhat between data sets.
To accurately classify peptides as cytoplasmic or luminal based on their measured iTRAQ ratios, a simple statistical model was developed. Based on the assumption that peptides must be derived from one of these two categories, the model fits two distributions to the data set representing each of the cytoplasmic or luminal fractions in an iterative manner (see “Experimental Procedures” for details). Fig. 4 displays the final fit of the distributions to two data sets described above under “Proteinase K Treatment Generated Two Populations of Tryptic Peptides with Distinct iTRAQ Ratio (Proteinase K-treated Versus Control) Distributions.” The green plots indicate a histogram of the number of peptides identified at each indicated iTRAQ ratio with ratios binned at 0.05-unit intervals. The red and blue plots in the figure represent the learned fits of the model for the cytoplasmic and luminal fractions, respectively. Probability scores of a given peptide being derived from either fraction are then calculated based on the relative contributions of each of the distributions for any measured iTRAQ ratio. Initial starting parameters for the distributions are derived from a training set of peptides of known topology (supplemental Table 2); curves representing the starting distributions are plotted for each fraction in the figure as dashed lines. The y axis for the starting distributions is scaled by a factor of 10 in the figure to more easily visualize their shape. For each data set, probabilities for peptides of known topology are fixed at 1.0 for their respective category. These peptides assist the model in learning optimal distributions for each data set. The use of the γ distribution for the luminal fraction was deliberately chosen to provide a steep descent in the left-hand portion of the curve to provide a sharper differentiation between the cytosolic and luminal fractions.
In Fig. 4, right (experiment 2), the iTRAQ ratios of the luminal fraction averaged around 0.9 (peak at around 0.8) instead of 1.0 as in experiment 1. Correspondingly the average of the cytoplasmic fraction was also lower. We think this shift reflected the slightly unevenness of sample amounts mixed together between control and proteinase K-treated groups. We could have normalized the second experiment by increasing all the iTRAQ ratios by a factor of ∼1.1 to shift the average iTRAQ ratio of the luminal peptides to 1.0. However, we decided not to do this for the following reasons. First, the drift, less than 15%, was fairly small. Second, more importantly, we want to show that the separation of peptides into two clusters based on their iTRAQ ratios does not rely on such normalization.
Global Topology Analysis of ZGM Proteins—
Using the statistical model, we obtained probabilities of being cytoplasmic or luminal for all 654 unique tryptic peptides detected in two independent studies (483 from experiment 1 and 484 from experiment 2) (supplemental Table 3). 95% (experiment 1) and 90% (experiment 2) of peptides had a calculated probability of 0.95 or greater to be either cytoplasmic or luminal. The percentage increased to 98% in experiment 1 and 95% in experiment 2 for peptides with a calculated probability of greater than 0.80. All peptides were separated into two categories, cytoplasmic (color-coded as red) if cytoplasmic probability was >0.50 or luminal (blue) if cytoplasmic probability was <0.50 (supplemental Tables 1, 2, and 3). The iTRAQ ratios (PK/CT) corresponding to a cytoplasmic probability at 0.50 are 0.57 in experiment 1 and 0.48 in experiment 2. 313 peptides were common in both experiments among which 308 peptides (∼98%) had consistent topology assignments and only five peptides (∼2%) had inconsistent topology assignments in two experiments. These peptides led to the identification of 285 non-redundant proteins together with their topology information derived from the corresponding iTRAQ ratios. The protein names, accession numbers, peptide sequences, their iTRAQ ratios in two independent studies, and corresponding cytoplasmic probabilities are summarized in supplemental Table 3. After a database and literature search, 73 highly likely contaminating mitochondrial and ribosomal proteins, indicated in supplemental Table 3, were excluded from further analysis. The membrane topology was analyzed for the remaining proteins by combining our experimental results with transmembrane helix prediction using computational software including TMHMM, TMpred, and SOSUI. The detailed topology analyses of a subset of 66 known and highly likely ZGM proteins are summarized in Table I. The remaining 146 proteins include a large number of proteins with uncharacterized functions or subcellular localization as well as proteins reported previously in another compartment or compartments along the secretory pathway but still possible to be genuine ZGM proteins. To validate the iTRAQ-based topology analysis approach, we compared our experimentally constrained models with published results when available. These results included both direct evidence from ZGMs and indirect evidence from other cellular membranes with the assumption that the membrane topology is conserved along the secretory pathway and in different cell types. In addition, the ZGM localization and iTRAQ-based topology assignment of several newly identified proteins were confirmed by Western blot analysis, including ENTP1, myosin Vc, pIgR, Rab27B, Rap1, Slp1, syntaxin 7 (Fig. 5), and SCAMP 1 (Fig. 6). Among the 87 proteins examined, 81 proteins are consistent with published topology models, five are inconsistent, and one shows mixed results. Essentially all the cytoplasmic topology assignments based on low iTRAQ ratios are correct. Although the majority (∼90%) of the luminal assignments are also correct, five exceptions have been found with reported cytoplasm-orientated proteins or domains having unchanged iTRAQ ratios. These included two ATPase subunits, the a1 subunit of vacuolar H+-ATPase and the α1 chain of sodium/potassium-transporting ATPase (22, 23), and three small GTPase, α subunit of guanine nucleotide-binding protein Gk, Ras homolog gene family member G, and Rac1. All but one peptide detected from these proteins showed protease protection in two independent experiments (supplemental Table 3). The exact mechanism for their resistance to protease digestion will require further investigation. Because all of these proteins have been reported on plasma membrane, one possibility is that they originated from the contaminating plasma membrane that was co-purified with ZGs as closed vesicles. In the case of synaptotagmin-like protein 1, as a member of the Rab27 effector family (24) with no predicted transmembrane domain, it is expected to be located on the cytoplasmic surface of ZGM. However, only three (of eight) detected peptides near the amino terminus appeared to be protease-sensitive. Interestingly the remaining five protected peptides are located within the two C2 domains containing an eight-stranded antiparallel β structure that binds membrane lipid very tightly (25). The strong membrane interaction and the unique antiparallel β structure of this region of Slp1 might explain the unusual protease protection observed in this study.
