

Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL/Apo-2L) has emerged as a promising anticancer agent. However, resistance to TRAIL is likely to be a major problem, and sensitization of cancer cells to TRAIL may therefore be an important anticancer strategy. In this study, we examined the effect of the epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor (TKI) gefitinib and a human epidermal receptor 2 (HER2)-TKI (M578440) on the sensitivity of human colorectal cancer (CRC) cell lines to recombinant human TRAIL (rhTRAIL). A synergistic interaction between rhTRAIL and gefitinib and rhTRAIL and M578440 was observed in both rhTRAIL-sensitive and resistant CRC cells. This synergy correlated with an increase in EGFR and HER2 activation after rhTRAIL treatment. Furthermore, treatment of CRC cells with rhTRAIL resulted in activation of the Src family kinases (SFK). Importantly, we found that rhTRAIL treatment induced shedding of transforming growth factor-α (TGF-α) that was dependent on SFK activity and the protease ADAM-17. Moreover, this shedding of TGF-α was critical for rhTRAIL-induced activation of EGFR. In support of this, SFK inhibitors and small interfering RNAs targeting ADAM-17 and TGF-α also sensitized CRC cells to rhTRAIL-mediated apoptosis. Taken together, our findings indicate that both rhTRAIL-sensitive and resistant CRC cells respond to rhTRAIL treatment by activating an EGFR/HER2-mediated survival response and that these cells can be sensitized to rhTRAIL using EGFR/HER2-targeted therapies. Furthermore, this acute response to rhTRAIL is regulated by SFK-mediated and ADAM-17-mediated shedding of TGF-α, such that targeting SFKs or inhibiting ADAM-17, in combination with rhTRAIL, may enhance the response of CRC tumors to rhTRAIL.
Introduction
The tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL) belong to the proapoptotic cytokines of the TNF superfamily (1). TRAIL can interact with five distinct type 1 transmembrane receptors, two of which are death receptors, TRAIL-R1/DR4 and TRAIL-R2/DR5, and three of which are decoy receptors, DcR-1/TRAIL-R3, DcR-2/TRAIL-R4, and osteoprotegerin. Ligation of functional receptors with TRAIL leads to formation of death-inducing signaling complexes (DISC). The intracellular death domain (DD) of these receptors recruits the Fas-associated DD (FADD)-containing protein, which in turn binds procaspase-8. After recruitment to the DISC, procaspase-8 is activated by autoproteolytic cleavage, resulting in initiation of an apoptotic cascade (2). Constitutive expression of death receptors and TRAIL has been observed in a wide range of human tissue types, including colorectal cancer (CRC; ref. 3). Furthermore, TRAIL has been shown to induce apoptosis in many cancer types with limited toxicities in normal tissues (4). Hence, various approaches have been developed to target the TRAIL receptors therapeutically, and several phase I studies are currently ongoing in solid tumors evaluating the effect of fully human agonist monoclonal antibodies (mAb) against TRAIL-R1/DR4 (such as mapatumumab, Human Genome Sciences, Inc.) and TRAIL-R2/DR5 (such as lexatumumab, Human Genome Sciences, Inc. and AMG655, Amgen) or agents that target both receptors (such as rhApo2L/TRAIL, Genentech). Inherent tumor resistance may be a major barrier for effective TRAIL-targeted therapy, so it is important to understand these resistance mechanisms and to identify agents that sensitize cancer cells to TRAIL-mediated apoptosis.
The human epidermal receptor (HER) family of receptor tyrosine kinases and their ligands are important regulators of tumor cell proliferation, survival, angiogenesis, and metastasis (5). The family comprises four members: HER1 [ErbB1/epidermal growth factor receptor (EGFR)], HER2 (ErbB2/Neu), HER3 (ErbB3), and HER4 (ErbB4; ref. 6). Seven ligands have been reported to bind EGFR, including the EGFR-specific ligands, EGF, transforming growth factor-α (TGF-α), amphiregulin, and epigen, and the ligands with dual specificity, heparin-binding EGF (HB-EGF), β-cellulin, and epiregulin (6). EGFR ligands are synthesized as transmembrane precursors that can be proteolytically cleaved by cell surface proteases, in particular members of the ADAM (a desintegrin and metalloprotease) family (7). ADAM-mediated ligand shedding results in enhanced autocrine, juxtacrine, and paracrine signaling. These ligands bind to EGFR resulting in the formation of homodimers or heterodimers, tyrosine kinase activation, receptor autophosphorylation, and activation of multiple downstream signaling cascades (6). As EGFR and HER2 are frequently aberrantly overexpressed, mutated, and/or activated in a wide range of human tumors, these receptors represent attractive targets for the treatment of cancer (8). This has resulted in the development of multiple anti-HER therapeutics, including the mAbs trastuzumab (directed against HER2) and cetuximab (directed against EGFR), as well as low molecular weight tyrosine kinase inhibitors (TKI) targeting EGFR (e.g., gefitinib, erlotinib) and HER2 (e.g., CP-724,714, M578440).
Recently, we have shown that CRC and non-small cell lung cancer cells exposed to different cytotoxic agents may respond to chemotherapy with an EGFR-mediated prosurvival response, which can be blocked by EGFR-targeted agents (9, 10). Furthermore, Chinnaiyan and colleagues reported that radiation-induced EGFR phosphorylation could be the mechanism underlying the synergism observed between erlotinib and radiation (11). Recently, several studies have shown that various members of the ADAM family, such as ADAM-17, ADAM-15, ADAM-12, ADAM-10, and ADAM-9, may be involved in EGFR activation after cytokine stimulation of various G protein-coupled receptors and oxidative or osmotic stress (12-14). Furthermore, cytoplasmic nonreceptor tyrosine kinases, such as PKC, Janus-activated kinase 2, and Src family kinases (SFK), have been shown to activate EGFR (15-17).
The aim of the present study was to investigate the role of the EGFR/HER2 survival pathway in regulating TRAIL-induced cytotoxicity in a panel of CRC cell lines.
Materials and Methods
Materials
All chemicals and reagents of Analar grade were obtained from BDH Laboratory Supplies, unless otherwise stated. Gefitinib, the HER2 selective TKI, M578440, and AZD0530 were provided by AstraZeneca. A 10 mmol/L working solution of gefitinib, M578440, and AZD0530 in DMSO was prepared, aliquotted, and stored at -20°C. PP2 was obtained from Calbiochem. Recombinant human TRAIL (rhTRAIL) was provided by Calbiochem. A 20 μg/mL stock solution of rhTRAIL was prepared in PBS/0.1% bovine serum albumin (BSA) and stored at -70°C. Recombinant TGF-α was purchased from Calbiochem and reconstituted to a concentration of 50 μg/mL in 10 mmol/L acetic acid containing 0.1% BSA and stored at -70°C. Recombinant NRG1 was obtained from R&D Systems and reconstituted to a concentration of 25 μg/mL in BPS containing 0.1% BSA and stored at -20°C.
Cell culture
All tissue culture material was obtained from Invitrogen, unless otherwise stated. HCT116-p53wt and HCT116-p53null isogenic human CRC cells were kindly provided by Bert Vogelstein (Johns Hopkins University) and maintained in McCoy’s 5A medium. LoVo CRC cells, supplied by AstraZeneca, were grown in DMEM. H630, RKO, and HT-29 human CRC cell lines were provided by the National Cancer Institute and maintained in DMEM. All medium was supplemented with 10% FCS, 50 μg/mL penicillin-streptomycin, 2 mmol/L l-glutamine, and 1 mmol/L sodium pyruvate (Invitrogen). All cells were grown in a humidified atmosphere with 5% CO2 at 37°C.