Table I.
Topology models of known and potential ZGM proteins derived from iTRAQ-based quantitative proteomics analysis
The table is organized by ZG membrane topology with the peripheral cytoplasmic membrane proteins appearing first followed by transmembrane proteins and peripheral luminal membrane proteins and ZG content last. Within each category, the proteins are sorted alphabetically.
| Protein name | Accession number | Unique peptidesa
|
Sequence coverage | No. of TM domainsb | Membrane topologyc | Topology confirmationd | |
|---|---|---|---|---|---|---|---|
| C | L | ||||||
| % | |||||||
| 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase | IPI00199394 | 3 | 0 | 7 | 0 | C | By similarity (37) |
| Brain acid-soluble protein 1 | IPI00231651 | 3 | 0 | 26 | Myristoyl | C | By similarity (38) |
| Dynactin1 | IPI00196703 | 1 | 0 | 5 | 0 | C | ZG (39) |
| Dynein light intermediate chain 1 | IPI00213552 | 2 | 0 | 9 | 0 | C | ZG (39) |
| Dynein heavy chain | IPI00327630 | 6 | 0 | 3 | 0 | C | ZG (39) |
| Myosin I heavy chain | IPI00393867 | 1 | 0 | 5 | 0 | C | |
| Myosin Vc | IPI00370405 | 10 | 0 | 6 | 0 | C | x |
| Myosin VIIb | IPI00208315 | 5 | 0 | 2 | 0 | C | |
| Myristoylated alanine-rich C-kinase substrate | IPI00480687 | 3 | 0 | 18 | Isoprenyl | C | By similarity (40) |
| Rab1A | IPI00421897 | 6 | 0 | 20 | Isoprenyl | C | |
| Rab2A | IPI00202570 | 3 | 0 | 16 | Isoprenyl | C | |
| Rab3D | IPI00213685 | 2 | 0 | 10 | Isoprenyl | C | x |
| Rab5B | IPI00564419 | 2 | 0 | 6 | Isoprenyl | C | |
| Rab11A | IPI00214434 | 2 | 0 | 9 | Isoprenyl | C | |
| Rab14 | IPI00196794 | 1 | 0 | 11 | Isoprenyl | C | |
| Rab27B | IPI00200425 | 2 | 0 | 15 | Isoprenyl | C | x |
| Rap1 | IPI00187747 | 1 | 0 | 5 | Isoprenyl | C | x |
| Synaptotagmin-like protein 1 | IPI00198909 | 3 | 5 | 28 | 0 | C* | x |
| Synaptotagmin-like protein 4 | IPI00207255 | 3 | 0 | 8 | 0 | C | |
| 4F2 light chain | IPI00204778 | 1 | 0 | 2 | 12 | C51–71L85–105C121–141L147–167C174–194L200–220C248–268L279–299C324–344L398–418C436–456L463–483C | By similarity (41) |
| 4F2 heavy chain | IPI00211616 | 1 | 6 | 15 | 1 | C76–98L | By similarity (41) |
| Aminopeptidase N | IPI00230862 | 0 | 4 | 6 | 1 | C12–34L | By similarity (42) |
| Amyloid β A4 protein precursor | IPI00558198 | 0 | 3 | 10 | 1 | L701–723C | By similarity (43) |
| Carboxypeptidase D precursor | IPI00213182 | 0 | 3 | 7 | 1 | L1298–1320C | By similarity (44) |
| Cation-chloride cotransporter 9 homolog | IPI00194878 | 1 | 0 | 5 | 12 | L44–66C74–96L117–139C150–172L182–204C232–254L277–299C305–327L367–389C578–600L 604–626C630–652L | By similarity (45) |
| CD59 glycoprotein | IPI00195173 | 0 | 3 | 19 | 2 | C7–26L103–122C | By similarity (46) |
| ENTP1 | IPI00206668 | 0 | 1 | 3 | 2 | C17–39L478–500C | x |
| Ectonucleotide pyrophosphatase | IPI00326462 | 0 | 8 | 10 | 1 | C23–45L | |
| Itmap1 | IPI00208448 | 0 | 5 | 15 | 1 | L573–595C | ZG (20) |
| γ-Glutamyltranspeptidase 1 precursor | IPI00206254 | 0 | 12 | 27 | 1 | C7–29L | |
| Mucin 1, transmembrane | IPI00373230 | 2 | 1 | 1 | 1 | L575–597C | |
| Nicastrin | IPI00192495 | 0 | 1 | 1 | 1 | L669–691C | By similarity (47) |
| Pantophysin | IPI00471762 | 0 | 2 | 13 | 3 | L117–139C149–171L210–232C | |
| Phospholemman | IPI00193828 | 1 | 0 | 13 | 1 | L35–57C | By similarity (48) |
| Polymeric immunoglobulin receptor | IPI00205255 | 0 | 4 | 7 | 1 | L644–666C | x |
| Presenilin 2 | IPI00327678 | 1 | 0 | 4 | 8 | C86–108L138–160C167–189L198–220C238–260L285–307C360–381L396–418C | By similarity (49) |
| Proteolipid protein 2 | IPI00358313 | 1 | 0 | 7 | 4 | C25–42L49–68C83–105L112–134C | By similarity (50) |
| SCAMP 1 | IPI00194179 | 3 | 0 | 15 | 4 | C154–176L181–203C216–238L258–280C | x; ZG (27) |
| SCAMP 2 | IPI00366649 | 2 | 0 | 9 | 4 | C154–176L181–203C216–238L261–283C | |