Cell viability assay
Cell viability was assessed by the tetrazolium dye [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma] assay (18). Cells were seeded at 2,000 to 4,000 per well in 96-well plates. Cells were treated with increasing doses of gefitinib or M578440 for 72 h. After treatment, cells were washed once with 1 × PBS and incubated with medium containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (0.5 mg/mL) for 3 h at 37°C. Culture medium with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was removed, and formazan crystals were reabsorbed in 200 μL DMSO (Sigma). Cell viability was determined by measuring the absorbance at 570 nm, using a microplate reader (Molecular Devices). IC50 was calculated using Prism software package. Each value is representative of at least three independent experiments.
Flow cytometric analysis and cell death measurement
Cells were seeded at a density of 1 × 105 cells per well in six-well plates. After treatment, DNA content was evaluated by propidium iodide (Sigma) staining of cells by resuspending them in 360 μL 0.1% FCS, 10 μg/mL propidium iodide, 0.25 mg/mL RNaseA in 1 × PBS. Samples were incubated for at least 30 min before analysis on the EPICS XL Flow Cytometer (Coulter). The extent of cell death was determined by evaluating the percentage of cells with DNA content of <2N.
Detection of cell surface EGFR and HER2 expression
Cells were seeded at a density of 1.5 × 105 per well in six-well plates. Twenty-four hours after seeding, cells were treated with increasing doses of rTRAIL for 24 h. Cells were trypsinized and resuspended in 100 μL of either a nonconjugated anti-EGFR mAb (Santa Cruz) or an FITC-conjugated anti-HER2 mAb (FITC, Santa Cruz) at a dilution of 1:20 and allowed to incubate in the dark for 60 min at 4°C. A mouse IgG1 (DAKO) was used as an isotype-matched control. Cells incubated with the anti-EGFR mAb were incubated in the dark for 60 min at 4°C with an FITC-labeled goat anti-mouse IgG secondary antibody (1:20; Sigma). After incubation with FITC-labeled antibody, the cells were washed thrice with ice-cold PBS and then resuspended in 300 μL 1% paraformaldehyde. Fluorescence was evaluated using the EPICS XL Flow Cytometer (Coulter).
Western blotting
Cells were harvested in ice-cold PBS, pelleted, and snap frozen in liquid nitrogen. Cell pellets were resuspended in radioimmunoprecipitation assay buffer [50 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS] with protease inhibitors (Roche Diagnostics GmbH), 1 mmol/L sodium orthovanadate (Sigma), and 10 mmol/L sodium fluoride. Cells were then lysed by passing them through a 25-gauge needle 10 times and centrifuged at 13,200 rpm/4°C for 20 min to remove cell debris. Protein concentration was determined using the bicinchoninic acid protein assay reagent (Pierce). Each protein sample (20-40 μg) was resolved on SDS-polyacrylamide gels (8%) and transferred to a PROTRAN BA 83 nitrocellulose membrane (Whatman, Schleicher & Schuell). Immunodetections were performed using anti-EGFR (Clone 13, PharMingen, BD Biosciences), anti-HER2 (Clone e2-4001, Neomarkers, Lab Vision Corp.), anti-HER3 (Clone 2C3, Neomarkers), caspase 8 (Alexis), and poly(ADP-ribose) polymerase (PARP; eBioscience) mouse mAbs in conjunction with a horseradish peroxidase (HRP)-conjugated antimouse secondary antibody (Amersham). Anti-phosphorylated EGFR (Tyr1068; Calbiochem), anti-phosphorylated HER2 (Tyr1248; Santa Cruz), anti-phosphorylated HER3 (Tyr1289; Cell Signaling), anti-phosphorylated Akt (Ser473, Cell Signaling), anti-Akt (Cell Signaling), anti-phosphorylated SFK (Tyr418; Biosource), anti-Src (Cell Signaling), anti-ADAM-17 (Pharmigen), and caspase-3 (Cell Signaling) rabbit polyclonal antibodies were used in conjunction with an HRP-conjugated antirabbit secondary antibody (Amersham). Equal loading was assessed using β-tubulin (Sigma), β-actin (Sigma), or glyceraldehyde-3-phosphate dehydrogenase (Biogenesis) mouse monoclonal primary antibodies. The Super Signal chemiluminescent system (Pierce) or ECL-plus (Amersham) were used for detection.
Small interfering RNA transfections
ADAM-17, TGF-α, and scramble control (SC) small interfering RNAs (siRNA) were obtained from Dharmacon. HCT116 and LoVo cells were seeded out in the appropriate media without penicillin-streptomycin. Twenty-four hours after seeding, siRNA transfections were performed on subconfluent cells incubated in unsupplemented Optimem using the oligofectamine reagent (both from Invitrogen) according to the manufacturer’s instructions. After 4 h, growth medium was added thrice; cells were treated with rTRAIL 1 h after this.
TGF-α ELISA
An equal number of HCT116 and LoVo cells were plated into 24-well plates and incubated for 24 h in penicillin-streptomycin-free media supplemented with 2% FCS. Cells were thereafter either transfected with SC, ADAM-17 siRNA, or TGF-α siRNA or preincubated with PP2 or AZD0530 for 4 h. After this, cells were treated with rTRAIL for 24 h, and TGF-α levels in conditioned medium were measured by ELISA according to the manufacturer’s instructions (Calbiochem).
Statistical analysis
Two-way ANOVA test was used to determine the significance of change in levels of apoptosis between different treatment groups. All changes in levels of apoptosis that are described as significant had P values that were <0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
Results
Sensitivity of CRC cells to rhTRAIL and correlation with constitutive expression of the TRAIL receptors
To investigate the sensitivity of CRC cells to rhTRAIL, HCT116-p53wt, HCT116-p53null, LoVo (p53wt), RKO (p53wt), HT29 (p53mut), and H630 (p53mut) CRC cells were exposed to increasing concentrations of rhTRAIL, and the levels of apoptosis induced was determined by flow cytometry (Fig. 1A). Although, p53wt HCT116 cells were slightly more sensitive to rhTRAIL than the p53null daughter cell line, the p53wt RKO and p53mutant HT29 cells were both resistant to rhTRAIL, whereas the p53wt LoVo and p53mutant H630 cell lines were sensitive to rhTRAIL. Collectively, these results suggest that p53 status does not determine sensitivity to rhTRAIL. To evaluate whether the sensitivity to rhTRAIL correlated with expression levels of its receptors, we determined the constitutive cell surface expression of DR4 and DR5 and the TRAIL decoy receptors DcR1 and DcR2 using flow cytometry (Fig. 1B). All six cell lines expressed DR5, with lowest expression in the LoVo cell line. DR4 was expressed in both HCT116 cell lines but at a lower level than DR5. DR4 was detectable but expressed at a low level in each of the other four cell lines. The H630 cell line was the only one to significantly express DcR1. DcR2 was expressed at very low levels or was undetectable in this cell line panel. Overall, sensitivity to rhTRAIL did not correlate with constitutive cell surface expression levels of the TRAIL receptors. Furthermore, the expression of these receptors did not correlate with p53 status.
Figure 1.