| SCAMP 3 | IPI00206037 | 1 | 0 | 4 | 4 | C172–194L199–221C234–256L276–298C | |
| SCAMP 4 | IPI00199345 | 1 | 0 | 6 | 4 | C43–65L71–90C103–125L145–167C | |
| Syntaxin 7 | IPI00569907 | 3 | 0 | 16 | 1 | C237–259L | x |
| Syntaxin 12 | IPI00208759 | 1 | 0 | 5 | 1 | C251–273 | |
| Transmembrane 4 superfamily member 6 | IPI00201753 | 0 | 2 | 8 | 4 | C127–149L164–186C199–221L318–340C | By similarity (51) |
| % | |||||||
| Transmembrane protein 63A | IPI00363369 | 2 | 2 | 6 | 10 | C46–68L144–166C190–212L420–442C465–487L505–527C561–583L613–635C665–687L697–719C | |
| VAMP 2 | IPI00204447 | 2 | 0 | 24 | 1 | C92–114L | |
| VAMP 8 | IPI00325975 | 1 | 0 | 9 | 1 | C76–98L | |
| Anionic trypsin 1 precursor | IPI00212767 | 0 | 1 | 8 | 0 | L, content | Digestive enzyme |
| ATPase family, AAA domain-containing protein 3 | IPI00562649 | 0 | 2 | 6 | 0 | L | |
| Carboxypeptidase A2 precursor | IPI00193391 | 0 | 3 | 9 | 0 | L, content | Digestive enzyme |
| Carboxypeptidase A3 precursor | IPI00373176 | 0 | 2 | 7 | 0 | L, content | Digestive enzyme |
| Cholesterol esterase | IPI00566859 | 0 | 11 | 16 | 0 | L, content | Digestive enzyme |
| Chymotrypsin B precursor | IPI00206309 | 0 | 3 | 17 | 0 | L, content | Digestive enzyme |
| Clusterin | IPI00198667 | 0 | 3 | 9 | 0 | L | In pancreatic juice (52) |
| Colipase | IPI00212799 | 0 | 2 | 16 | 0 | L | |
| Dipeptidase 1 | IPI00563552 | 0 | 5 | 9 | GPI | L | ZG (53) |
| Elastase 1 precursor | IPI00327729 | 0 | 1 | 8 | 0 | L, content | Digestive enzyme |
| GP2 | IPI00734632 | 0 | 14 | 28 | GPI | L | |
| GP3 | IPI00231487 | 0 | 9 | 17 | GPI | L | ZG (19) |
| Mast cell protease 1 precursor | IPI00188478 | 0 | 2 | 12 | 0 | L | Secreted protease |
| Mast cell protease 3 precursor | IPI00197397 | 0 | 2 | 12 | 0 | L | Secreted protease |
| Pancreatic α-amylase | IPI00211904 | 0 | 10 | 24 | 0 | L, content | x, digestive enzyme |
| Pancreatic lipase-related protein 1 | IPI00212662 | 0 | 4 | 15 | 0 | L | |
| Pancreatic triacylglycerol lipase | IPI00198916 | 0 | 8 | 13 | 0 | L, content | Digestive enzyme |
| Syncollin | IPI00212367 | 0 | 4 | 20 | 0 | L | ZG (21) |
| ZG16 | IPI00194721 | 0 | 3 | 26 | 0 | L | |
The numbers of unique peptides detected from each protein are reported in two separate columns according to their corresponding cytoplasmic (C) and luminal (L) assignment. Extra information supporting the identification of the potential ZGM proteins with single unique peptide are summarized in supplemental Table 4.
TMHMM, SOSUI, and TMpred softwares were used to predict the TM helixes of identified proteins. For known lipid-anchored proteins, the specific lipid modifications are listed. GPI, glycosylphosphatidylinositol.
A full topology model was developed for each protein by combining the predicted TM domains with the iTRAQ-based topology assignment. For proteins with no TM domains, the assigned sidedness in bold, cytoplasm (C) or lumen (L), is reported. For transmembrane proteins, the topology models are presented with transmembrane regions labeled by starting and ending amino acid positions and the extramembrane loops labeled by their predicted or measured sidedness, cytoplasm (C) or lumen (L). The extramembrane loops mapped with iTRAQ-labeled peptides are indicated with bold underlined C or bold underlined L. *, Slp1 has mixed assignments, and for more detail see “Results.”
The independent experimental evidence supporting the topology models are summarized in this column: “ZG” indicates evidence directly obtained from ZG membranes, whereas “by similarity” indicates evidence from other cellular membranes in other cell types. The references of the corresponding literature are listed, and “x” indicates the Western blotting confirmations by our own results.
Fig. 5.