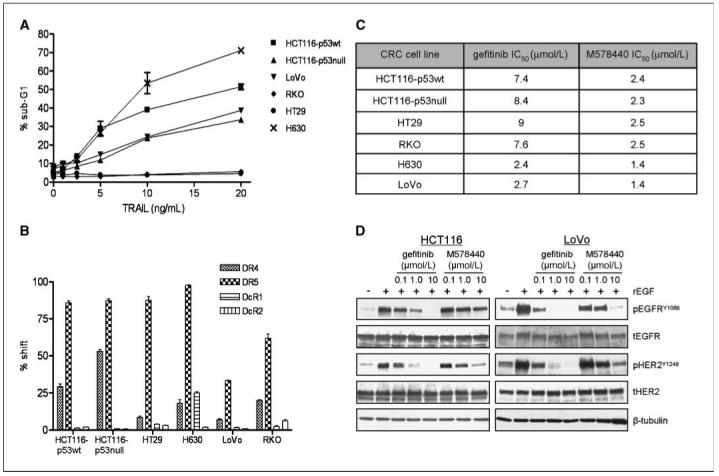
TRAIL receptor expression and sensitivity of a panel of CRC cells to rhTRAIL, gefitinib, and M578440. A, rhTRAIL-induced apoptosis in HCT116-p53wt, HCT116-p53null, HT29, RKO, H630, and LoVo as determined by flow cytometry. The DNA-content of propidium iodide-stained cells was analyzed after 24-h treatment with rhTRAIL, and the extent of cell death was determined by evaluating the percentage of cells with DNA content <2N. B, DR4, DR5, DcR1, and DcR2 cell membrane expression was assessed by flow cytometry using receptor-specific phycoerythrin-conjugated mAbs. Expression was compared with a nonspecific isotype-matched control antibody. C, cells were treated with increasing doses of gefitinib or M578440 for 72 h, and cell viability was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide assay. IC50 was calculated using Prism software package. Representative values of at least three independent experiments. D, effect of EGF, gefitinib, and M578440 on pEGFR and pHER2. LoVo and HCT116 CRC cells were incubated with 0.1, 1, and 10 μmol/L of gefitinib or M578440 for 6 h followed by EGF stimulation (100 ng/mL) for 15 min and pEGFRY1068, total EGFR (tEGFR), pHER2Y1248, and tHER2 levels were determined by Western blotting. Equal loading was assessed by probing for β-tubulin.
Sensitivity of human CRC cells to EGFR-TKI and HER2-TKI
To assess the sensitivity of the CRC cell lines to EGFR and HER2 inhibition, we examined the antiproliferative activity of the EGFR-selective TKI gefitinib and the HER2-selective TKI M578440 in the cell line panel. M578440 inhibits the HER2 kinase in vitro with an IC50 of 2 nmol/L compared with 140 nmol/L for EGFR (19), whereas gefitinib has been found to inhibit EGFR kinase activity with an IC50 of 33 nmol/L compared with 3.7 to 10 μmol/L for HER2 (20). The IC50 concentrations of gefitinib ranged from 2.4 to 9 μmol/L, whereas the IC50 concentrations of M578440 were more uniform across the panel, ranging from 1.4 to 2.5 μmol/L (Fig. 1C). We also determined the effect of gefitinib and M578440 on EGF-induced EGFR and HER2 activation in LoVo and HCT116 CRC cells. In both cell lines, rEGF treatment resulted in activation of both EGFR and HER2, suggesting crosstalk between EGFR and HER2 in these cell lines (Fig. 1D). In rEGF stimulated LoVo cells, inhibition of EGFR activity with 1 μmol/L gefitinib also led to a complete inhibition of HER2 phosphorylation. Furthermore, inhibition of HER2 led to decreased EGFR phosphorylation, suggesting the formation of EGFR/HER2 heterodimers in response to rEGF stimulation. Although gefitinib treatment potently inhibited both EGFR and HER2 activation in response to rEGF in the HCT116 cell line, EGFR activity was much less inhibited by M578440 (Fig. 1D). So, in the HCT116 cell line, it seems that, whereas HER2 is reliant on heterodimerization with EGFR for activation after EGF stimulation, EGFR activation is not solely dependent on HER2 activity, presumably because EGFR can efficiently homodimerize and/or heterodimerize with other HER receptors in this cell line after EGF treatment.
Effect of gefitinib and M578440 on rhTRAIL-induced apoptosis
To determine whether inhibition of EGFR and HER2 would sensitize CRC cells to rhTRAIL, we assessed the effect of gefitinib and M578440 on rhTRAIL-induced apoptosis (Fig. 2 and Supplementary Fig. S1). CRC cells were exposed to each TKI for 24 hours, followed by rhTRAIL for 24 hours. Gefitinib alone had no effect on apoptosis in the HCT116 and HT29 cell lines but did moderately increase cell death in H630 and LoVo cells. Treatment with M578440 alone induced significant levels of apoptosis in the HCT116 cell lines but had no or little effect on apoptosis in the other cell lines. Both gefitinib and M578440 sensitized HCT116-p53wt cells to apoptosis induced by rhTRAIL, as indicated by the percentage of cells in the sub-G0-G1 apoptotic fraction (Fig. 2A andC). C). Gefitinib and M578440 also sensitized the p53 null HCT116 daughter cell line to rhTRAIL, indicating that this interaction is independent of the p53 status (Fig. 2A and C). Representative histograms are presented in Fig. 2B. Consistent with the HCT116 cell lines, gefitinib and M578440 sensitized both LoVo and H630 cell lines to apoptosis induced by rhTRAIL (Fig. 2A and C and Supplementary Fig. S1). Interestingly, inhibition of EGFR and HER2 activity sensitized the rhTRAIL-resistant HT29 cell line to rhTRAIL (Supplementary Fig. S1). These results were confirmed by Western blot analysis, which indicated that pretreatment of HCT116-p53wt and LoVo cells with either gefitinib or M578440 enhanced PARP cleavage (a marker of apoptosis) induced by rhTRAIL (Fig. 2D). Furthermore, there was evidence of enhanced rhTRAIL-induced caspase-8 activation in LoVo cells after pretreatment with gefitinib or M578440 and in HCT116-p53wt cells after pretreatment with M578440. Collectively, these data indicate that EGFR and HER2 are important regulators of CRC cell death induced by rhTRAIL.
Figure 2.
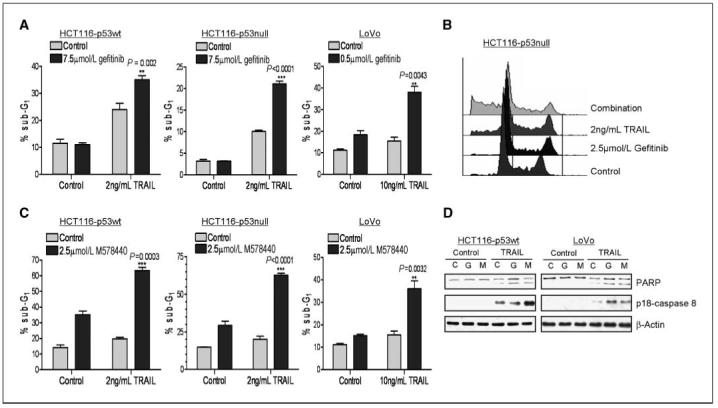
Effect of gefitinib and M578440 on rhTRAIL-induced apoptosis. Determination of apoptosis in CRC cells treated with the combination of gefitinib (A and B) or M578440 (C) with rhTRAIL in HCT116-p53wt, HCT116 p53 null, and LoVo cells. Cells were preincubated with gefitinib or M578440 for 24 h, followed by rhTRAIL for 24 h. The cell cycle status of the cells was monitored by flow cytometry after propidium iodide staining. Percentages of cells in sub-G1 phase are given. Representative results of at least two independent experiments. D, Western blot analysis of PARP cleavage and caspase-8 activity (monitored by emergence of the active p18-subunit) in HCT116-p53wt and LoVo cells pretreated with gefitinib (G, 7.5 μmol/L gefitinib and 0.4 μmol/L gefitinib in HCT116 and LoVo cells respectively) or M578440 (M, 2.5 μmol/L M578440 and 0.4 μmol/L M578440 in HCT116 and LoVo cells, respectively) for 24 h before treatment with rhTRAIL (2.5 and 7.5 ng/mL rhTRAIL in HCT116 and LoVo cells, respectively) for 6 h.