Western blot confirmation of the iTRAQ-based topology assignment to several newly identified ZGM proteins. 10 μg of proteins from total pancreatic lysate (Total), purified ZGs (ZG), ZG content (Content), ZGM, and ZGM from proteinase K (ZGM (PK))-treated ZGs were analyzed by Western blot. The antibodies used are anti-ENTP1 raised against amino acids 256–340, anti-Rap1 against a peptide near the carboxyl terminus, anti-Slp1 antibody raised against amino-terminal peptide MAHGPKPETEGLLDLS, anti-syntaxin 7 antibody raised against the recombinant cytoplasmic domain of rat protein containing amino acids 1–236, and anti-Rab27B and anti-myosin Vc antibodies raised against recombinant proteins of Rab27B and myosin Vc tail domain, respectively. Rabbit polyclonal antiserum against purified secretory component was used to detect pIgR.
Fig. 6.

The topology model of a multipass transmembrane protein, SCAMP 1. A, amino acid sequence of the SCAMP 1 with predicted transmembrane (TM) helixes labeled in red and detected cytoplasmic peptides in orange. B, iTRAQ ratios for the three detected peptides from two independent experiments. C, the topology model of SCAMP 1 derived from iTRAQ ratios and the TM helix prediction by TMHMM. The plot was produced using the transmembrane sequence display software TOPO2. D, Western blot confirmation of the topology of SCAMP 1. The antibody used was raised against the carboxyl-terminal 13 amino acids of SCAMP 1. C.I., confidence interval; PK, proteinase K.
Examples of Topology Models Derived from iTRAQ-based Quantitative Proteomics Analysis—
Previously we reported the identification of 101 proteins in our initial proteomics analysis of ZGM (3). Our current study significantly extended the list of potential ZGM proteins, especially intrinsic membrane proteins, and provided topology information for known and newly identified ZGM proteins. For example, SCAMPs 1–4 have been reported on other secretory and endocytic carriers (26). Here we confirmed the presence of SCAMPs 1–4 (however, with only one unique peptide for SCAMPs 3 and 4) on ZGM. Moreover based on the iTRAQ ratios of three unique peptides from SCAMP 1 (Fig. 6B), we developed a membrane topology model of SCAMP 1 on ZGM (Fig. 6C). The ZGM localization of SCAMP 1 and the cytoplasmic orientation of its carboxyl terminus were confirmed by Western blotting analysis using an anti-SCAMP 1 antibody raised against the carboxyl-terminal 13 amino acids (Fig. 6D). Similar topology models were derived for SCAMPs 2, 3, and 4 based on the low iTRAQ ratios of the detected peptides (Table I and supplemental Table 3). Our iTRAQ-based topology model of SCAMP 1 on the pancreatic ZGM is consistent with the published report of SCAMP 1 on the parotid ZGM using conventional protease protection analysis (27).
Because the iTRAQ-based quantification is at the peptide level, this technique can map peptides at both amino- and carboxyl-termini as demonstrated in Fig. 6C. Moreover it can also map peptides to both cytoplasmic and luminal loops of the same transmembrane proteins and therefore has the potential to validate software-based transmembrane helix predications. Such examples include 4F2 heavy chain, mucin 1, and transmembrane protein 63A (Tm63A) (Table I). As shown in Fig. 7, A and B, four unique peptides were identified from Tm63A. Two of the four peptides with higher iTRAQ ratios were mapped to a large hydrophilic loop, and the other two with very low iTRAQ ratios were mapped to the carboxyl terminus (Fig. 7, B and C). Our results indicated that the carboxyl terminus of Tm63A is facing the cytoplasm, whereas the large hydrophilic loop between positions 200 and 400 is highly likely in the lumen. This implies that there should be an odd number of complete transmembrane helixes in between the carboxyl terminus and the loop. Interestingly although very consistent in predicting the first three transmembrane helixes between amino acids 1 and 200, five widely used topology prediction softwares diverged on the predicted numbers of transmembrane helixes from amino acid 400 on. Although SOSUI, TopPred, and TMpred all predicted seven helixes, TMHMM predicted eight, and HMMTOP predicted six transmembrane helixes. Our results supported the presence of seven transmembrane helixes, and a topology model was proposed accordingly as shown in Fig. 7C.
Fig. 7.

The topology model of a multipass transmembrane protein, Tm63A, with unknown function. A, amino acid sequence of Tm63A with predicted TM helixes labeled in red, detected cytoplasmic peptides in orange, and luminal peptides in blue. B, iTRAQ ratios for the four detected peptides from two independent experiments. C, the topology model of Tm63A developed based on iTRAQ ratios and the TM helix prediction by SOSUI and TMpred. The plot was produced using the transmembrane sequence display software, TOPO2. C.I., confidence interval.
Because the ZGM lipid bilayer acts as a physical barrier for proteinase K, we expected that membrane-embedded peptides would have remained on ZGM even after proteinase K cleavage on their cytoplasmic sides. If these peptides could be detected after trypsin digestion, they should have the following unique features: 1) localization in or with close proximity to ZGM, 2) semitryptic peptides with the cytoplasmic end of the peptide having nonspecific cleavage (presumably by proteinase K) and the other end having tryptic cleavage, and 3) high enrichment in proteinase K-treated samples, in other words having high iTRAQ ratios (PK/CT) despite exposure to cytoplasm. Attempting to increase our ability to detect membrane-embedded peptides, we searched for such semitryptic peptides, and one peptide (and another peptide with three additional amino acids due to one trypsin miscleavage) was identified with high confidence and an unusually high iTRAQ ratio (>3.0). The annotated MS/MS spectrum and the deduced peptide sequence are shown in supplemental Fig. 2A. Interestingly this peptide led to the identification of presenilin 2 for the first time on ZGM. Presenilin 2 is a hydrophobic protein with at least eight transmembrane helixes and is a subunit of the γ-secretase complex. Two other subunits of the γ-secretase, presenilin 1 and nicastrin (28, 29), were also identified in this study (Table I and supplemental Table 3). The ZG localization of presenilin 2 was confirmed by immunostaining of isolated ZGs with specific antibody (data not shown). The topology model of presenilin 2 and the location of the detected semitryptic peptides are shown in supplemental Fig. 2B.
DISCUSSION
Recent large scale topology-mapping studies (5, 6) have required exogenous expression of a large number of fusion proteins, which is still a daunting task in mammalian systems. In the current study, we developed an iTRAQ-based quantitative proteomics approach and applied it to global topology analysis of ZGM proteins. Our results from multiple experiments demonstrated the reproducibility and reliability of this approach for systematic topology analysis. This method is readily applicable to other secretory granules and subcellular organelles and has several advantages. 1) It allows analysis of a large number of endogenous proteins in a single experiment without the construction and expression of fusion proteins. 2) The topology information is not limited to the amino or carboxyl termini of the proteins. 3) It is capable of mapping both cytoplasmic and luminal domains from the same transmembrane proteins and therefore constrain transmembrane helix prediction. 4) An iTRAQ-based quantitative measurement conveying topological information is associated with every identified peptide. 5) A rigorous statistical approach has been developed to model the iTRAQ ratio distributions and assign topology probabilities to each peptide.
Despite the advantages discussed above, this method does have some limitations and requires caution during implementation. Based on our time course study, the experimental condition needs to be carefully tested to achieve sufficient removal of cytoplasm-orientated protein domains while preventing organelle membrane disruption and consequent digestion of luminal proteins. Using a training set of proteins with known topology has turned out to be a very valuable approach for optimizing the digestion conditions. In addition to the digestion time, the amount of proteinase K may also be adjusted to optimize digestion if necessary. In our topology analysis, essentially all the low iTRAQ ratios corresponded to cytoplasm-orientated peptides. However, the opposite conclusion is not always true, and several potentially cytoplasm-orientated protein domains were found with unchanged iTRAQ ratios. The exact mechanism for their resistance to protease digestion requires further investigation. Although luminal peptide assignment, estimated to be around 90% in our case, is less accurate compared with cytoplasmic peptide assignment, essentially 100%, it still outperforms the widely used computational algorithms that have up to around 80% overall accuracy for predicting membrane protein topology (30). In a conventional protease protection assay, non-ionic detergent such as Triton X-100 is commonly used to confirm the lipid bilayer-dependent protection. However, this approach is not directly applicable in our experiments because complete solubilization of ZGs will prevent the separation of ZGM proteins from overwhelmingly abundant ZG content proteins. It may be possible to distinguish membrane barrier-dependent protections from conformation-specific protections by using mild chaotropes or ionophore to partially solubilize ZGs. Such a procedure is currently being developed in our laboratory. Because trypsin was used in the second digestion in our method and some extramembrane loops contain no or only one trypsin cleavage site, the sequence coverage was relatively low for transmembrane proteins. In future studies, multiple analyses with different protease digestions could be combined to increase sequence coverage.
The experimentally constrained topology map of ZGM proteins presented in this study allows better understanding of the organization of these proteins across the lipid bilayer and may provide insight into their potential functions. For example, the proteins regulating vesicular trafficking are all located on the cytoplasmic surface of the ZGM. These include multiple Rab proteins and their potential effectors, Slp1 and Slp4 (24), and molecular motor proteins, myosins and dynein together with its adaptor, dynactin 1 (31). Furthermore the SNARE proteins, syntaxins and VAMPs, have one transmembrane domain and the rest of protein facing the cytoplasm to mediate ZG docking and membrane fusion. The newly identified cytoplasm-orientated ZGM proteins also include myristoylated alanine-rich protein kinase C substrate, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase, and phospholemman. Their roles on the ZGM are not well understood, but they likely play a role in mediating the action of known regulators of ZG secretion such as cAMP-dependent protein kinase and protein kinase C (32). Proteins on the luminal side of ZGM include digestive enzymes and abundant ZG matrix proteins such as GP2, GP3, syncollin, and ZG16. Some of these abundant lumen-orientated proteins could play a role in zymogen sorting and ZG formation (4, 33), but their exact functions are not clear. Among the multipass transmembrane proteins on the ZGM are transporters, membrane-bound enzymes, and several isoforms of SCAMPs as well as polymeric immunoglobulin receptor and pantophysin. Interestingly the amyloid protein precursor and several subunits of γ-secretase including nicastrin and presenilins 1 and 2 were also found associated with the ZGM. The functions of most of these transmembrane proteins are currently not clear and will be the targets of intensive investigation in future studies. Among these proteins, several single pass transmembrane proteins with the majority of the sequences in ZG lumen are of special interest. If receptor-mediated transport is believed to be the mechanism of ZG budding from trans-Golgi network by topology homology with known cargo receptors such as mannose 6-phosphate receptors, ERGIC53, and VIP36 family proteins (34, 35), these proteins may include the candidate cargo receptor for zymogen sorting. In addition to the functional implications, the topology map of ZGM proteins also provides a foundation for subsequent analysis of a protein-protein interaction network on ZGM. For example, the topological organization restricts the Rab-interacting proteins on the cytoplasmic surface of ZGM. In fact, two cytoplasm-orientated proteins, Slp1 and myosin Vc, have been found to interact with two Rab proteins on ZGM.2
In addition to the topology analysis reported here, we carried out two additional proteomics analyses on purified ZGM, one using 1D SDS-PAGE combined with 1D LC-MALDI-MS/MS and the other using 2D LC-ESI-MS/MS on an LTQ-Orbitrap (data will be published separately). Altogether over 300 proteins were identified from purified ZGMs with high confidence. This represents a significant extension of identified ZGM proteins. It is worth noting that although increased instrument sensitivity allowed identification of many more low abundance ZGM proteins it also uncovered more contaminating proteins. The confirmation of identified proteins on ZGM will continue to be a major task in the foreseeable future. Indicative of the intense interest in understanding the ZGM proteome, a very recent study using 1D SDS-PAGE coupled with 1D LC-MS/MS identified, using less stringent criteria and a more redundant database, 371 proteins from both ZG membrane and content (36). A preliminary comparison indicated a large degree of overlap between our studies and these data. A detailed comparison is out of the scope of this study and will be published elsewhere.