Effect of rhTRAIL on EGFR and HER2 activation
To elucidate the mechanisms involved in regulating the interaction between HER-targeted TKIs and rhTRAIL, we examined the effect of rhTRAIL on EGFR and HER2 phosphorylation in the HCT116-p53wt, HCT116-p53null, LoVo, and H630 cell lines. EGFR phosphorylation was measured using a phosphorylated-specific EGFR antibody directed against phosphorylated Y1068 that reflects the activation state of the receptor (21). The HER2Y1248 autophosphorylation site has been shown to couple the receptor to the RAS-MAPK-Jun pathway has been closely linked to the oncogenic potential of HER2 (22). In all four cell lines, a dose-dependent increase in EGFR and HER2 phosphorylation was observed 24 hours after rhTRAIL exposure (Fig. 3A). There was no change in total EGFR or HER2 expression levels after treatment with rhTRAIL, indicating that the receptors are activated in response to rhTRAIL (Fig. 3A). siRNA-mediated silencing of DR4 and DR5 abrogated rhTRAIL-induced phosphorylation of EGFR and HER2 (data not shown), indicating that the activation of EGFR and HER2 by rhTRAIL was dependent on the TRAIL death receptors and not on the decoy receptors, which are in any case expressed at very low levels (if at all) in these models (Fig. 1B). The effect of rhTRAIL on HER3 expression and activation was also assessed (Supplementary Fig. S2). In contrast to EGFR and HER2, HER3 was not activated in response to rhTRAIL treatment in either cell line. We also assessed cell surface expression of EGFR and HER2 after treatment with rhTRAIL in the HCT116-p53 wt and LoVo cell lines (Fig. 3B). Interestingly, a significant decrease in cell surface expression of EGFR and HER2 was observed in HCT116-p53wt cell line at higher doses of rhTRAIL. Consistent with the findings of others (23), we have found that receptor internalization is triggered after ligand-mediated HER activation in these cell line models (data not shown). The LoVo cell line also exhibited a significant dose-dependent decrease in EGFR cell surface expression in response to rhTRAIL, but no change in HER2 cell surface expression was observed; however, HER2 cell surface expression is relatively low in this cell line. Taken together, these results indicate that rhTRAIL activates EGFR and HER2 in CRC cells.
Figure 3.
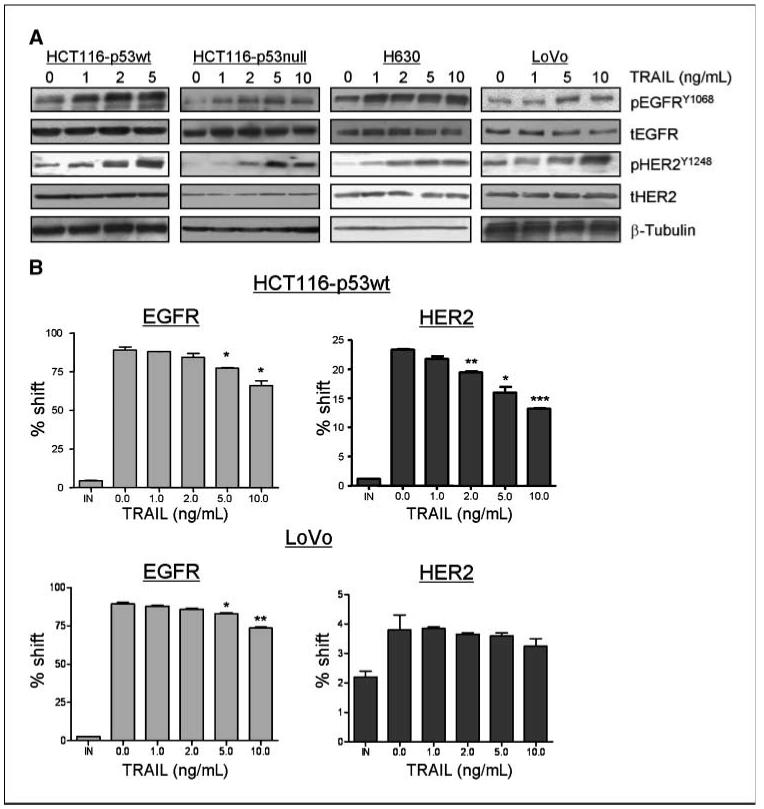
Effect of rhTRAIL on EGFR and HER2 activation and expression. A, HCT116-p53wt, HCT116-p53null, H630, and LoVo cells were treated with increasing doses of rhTRAIL for 24 h and pEGFRY1068, tEGFR, pHER2Y1248, and tHER2 levels were determined by Western blotting. Equal loading was assessed by probing for β-tubulin. B, HCT116-p53wt and LoVo cells were treated with increasing doses of rhTRAIL for 24 h, and EGFR and HER2 cell surface expression was determined by flow cytometry.
The role of Src family kinases in regulating rhTRAIL-induced EGFR activation
Cytoplasmic nonreceptor tyrosine kinases, such as c-Src and Yes, have been shown to activate the EGFR in response to a variety of stimuli (24-26). To gain insight into the mechanism of increased EGFR and HER2 activation in CRC cells treated with rhTRAIL, we examined the effect of rhTRAIL on SFK expression and phosphorylation in HCT116-p53wt and LoVo CRC cells. c-Src activation was measured using a phosphorylated-specific antibody directed against the Y418 autophosphorylation site, which represents fully catalytic active c-Src (27). As the region containing Y418 is highly conserved in other related Src family kinases, this antibody may also react with Fyn and Yes. In HCT116-p53wt (Fig. 4A, left) and LoVo (Fig. 4A, right), increased SFK activation correlated with activation of EGFR and HER2 after 24 hours treatment with rhTRAIL.
Figure 4.
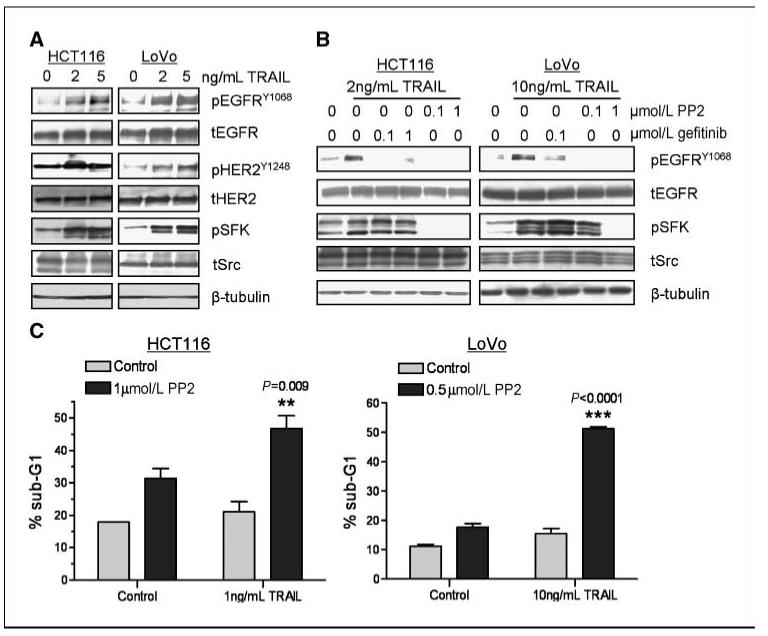
Role of SFKs in rhTRAIL-induced EGFR/HER 2 activation and apoptosis. A, HCT116-p53wt and LoVo cells were incubated with 2 or 5 ng/mL rhTRAIL for 24 h, and pEGFRY1068, tEGFR, pHER2Y1248, tHER2, pSFKY418, and tSrc expression were detected by Western blotting. Equal loading was assessed by probing for β-tubulin. B, HCT116-p53wt and LoVo cells were preincubated with the SFK inhibitor PP2 or gefitinib for 4 h followed by treatment with rhTRAIL for 24 h as indicated. Phosphorylated EGFRY1068, tEGFR, pSFKY418, and tSrc expression were visualized by Western blotting. Equal loading was assessed by probing for β-tubulin. C, HCT116-p53wt and LoVo cells were treated with no drug (control), PP2 alone, rhTRAIL alone, or PP2 in combination with rhTRAIL. Cells were exposed to PP2 for 24 h followed by rhTRAIL for 24 h, and the cell cycle status of the cells was monitored by flow cytometry after propidium iodide staining. Percentages of cells in sub-G0-G1 phase are given. Representative results of two independent experiments.
To investigate the sequence of EGFR and SFK activation after rhTRAIL treatment, we examined the relative effects of the SFK inhibitor PP2 and gefitinib on rhTRAIL-induced EGFR and SFK phosphorylation, respectively (Fig. 4B). Treatment with 1 μmol/L PP2 inhibited not only the increase in SFK phosphorylation after treatment with rhTRAIL but also inhibited rhTRAIL-induced EGFR and HER2 phosphorylation in HCT116-p53wt and LoVo cells. In contrast, although treatment with gefitinib inhibited rhTRAIL-induced EGFR phosphorylation, it had no significant effect on SFK activation after rhTRAIL treatment in these cell lines. Taken together, these results suggest that SFKs act upstream of EGFR and play a role in the activation of EGFR after rhTRAIL treatment in HCT116-p53wt and LoVo cells. Furthermore, pretreatment with PP2 resulted in a significant supraadditive increase in rhTRAIL-induced apoptosis in LoVo and HCT116 cells compared with cells treated with rhTRAIL or PP2 alone (Fig. 4C). To provide further support for the importance of SFKs in regulating rhTRAIL-induced apoptosis, we used AZD0530, which is a more specific SFK inhibitor than PP2. PP2 can also inhibit platelet-derived growth factor receptor (PDGFR) with similar affinity as Src (PP2-Src IC50 = 1.4 μmol/L and PP2-PDGFR IC50 =1.5 μmol/L; ref. 28; AZD0530-Src IC50 = 0.0027 μmol/L and AZD0530-PDGFRα and PDGFRβ, IC50 >5 μmol/L; personal communication from AstraZeneca). We found that AZD0530 inhibited constitutive SFK phosphorylation at concentrations of 0.1 μmol/L and above (Supplementary Fig. S3A). Furthermore, EGFR phosphorylation was also down-regulated after AZD0530 treatment, again indicating the dependence of EGFR activity on SFK activity in these cell line models (Supplementary Fig. S3A). Moreover, both HCT116 and LoVo cells were sensitized to rhTRAIL after treatment with AZD0530 (Supplementary Fig. S3B), similar to the results obtained with PP2.
Role of metalloproteases and ADAM-17 in rhTRAIL-induced EGFR activation
Members of the zinc protease family and metzincin subfamily (ADAMs) have been implicated in regulating EGFR activity by cleaving and promoting the release of its ligands, such as TGF-α, amphiregulin, and HB-EGF from the cell membrane (12, 14). We investigated the possibility of metalloprotease involvement in EGFR activation after rhTRAIL treatment in HCT116-p53wt and LoVo cells. Treatment with 20 μmol/L GM6001, a broad-spectrum hydroxamic acid matrix metalloproteinase (MMP) and ADAM inhibitor, resulted in inhibition of rhTRAIL-induced activation of EGFR in both cell lines, suggesting that metalloproteases are involved in rhTRAIL-induced activation of EGFR (Fig. 5A). EGFR has been reported to be coexpressed with ADAM-17 in cancer cells and endothelial cells of CRC tumors (29). Moreover, ADAM-17 has been suggested to play a central role in the shedding of EGFR ligands (7). Hence, we investigated whether ADAM-17 played a role in rhTRAIL-induced EGFR activation using an ADAM-17-specific siRNA (AD17 siRNA). AD17 siRNA down-regulated ADAM-17 expression at concentrations of ≥1 nmol/L (Fig. 5B). Importantly, we found that silencing of ADAM-17 attenuated rhTRAIL-induced EGFR and HER2 activation in both HCT116-p53wt and LoVo cells (Fig. 5C). As a single agent, AD17 siRNA had no significant effect on apoptosis in either cell line (Fig. 5D, left). However, pretreatment with AD17 siRNA for 24 hours, followed by rhTRAIL for 24 hours, resulted in a significant increase in apoptosis compared with rhTRAIL alone. Taken together, these results suggest that rhTRAIL-induced activation of EGFR/HER2 is ADAM-17-dependent and that by inhibiting ADAM-17, EGFR/HER2-mediated prosurvival signaling in response to rhTRAIL is inhibited and rhTRAIL-induced apoptosis is synergistically enhanced.
Figure 5.
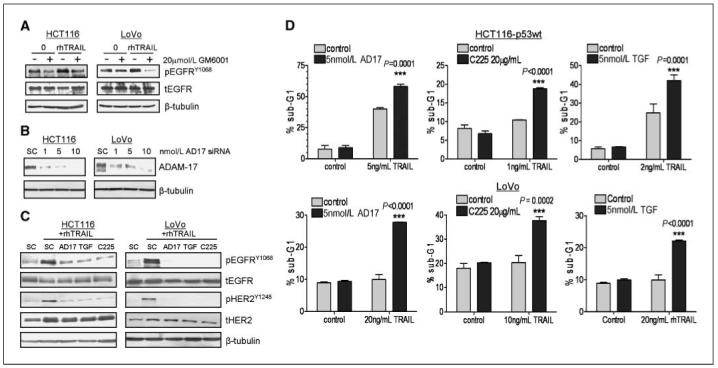
Role of ADAM-17 and TGF-α in rhTRAIL-induced EGFR activation and apoptosis. A, CRC cells were preincubated with 20 μmol/L GM6001 for 4 h followed by treatment with rhTRAIL for 24 h as indicated. EGFR phosphorylation and expression were measured by Western blotting. Equal loading was assessed by probing for β-tubulin. B, LoVo and HCT116 cells were transfected with 5 nmol/L ADAM-17 (AD17 siRNA) or 5 nmol/L SC siRNA and ADAM-17 expression analyzed by Western blot. Equal loading was assessed by probing for β-tubulin. C, LoVo and HCT116 cells were transfected with 5 nmol/L ADAM-17 (AD17 siRNA), 5 nmol/L TGF-α (TGF siRNA), 5 nmol/L SC siRNA or preincubated with 20 μg/mL C225 followed by rhTRAIL treatment for 24 h. Phosphorylated EGFRY1068, pHER2Y1248, tEGFR, and tHER2 expressions were detected by Western blotting. Equal loading was assessed by probing for β-tubulin. D, determination of apoptosis in CRC cells treated with combinations of 5 nmol/L AD17 siRNA (left), 20 μg/mL C225 (middle), or 5 nmol/L TGF-α siRNA (right) with rhTRAIL in HCT116 and LoVo CRC cell lines. Cells were transfected with siRNA or preincubated with C225 for 24 h before treatment with rhTRAIL for 24 h. The cell cycle status of the cells was monitored by flow cytometry after propidium iodide staining. Percentages of cells in sub-G0-G1 phase are given. Representative results of two independent experiments.