In summary, we reported here a new quantitative proteomics approach to conduct global topology analysis of ZGM proteins by combining a protease protection assay with iTRAQ-based quantification. In addition, a statistical mixture model was developed to provide probabilities of assigned topology for each identified peptides based on their iTRAQ ratios. By implementing this approach, we presented, for the first time, an experimentally constrained, comprehensive topology map of ZG membrane proteins. This model provides a firm foundation for developing a higher order architecture model of ZGM and for future functional studies of each individual ZGM protein.
Supplementary Material
Acknowledgments
We gratefully acknowledge Drs. T. Izumi, M. McNiven, D. Castle, S. Catz, R. Cheney, and C. Okamato for generously providing antibodies. We thank Dr. S. A. Ernst for assistance with the to-be-published immunolocalization studies mentioned here.
Footnotes
Published, MCP Papers in Press, August 4, 2008, DOI 10.1074/mcp.M700575-MCP200
The abbreviations used are: ZG, zymogen granule; ENTP1, ectonucleoside triphosphate diphosphohydrolase 1; iTRAQ, isobaric tag for relative and absolute quantification; pIgR, polymeric immunoglobulin receptor; SCX, strong cation exchange; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; TMHMM, Transmembrane Hidden Markov Model; VAMP, vesicle-associated membrane protein; ZGM, zymogen granule membrane; TM, transmembrane; Slp, synaptotagmin-like protein; 2D, two-dimensional; FPR, false positive rate; PK/CT, ratio between the average of two proteinase K-treated samples and that of two control samples; SCAMP, secretory carrier membrane protein; Tm63A, transmembrane protein 63A; 1D, one-dimensional.
X. Chen and J. A. Williams, unpublished data.
This work was supported, in whole or in part, by National Institutes of Health Grants P41 RR018627 from the National Resource for Proteomics and Pathways (to P. C. A.), R37 DK41122 (to J. A. W.), P30 DK34933 (to the Michigan Gastrointestinal Peptide Center through its core facilities), and P60 DK20572 (to the Michigan Diabetes Research and Training Center through its Morphology and Image Analysis Core). This work was also supported by a Pilot Project award (to X. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Williams, J. A. ( 2001) Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu. Rev. Physiol. 63, 77–97 [DOI] [PubMed] [Google Scholar]
- 2.Palade, G. ( 1975) Intracellular aspects of the process of protein synthesis. Science 189, 347–358 [DOI] [PubMed] [Google Scholar]
- 3.Chen, X., Walker, A. K., Strahler, J. R., Simon, E. S., Tomanicek-Volk, S. L., Nelson, B. B., Hurley, M. C., Ernst, S. A., Williams, J. A., and Andrews, P. C. ( 2006) Organellar proteomics: analysis of pancreatic zymogen granule membranes. Mol. Cell. Proteomics 5, 306–312 [DOI] [PubMed] [Google Scholar]
- 4.Antonin, W., Wagner, M., Riedel, D., Brose, N., and Jahn, R. ( 2002) Loss of the zymogen granule protein syncollin affects pancreatic protein synthesis and transport but not secretion. Mol. Cell. Biol. 22, 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim, H., Melen, K., Osterberg, M., and von Heijne, G. ( 2006) A global topology map of the Saccharomyces cerevisiae membrane proteome. Proc. Natl. Acad. Sci. U. S. A. 103, 11142–11147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daley, D. O., Rapp, M., Granseth, E., Melen, K., Drew, D., and von Heijne, G. ( 2005) Global topology analysis of the Escherichia coli inner membrane proteome. Science 308, 1321–1323 [DOI] [PubMed] [Google Scholar]
- 7.Lorenz, H., Hailey, D. W., and Lippincott-Schwartz, J. ( 2006) Fluorescence protease protection of GFP chimeras to reveal protein topology and subcellular localization. Nat. Methods 3, 205–210 [DOI] [PubMed] [Google Scholar]
- 8.Wu, C. C., MacCoss, M. J., Howell, K. E., and Yates, J. R., III ( 2003) A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 21, 532–538 [DOI] [PubMed] [Google Scholar]
- 9.Wu, C. C., and Yates, J. R., III ( 2003) The application of mass spectrometry to membrane proteomics. Nat. Biotechnol. 21, 262–267 [DOI] [PubMed] [Google Scholar]
- 10.Chen, X., and Andrews, P. C. ( 2008) Purification and proteomics analysis of pancreatic zymogen granule membranes. Methods Mol. Biol. 432, 275–287 [DOI] [PubMed] [Google Scholar]