Effect of TGF-α on rhTRAIL-induced EGFR/HER2 activation and apoptosis
To determine whether rhTRAIL-induced EGFR and HER2 activation in CRC cells is ligand-dependent, we examined the effect of the anti-EGFR mouse-human chimeric mAb cetuximab (C225) on rhTRAIL-induced EGFR and HER2 phosphorylation (Fig. 5C). Cetuximab binds to the receptor with affinity comparable with the natural ligands and competes for ligand binding thereby inhibiting EGFR activation (30). In HCT116-p53wt and LoVo cells, we found that C225 completely inhibited rhTRAIL-induced EGFR and HER2 phosphorylation, suggesting that the activation of EGFR and HER2 after rhTRAIL is related to binding of EGFR ligands to the EGFR (Fig. 5C). As ADAM-17 has emerged as a major sheddase for TGF-α, we also determined the effect of TGF-α gene silencing on EGFR and HER2 phosphorylation in LoVo and HCT116 cells. Using quantitative real-time PCR, we found a decrease of 70% to 80% in TGF-α mRNA expression after transfection with 5 nmol/L TGF-α siRNA in the LoVo and HCT116 cell lines (data not shown). Transfection with 5 nmol/L TGF-α siRNA (TGF) inhibited EGFR and HER2 activation after rhTRAIL treatment in LoVo and HCT116 cells, and this down-regulation was similar to that observed when ADAM-17 was silenced (Fig. 5C).
We next directly investigated the importance of ligands and, in particular, TGF-α in regulating resistance to rhTRAIL using C225 and TGF-α-targeted siRNA (Fig. 5D, middle and right). In HCT116 and LoVo cells, pretreatment with C225 for 24 hours, followed by rhTRAIL for 24 hours, resulted in a significant supraadditive/synergistic increase in apoptosis compared with cells treated with rhTRAIL or C225 alone, as determined by flow cytometry (Fig. 5D, middle). Furthermore, a statistically significant supraadditive increase in apoptosis was observed when TGF-α siRNA was combined with rhTRAIL in the LoVo and HCT116 cells (Fig. 5D, right), indicative of a synergistic interaction. Moreover, treatment of HCT116 and LoVo cells with exogenous TGF-α abrogated rhTRAIL-induced PARP cleavage and caspase-3 and caspase-8 activation (Supplementary Fig. S4A). In addition, the apoptosis induced by combined treatment with rhTRAIL and the SFK inhibitor AZD0530 was also abrogated by coincubation with exogenous TGF-α (Supplementary Fig. S4B). These results indicate that TGF-α is an important mediator of resistance to rhTRAIL in CRC cells.
Finally, we used an ELISA assay to directly assess shedding of TGF-α into cell culture medium after rhTRAIL treatment in LoVo and HCT116-p53wt CRC cells (Fig. 6). In both cell lines, rhTRAIL induced TGF-α shedding, and this was attenuated when cells were cotransfected with ADAM-17-targeted siRNA (Fig. 6, middle). Furthermore, rhTRAIL-induced TGF-α shedding was abrogated in cells cotreated with the SFK inhibitors PP2 (Fig. 6, right) and AZD0530 (Supplementary Fig. S3C). Transfection with TGF-α siRNA also significantly down-regulated TGF-α shedding from both nontreated and rhTRAIL-treated cells, indicating both the specificity of the ELISA assay and the effectiveness of the TGF-α-targeted siRNA (Fig. 6, left). Collectively, these data suggest that TGF-α is required for rhTRAIL-induced EGFR activation and EGFR/HER2 dimerization and that this process is mediated via SFKs and ADAM-17.
Figure 6.
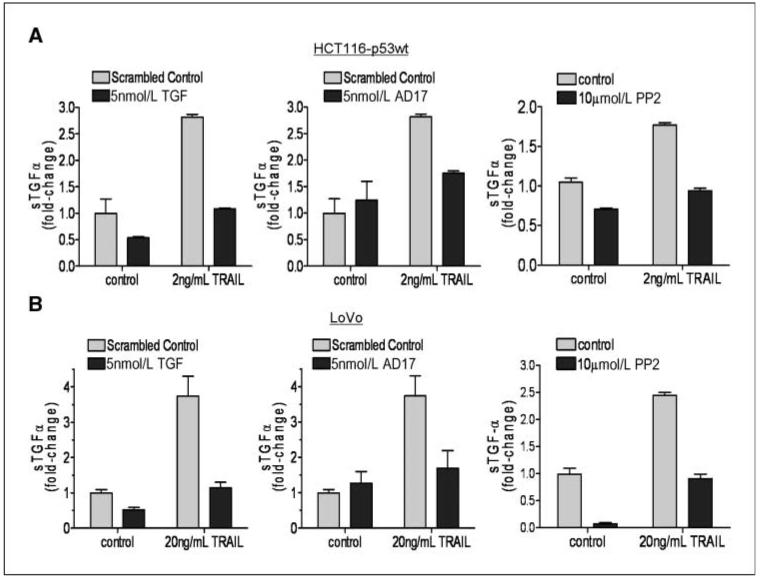
Role of SFKs and ADAM-17 in rhTRAIL-induced TGF-α shedding. LoVo and HCT116 cells were transfected with 5 nmol/L ADAM-17, 5 nmol/L TGF-α, or 5 nmol/L SC siRNAs or preincubated with PP2 for 4 h and then treated with rhTRAIL for 24 h. Shedding of TGF-α into the medium was measured using a TGF-α ELISA kit.
Discussion
Due to its apoptosis-inducing capability, selectivity for tumor cells and absence of significant adverse side effects, TRAIL-targeted therapies have emerged as promising anticancer agents. Various approaches have been developed to therapeutically target the TRAIL receptors, and several phase I studies evaluating the effect of fully human agonist mAbs against the TRAIL receptors DR4 and DR5 are currently ongoing. However, inherent tumor resistant may be a major barrier to effective TRAIL-targeted therapy (31, 32), indicating that sensitization of cancer cells to the TRAIL-induced apoptotic pathway is likely to be an important anticancer strategy. Initially, we examined the sensitivity of a panel of CRC cell lines to rhTRAIL as a single agent. We found that cell surface expression levels of TRAIL receptors did not correlate with sensitivity to rhTRAIL. Furthermore, although we found the HCT116-p53wt cell line to be slightly more sensitive to rhTRAIL compared with its p53 null counterpart, there was no clear correlation between p53 status and sensitivity to rhTRAIL.
Previously, we reported that down-regulation of c-FLIP after chemotherapy treatment, sensitizes CRC cells to rhTRAIL-induced apoptosis in vitro (33). Furthermore, previous reports have shown that genistein, a broad-spectrum tyrosine kinase inhibitor, and gefitinib sensitize human lung, gastric, and bladder cancer cells to TRAIL (34-36). So, we decided to evaluate the effect of gefitinib on rhTRAIL-induced cytotoxicity in CRC cell lines. We also studied the role of HER2 using the selective HER2-TKI M578440. Interestingly, pretreating rhTRAIL-sensitive and rhTRAIL-resistant cell lines to gefitinib or M578440 markedly sensitized them to rhTRAIL-induced apoptosis, indicating that inhibition of EGFR and HER2 may be a promising strategy for sensitizing CRC cells to TRAIL agonists. To elucidate the mechanism underlying these interactions, we examined rhTRAIL-induced EGFR and HER2 expression and phosphorylation. Importantly, the synergistic nature of the interactions between rhTRAIL and the EGFR-targeted and HER2-targeted TKIs correlated with a dose-dependent increase in EGFR and HER2 phosphorylation after rhTRAIL treatment. Of note, HER3 was not activated in response to rhTRAIL in these model systems. We have previously shown that chemotherapy-induced EGFR activation predicts the effectiveness of combined treatment with chemotherapy and EGFR-targeted therapies (9, 10). We found that CRC cell lines that activated EGFR in response to chemotherapy were sensitized to EGFR-targeted therapies. Our current findings suggest that rhTRAIL also induces an EGFR/HER2-mediated survival response in CRC cells and that, by blocking this response, rhTRAIL sensitivity is enhanced.
The nonreceptor tyrosine kinases, c-Src and c-Yes, have been found to activate the EGFR after osmotic stress and upon stimulation of various G protein-coupled receptors (GPCR; refs. 16, 25). The SFKs comprise nine members, some of which are ubiquitously expressed (c-Src, c-Fyn, c-Yes) and some of which display a more restricted expression (Blk, Yrk, Fgr, Hck, Lck, and Lyn; ref. 37). In particular, c-Src is overexpressed in >70% of CRCs and frequently activated in the advanced disease setting (38, 39). Apart from its interaction with growth factor receptors, such as EGFR and vascular endothelial growth factor receptor, c-Src plays an important role in cell adhesion, migration, and metastasis (39, 40). Initially, we examined the effect of rhTRAIL on SFK activation and found that increased EGFR and HER2 activation was associated with increased SFK activation after treatment with rhTRAIL. Interestingly, similar results were obtained with the agonistic Fas mAb CH-11: the synergistic interaction observed between CH-11 and gefitinib or M578440 correlated with a dose-dependent increase in EGFR, HER2, and SFK phosphorylation after CH-11 treatment (data not shown), indicating that activation of SFK, EGFR, and HER2 activation may be a common prosurvival response after death receptor ligation. Furthermore, using the SFK inhibitors PP2 and AZD0530, we showed that rhTRAIL-induced EGFR activation is dependent on SFK activity in these cell lines. Moreover, as gefitinib had no effect on TRAIL-induced SFK phosphorylation, we can conclude that SFKs are upstream activators of EGFR after treatment with rhTRAIL in these cell lines. In support of this, we also found a highly synergistic interaction between SFK inhibitors and rhTRAIL in our cell line panel. Taken together, it seems that the combination of SFK inhibition with rhTRAIL is a very promising anticancer strategy, consistent with the concept that SFKs are central to multiple oncogenic prosurvival signal transduction pathways.
Several reports have showed that, after GPCR stimulation, ADAMs can induce EGFR activation via the shedding of different EGFR ligands (25, 41). We found that rhTRAIL-induced EGFR phosphorylation in CRC cells was inhibited by the broad-spectrum MMP-ADAM inhibitor GM6001. These results suggested that ADAMs may regulate EGFR activation after rhTRAIL treatment. Several studies have shown that different ADAMs are key regulators of ectodomain shedding of the seven ligands of EGFR (7). ADAM-17 is frequently overexpressed in solid tumors, such as CRC, breast cancer, and hepatocellular carcinoma, and this protease plays a central role in the shedding of EGFR ligands (42). Furthermore, there is published evidence of an association between c-Src and ADAM-17 after GPCR stimulation in head and neck carcinoma cells (43). Hence, we determined the effect of ADAM-17 gene silencing on EGFR activation after rhTRAIL treatment. In both LoVo and HCT116 CRC cell lines, ADAM-17 silencing inhibited rhTRAIL-induced EGFR activation. In addition to this, we investigated the interaction between ADAM-17 siRNA and rhTRAIL in CRC cells and found a synergistic activation of apoptosis in the LoVo and HCT116 cells after cotreatment with ADAM-17 siRNA and rhTRAIL. Taken together, our data suggest that ADAM-17 is an important regulator of EGFR activation in response to rhTRAIL in CRC cells and that targeting this ADAM in conjunction with rhTRAIL may have therapeutic potential.
To further assess whether rhTRAIL-induced EGFR activation is a ligand-dependent process, we used the mAb cetuximab (C225), which competes for ligand binding to the EGFR. We found that C225 completely inhibited rhTRAIL-induced EGFR phosphorylation in LoVo and HCT116 cell lines, suggesting that rhTRAIL-induced EGFR activation is due to binding of EGFR ligands to the receptor. As ADAM-17 is the main sheddase for TGF-α, we determined the role of TGF-α in regulating rhTRAIL-induced EGFR activation in HCT116 and LoVo cell lines and found that TGF-α siRNA inhibited rhTRAIL-induced EGFR activation. In addition, we found that rhTRAIL-induced TGF-α shedding was abolished when cells were cotreated with SFK inhibitors or cotransfected with ADAM-17 siRNA, indicating that increased EGFR activity after rhTRAIL treatment is induced through activation of SFKs and ADAM-17, which in turn induces shedding of TGF-α. Furthermore, the combination of TGF-α siRNA or cetuximab with rhTRAIL resulted in synergistic increases in apoptosis, and addition of exogenous TGF-α inhibited rhTRAIL-induced apoptosis.
In conclusion, our findings indicate that CRC cells respond to rhTRAIL with an EGFR/HER2-mediated survival response and that, by blocking this response, apoptosis is enhanced. These data provide a preclinical rationale for the ongoing phase I study with the EGFR human mAb Panitumumab in combination with the DR5 agonistic antibody AMG655 in patients with solid tumors. Furthermore, we provide strong evidence that rhTRAIL induces EGFR activation via a ligand-dependent mechanism in CRC, and we have identified SFKs, ADAM-17, and TGF-α as critical mediators of EGFR/HER2 activation after rhTRAIL treatment. We propose a model in which rhTRAIL treatment results in SFK activation that in turn activates ADAM-17-mediated shedding of TGF-α. TGF-α then activates EGFR/HER2 prosurvival signaling in an autocrine and paracrine fashion. Thus, inhibiting EGFR/HER2, SFKs, or specific ADAMs (in particular ADAM-17) may have therapeutic potential for sensitizing CRC tumors to TRAIL-targeted therapies.
Supplementary Material
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: Cancer Research United Kingdom, Medical Research Council, and Ulster Cancer Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest
P.G. Johnston: directorship, Almac Diagnostics (Founding Director and shareholder); shareholdings, Fusion Antibodies, GlaxoSmithKline (GSK); consultancy, AstraZeneca, Pfizer, Roche Pharmaceuticals, Merck, Amgen, Bristol Myers Squibb, Ortho Biotech; contracted research, AstraZeneca, Amgen. The other authors disclosed no potential conflicts of interest.