- 11.Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. ( 1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 12.Peng, J., Elias, J. E., Thoreen, C. C., Licklider, L. J., and Gygi, S. P. ( 2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 13.Elias, J. E., and Gygi, S. P. ( 2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 14.Huttlin, E. L., Hegeman, A. D., Harms, A. C., and Sussman, M. R. ( 2007) Prediction of error associated with false-positive rate determination for peptide identification in large-scale proteomics experiments using a combined reverse and forward peptide sequence database strategy. J. Proteome Res. 6, 392–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. ( 2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 16.Nesvizhskii, A. I., and Aebersold, R. ( 2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 17.Marelli, M., Smith, J. J., Jung, S., Yi, E., Nesvizhskii, A. I., Christmas, R. H., Saleem, R. A., Tam, Y. Y., Fagarasanu, A., Goodlett, D. R., Aebersold, R., Rachubinski, R. A., and Aitchison, J. D. ( 2004) Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol. 167, 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 19.Wagner, A. C., Wishart, M. J., Mulders, S. M., Blevins, P. M., Andrews, P. C., Lowe, A. W., and Williams, J. A. ( 1994) GP-3, a newly characterized glycoprotein on the inner surface of the zymogen granule membrane, undergoes regulated secretion. J. Biol. Chem. 269, 9099–9104 [PubMed] [Google Scholar]
- 20.Imamura, T., Asada, M., Vogt, S. K., Rudnick, D. A., Lowe, M. E., and Muglia, L. J. ( 2002) Protection from pancreatitis by the zymogen granule membrane protein integral membrane-associated protein-1. J. Biol. Chem. 277, 50725–50733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An, S. J., Hansen, N. J., Hodel, A., Jahn, R., and Edwardson, J. M. ( 2000) Analysis of the association of syncollin with the membrane of the pancreatic zymogen granule. J. Biol. Chem. 275, 11306–11311 [DOI] [PubMed] [Google Scholar]
- 22.Wieczorek, H., Grber, G., Harvey, W. R., Huss, M., Merzendorfer, H., and Zeiske, W. ( 2000) Structure and regulation of insect plasma membrane H+V-ATPase. J. Exp. Biol. 203, 127–135 [DOI] [PubMed] [Google Scholar]
- 23.Stokes, D. L., Taylor, W. R., and Green, N. M. ( 1994) Structure, transmembrane topology and helix packing of P-type ion pumps. FEBS Lett. 346, 32–38 [DOI] [PubMed] [Google Scholar]
- 24.Fukuda, M. ( 2005) Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J. Biochem. ( Tokyo) 137, 9–16 [DOI] [PubMed] [Google Scholar]
- 25.DiNitto, J. P., Cronin, T. C., and Lambright, D. G. ( 2003) Membrane recognition and targeting by lipid-binding domains. Sci. STKE 2003, re16. [DOI] [PubMed] [Google Scholar]
- 26.Castle, A., and Castle, D. ( 2005) Ubiquitously expressed secretory carrier membrane proteins (SCAMPs) 1–4 mark different pathways and exhibit limited constitutive trafficking to and from the cell surface. J. Cell Sci. 118, 3769–3780 [DOI] [PubMed] [Google Scholar]
- 27.Hubbard, C., Singleton, D., Rauch, M., Jayasinghe, S., Cafiso, D., and Castle, D. ( 2000) The secretory carrier membrane protein family: structure and membrane topology. Mol. Biol. Cell 11, 2933–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiner, H., and Haass, C. ( 2000) Intramembrane proteolysis by presenilins. Nat. Rev. Mol. Cell Biol. 1, 217–224 [DOI] [PubMed] [Google Scholar]
- 29.Brunkan, A. L., and Goate, A. M. ( 2005) Presenilin function and γ-secretase activity. J. Neurochem. 93, 769–792 [DOI] [PubMed] [Google Scholar]
- 30.Bernsel, A., Viklund, H., Falk, J., Lindahl, E., von Heijne, G., and Elofsson, A. ( 2008) Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U. S. A. 105, 7177–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroer, T. A. ( 2004) Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779 [DOI] [PubMed] [Google Scholar]
- 32.Wishart, M. J., Groblewski, G., Goke, B. J., Wagner, A. C., and Williams, J. A. ( 1994) Secretagogue regulation of pancreatic acinar cell protein phosphorylation shown by large-scale 2D-PAGE. Am. J. Physiol. 267, G676–G686 [DOI] [PubMed] [Google Scholar]
- 33.Schmidt, K., Dartsch, H., Linder, D., Kern, H. F., and Kleene, R. ( 2000) A submembranous matrix of proteoglycans on zymogen granule membranes is involved in granule formation in rat pancreatic acinar cells. J. Cell Sci. 113, 2233–2242 [DOI] [PubMed] [Google Scholar]
- 34.Mazella, J. ( 2001) Sortilin/neurotensin receptor-3: a new tool to investigate neurotensin signaling and cellular trafficking? Cell. Signal. 13, 1–6 [DOI] [PubMed] [Google Scholar]
- 35.Hauri, H., Appenzeller, C., Kuhn, F., and Nufer, O. ( 2000) Lectins and traffic in the secretory pathway. FEBS Lett. 476, 32–37 [DOI] [PubMed] [Google Scholar]
- 36.Rindler, M. J., Xu, C. F., Gumper, I., Smith, N. N., and Neubert, T. A. ( 2007) Proteomic analysis of pancreatic zymogen granules: identification of new granule proteins. J. Proteome Res. 6, 2978–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogel, U. S., and Thompson, R. J. ( 1988) Molecular structure, localization, and possible functions of the myelin-associated enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase. J. Neurochem. 50, 1667–1677 [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto, Y., Sokawa, Y., and Maekawa, S. ( 1997) Biochemical evidence for the presence of NAP-22, a novel acidic calmodulin binding protein, in the synaptic vesicles of rat brain. Neurosci. Lett. 224, 127–130 [DOI] [PubMed] [Google Scholar]
- 39.Kraemer, J., Schmitz, F., and Drenckhahn, D. ( 1999) Cytoplasmic dynein and dynactin as likely candidates for microtubule-dependent apical targeting of pancreatic zymogen granules. Eur J. Cell Biol. 78, 265–277 [DOI] [PubMed] [Google Scholar]
- 40.Aderem, A. ( 1992) The MARCKS brothers: a family of protein kinase C substrates. Cell 71, 713–716 [DOI] [PubMed] [Google Scholar]
- 41.Wagner, C. A., Lang, F., and Broer, S. ( 2001) Function and structure of heterodimeric amino acid transporters. Am. J. Physiol. 281, C1077–C1093 [DOI] [PubMed] [Google Scholar]
- 42.Kenny, A. J., and Maroux, S. ( 1982) Topology of microvillar membrance hydrolases of kidney and intestine. Physiol. Rev. 62, 91–128 [DOI] [PubMed] [Google Scholar]
- 43.Laudon, H., Winblad, B., and Naslund, J. ( 2007) The Alzheimer's disease-associated gamma-secretase complex: functional domains in the presenilin 1 protein. Physiol. Behav. 92, 115–120 [DOI] [PubMed] [Google Scholar]
- 44.Kalinina, E., Varlamov, O., and Fricker, L. D. ( 2002) Analysis of the carboxypeptidase D cytoplasmic domain: Implications in intracellular trafficking. J. Cell. Biochem. 85, 101–111 [PubMed] [Google Scholar]
- 45.Hebert, S. C., Mount, D. B., and Gamba, G. ( 2004) Molecular physiology of cation-coupled Cl− cotransport: the SLC12 family. Pfluegers Arch. Eur. J. Physiol. 447, 580–593 [DOI] [PubMed] [Google Scholar]
- 46.Kieffer, B., Driscoll, P. C., Campbell, I. D., Willis, A. C., van der Merwe, P. A., and Davis, S. J. ( 1994) Three-dimensional solution structure of the extracellular region of the complement regulatory protein CD59, a new cell-surface protein domain related to snake venom neurotoxins. Biochemistry 33, 4471–4482 [PubMed] [Google Scholar]
- 47.Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., Song, Y. Q., Rogaeva, E., Chen, F., Kawarai, T., Supala, A., Levesque, L., Yu, H., Yang, D. S., Holmes, E., Milman, P., Liang, Y., Zhang, D. M., Xu, D. H., Sato, C., Rogaev, E., Smith, M., Janus, C., Zhang, Y., Aebersold, R., Farrer, L. S., Sorbi, S., Bruni, A., Fraser, P., and St George-Hyslop, P. ( 2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407, 48–54 [DOI] [PubMed] [Google Scholar]
- 48.Chen, Z., Jones, L. R., O'Brian, J. J., Moorman, J. R., and Cala, S. E. ( 1998) Structural domains in phospholemman: a possible role for the carboxyl terminus in channel inactivation. Circ. Res. 82, 367–374 [DOI] [PubMed] [Google Scholar]
- 49.Spasic, D., Tolia, A., Dillen, K., Baert, V., De Strooper, B., Vrijens, S., and Annaert, W. ( 2006) Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 281, 26569–26577 [DOI] [PubMed] [Google Scholar]
- 50.Breitwieser, G. E., McLenithan, J. C., Cortese, J. F., Shields, J. M., Oliva, M. M., Majewski, J. L., Machamer, C. E., and Yang, V. W. ( 1997) Colonic epithelium-enriched protein A4 is a proteolipid that exhibits ion channel characteristics. Am. J. Physiol. 272, C957–C965 [DOI] [PubMed] [Google Scholar]
- 51.Maecker, H. T., Todd, S. C., and Levy, S. ( 1997) The tetraspanin superfamily: molecular facilitators. FASEB J. 11, 428–442 [PubMed] [Google Scholar]
- 52.Yokoyama, M., Matsue, H., Muramoto, K., Sasaki, M., Ono, K., and Endo, M. ( 1988) Isolation and characterization of sulfated glycoprotein from human pancreatic juice. Biochim. Biophys. Acta 967, 34–42 [DOI] [PubMed] [Google Scholar]
- 53.Hooper, N. M., Cook, S., Laine, J., and Lebel, D. ( 1997) Identification of membrane dipeptidase as a major glycosyl-phosphatidylinositol-anchored protein of the pancreatic zymogen granule membrane, and evidence for its release by phospholipase A. Biochem. J. 324, 151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.