Publisher's Disclaimer: The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Curtin JF, Cotter TG. Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003;15:983–92. doi: 10.1016/s0898-6568(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 2.Kischkel FC, Lawrence DA, Chuntharapai A, et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–20. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 3.van Geelen CM, Westra JL, de Vries EG, et al. Prognostic significance of tumor necrosis factor-related apoptosis-inducing ligand and its receptors in adjuvantly treated stage III colon cancer patients. J Clin Oncol. 2006;24:4998–5004. doi: 10.1200/JCO.2006.06.8809. [DOI] [PubMed] [Google Scholar]
- 4.Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–70. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 6.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–67. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–65. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 9.Van Schaeybroeck S, Karaiskou-McCaul A, Kelly D, et al. Epidermal growth factor receptor activity determines response of colorectal cancer cells to gefitinib alone and in combination with chemotherapy. Clin Cancer Res. 2005;11:7480–9. doi: 10.1158/1078-0432.CCR-05-0328. [DOI] [PubMed] [Google Scholar]
- 10.Van Schaeybroeck S, Kyula J, Kelly DM, et al. Chemotherapy-induced epidermal growth factor receptor activation determines response to combined gefitinib/chemotherapy treatment in non-small cell lung cancer cells. Mol Cancer Ther. 2006;5:1154–65. doi: 10.1158/1535-7163.MCT-05-0446. [DOI] [PubMed] [Google Scholar]
- 11.Chinnaiyan P, Huang S, Vallabhaneni G, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Res. 2005;65:3328–35. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 12.Fischer OM, Hart S, Gschwind A, Prenzel N, Ullrich A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol Cell Biol. 2004;24:5172–83. doi: 10.1128/MCB.24.12.5172-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh Y, Joh T, Tanida S, et al. IL-8 promotes cell proliferation and migration through metalloproteinase-cleavage proHB-EGF in human colon carcinoma cells. Cytokine. 2005;29:275–82. doi: 10.1016/j.cyto.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Schafer B, Marg B, Gschwind A, Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004;279:47929–38. doi: 10.1074/jbc.M400129200. [DOI] [PubMed] [Google Scholar]
- 15.Kishida O, Miyazaki Y, Murayama Y, et al. Gefitinib (“Iressa,” ZD1839) inhibits SN38-triggered EGF signals and IL-8 production in gastric cancer cells. Cancer Chemother Pharmacol. 2005;55:393–403. doi: 10.1007/s00280-004-0904-0. [DOI] [PubMed] [Google Scholar]
- 16.Reinehr R, Becker S, Hongen A, Haussinger D. The Src family kinase Yes triggers hyperosmotic activation of the epidermal growth factor receptor and CD95. J Biol Chem. 2004;279:23977–87. doi: 10.1074/jbc.M401519200. [DOI] [PubMed] [Google Scholar]
- 17.Yamauchi T, Ueki K, Tobe K, et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature. 1997;390:91–6. doi: 10.1038/36369. [DOI] [PubMed] [Google Scholar]
- 18.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.Ballard P, Bradbury RH, Hennequin LF, et al. 5-Substituted 4-anilinoquinazolines as potent, selective and orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg Med Chem Lett. 2005;15:4226–9. doi: 10.1016/j.bmcl.2005.06.068. [DOI] [PubMed] [Google Scholar]
- 20.Wakeling AE, Guy SP, Woodburn JR, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–54. [PubMed] [Google Scholar]
- 21.Helin K, Velu T, Martin P, et al. The biological activity of the human epidermal growth factor receptor is positively regulated by its C-terminal tyrosines. Oncogene. 1991;6:825–32. [PubMed] [Google Scholar]
- 22.Ben-Levy R, Paterson HF, Marshall CJ, Yarden Y. A single autophosphorylation site confers oncogenicity to the Neu/ErbB-2 receptor and enables coupling to the MAP kinase pathway. EMBO J. 1994;13:3302–11. doi: 10.1002/j.1460-2075.1994.tb06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiley HS. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res. 2003;284:78–88. doi: 10.1016/s0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 24.Kitagawa D, Tanemura S, Ohata S, et al. Activation of extracellular signal-regulated kinase by ultraviolet is mediated through Src-dependent epidermal growth factor receptor phosphorylation. Its implication in an anti-apoptotic function. J Biol Chem. 2002;277:366–71. doi: 10.1074/jbc.M107110200. [DOI] [PubMed] [Google Scholar]
- 25.Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. 2004;279:20927–34. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- 26.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am J Physiol Renal Physiol. 2004;286:F858–65. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 27.Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19:5620–35. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 28.Blake RA, Garcia-Paramio P, Parker PJ, Courtneidge SA. Src promotes PKCδ degradation. Cell Growth Differ. 1999;10:231–41. [PubMed] [Google Scholar]
- 29.Blanchot-Jossic F, Jarry A, Masson D, et al. Up-regulated expression of ADAM17 in human colon carcinoma: coexpression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156–63. doi: 10.1002/path.1814. [DOI] [PubMed] [Google Scholar]
- 30.Goldberg RM. Cetuximab. Nat Rev Drug Discov. 2005;(Suppl):S10–1. doi: 10.1038/nrd1728. [DOI] [PubMed] [Google Scholar]
- 31.Malhi H, Gores GJ. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene. 2006;25:7333–5. doi: 10.1038/sj.onc.1209765. [DOI] [PubMed] [Google Scholar]
- 32.Llobet D, Eritja N, Encinas M, et al. CK2 controls TRAIL and Fas sensitivity by regulating FLIP levels in endometrial carcinoma cells. Oncogene. 2008;27:2513–24. doi: 10.1038/sj.onc.1210924. [DOI] [PubMed] [Google Scholar]
- 33.Galligan L, Longley DB, McEwan M, et al. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005;4:2026–36. doi: 10.1158/1535-7163.MCT-05-0262. [DOI] [PubMed] [Google Scholar]
- 34.Jin CY, Park C, Cheong J, et al. Genistein sensitizes TRAIL-resistant human gastric adenocarcinoma AGS cells through activation of caspase-3. Cancer Lett. 2007;257:56–64. doi: 10.1016/j.canlet.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 35.Park SY, Seol DW. Regulation of Akt by EGF-R inhibitors, a possible mechanism of EGF-R inhibitor-enhanced TRAIL-induced apoptosis. Biochem Biophys Res Commun. 2002;295:515–8. doi: 10.1016/s0006-291x(02)00719-2. [DOI] [PubMed] [Google Scholar]
- 36.Shrader M, Pino MS, Lashinger L, et al. Gefitinib reverses TRAIL resistance in human bladder cancer cell lines via inhibition of AKT-mediated X-linked inhibitor of apoptosis protein expression. Cancer Res. 2007;67:1430–5. doi: 10.1158/0008-5472.CAN-06-1224. [DOI] [PubMed] [Google Scholar]
- 37.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–30. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 38.Bolen JB, Veillette A, Schwartz AM, Deseau V, Rosen N. Analysis of pp60c-src in human colon carcinoma and normal human colon mucosal cells. Oncogene Res. 1987;1:149–68. [PubMed] [Google Scholar]
- 39.Irby RB, Yeatman TJ. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002;62:2669–74. [PubMed] [Google Scholar]
- 40.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–46. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 41.Tanida S, Joh T, Itoh K, et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology. 2004;127:559–69. doi: 10.1053/j.gastro.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 42.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q, Thomas SM, Lui VW, et al. Phosphorylation of TNF-α converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A. 2006;103:6901–6. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.