

Abstract
Purpose
External beam radiation therapy is often used as in an attempt to cure localized prostate cancer (PCa), but is only palliative against disseminated disease. Raf Kinase Inhibitory Protein (RKIP) is a metastasis suppressor whose expression is reduced in approximately 50% of localized PCa tissues and is absent in metastases. Chemotherapeutic agents have been shown to induce tumor apoptosis through induction of RKIP expression. Our goal was to test if radiation therapy similarly induces apoptosis through induction of RKIP expression.
Methods
The C4-2B PCa cell line was engineered to over express or under express RKIP. The engineered cells were tested for apoptosis in cell culture and tumor regression in mice following radiation treatment.
Results
Radiation induced both RKIP expression and apoptosis of PCa cells. Over expression of RKIP sensitized PCa cells to radiation-induced apoptosis; whereas, short-hairpin targeting of RKIP, so that radiation could not induce RKIP expression, protected cells from radiation-induced apoptosis. In a murine model, knockdown of RKIP in PCa cells diminished radiation-induced apoptosis. Molecular concept mapping of genes altered upon manipulation of RKIP expression revealed that an inverse correlation with the concept of genes altered by irradiation.
Conclusion
The data presented here indicate that the loss of RKIP, as seen in primary PCa tumors and metastases, confers protection against radiation-induced apoptosis. Therefore, it is conceivable that loss of RKIP confers a growth advantage upon PCa cells at distant sites since loss of RKIP would decrease apoptosis, favoring proliferation.
Keywords: RKIP, ionizing radiation, apoptosis, prostate cancer, radioresistance
Introduction
The phosphotidylethanolamine binding protein (PEBP) family is highly conserved and does not share sequence homology to other proteins of known function (1). Yeung, et al. identified a PEBP homolog that is a competitive inhibitor of Raf/MEK/ERK signaling (2, 3) and named it Raf Kinase Inhibitory protein (RKIP). RKIP is expressed in prostate, brain, liver, lung, testis, muscle, and stomach (4, 5).
RKIP is involved in many cellular activities (reviewed in (6)). Many of the pathways RKIP is involved in are often dysregulated in cancer; therefore, it was postulated that RKIP plays a role in cancer progression. We demonstrated that RKIP was abundant in non-cancerous prostate tissue, decreased in primary PCa, and was weak or absent in PCa metastases (7) and that increasing RKIP in human PCa cells inhibits their ability to metastasize (8). Taken together, these results demonstrated that RKIP is a metastasis suppressor in PCa.
The mechanism through which RKIP contributes to PCa metastasis is not known. Loss of RKIP could promote metastasis through effects on intravasation, extravasation, or growth of the metastasis at distant sites. Chatterjee, et al. (9) suggested that RKIP can regulate apoptosis and, therefore, regulate growth. They demonstrated that DNA damaging agents 9-Nitrocamptothecin (9NC), cisplatin, and etoposide upregulate RKIP and induce apoptosis. Genetic knock-down of RKIP reduced the apoptotic response to the drugs. These data indicated that RKIP regulates apoptosis, suggesting that PCa cells that have lost RKIP during metastasis may have a survival advantage at distant sites since RKIP is not present to promote apoptosis.
Radiation therapy is a widely used modality for the treatment of PCa. Prostate radiotherapy can be used as curative treatment for clinically localized PCa. Recurrence after radiation happens in approximately 30% of cases depending on the disease stage (10). Palliative radiation therapy may be used if the disease is wide-spread. Radiation induces DNA damage, causing tumor cells to undergo apoptosis (11–14). Since chemical DNA damaging agents promote apoptosis through increasing RKIP levels, we investigated whether RKIP contributes to radiation-induced apoptosis and if loss of RKIP confers radioresistance to cancer cells.
Materials and Methods
Cell culture
C4-2B cells, isolated from bone-metastasizing LNCaP cells (15, 16), were maintained in T-medium (80% DMEM-20% Ham’s F12 (GIBCO), 5ug/ml Insulin, 13.6 pg/ml triiodothyronine, 5ug/ml transferrin, 0.25 μg/ml biotin, 25 μg/ml adenine (Sigma, St. Louis, MO), 1× penicillin/streptomycin and 10% FBS). Media for C4-2B cells expressing vector (pBP), fRKIP (over expressing RKIP) or shRKIP (under expressing RKIP) were supplemented with 0.5 μg/ml puromycin.
RKIP constructs
The RKIP gene was cloned into the CMV-FLAG vector (Sigma), then subcloned into pBP (pBABE) by the Yeung laboratory. Retrovirus resulting from a transfection of this plasmid into φNX-A cells was isolated as previously described (17) was used to infect C4-2B PCa cells. Infected cells were selected with puromycin. An RKIP-specific shRNA expression construct was generated by cloning the annealed oligonucleotides (5′GATCCCCGATTCAGGGAAGCTCTACATTCAAGAGATGTAGAGCTTCCCTGAATCTTTTTA-3′ and 5′ AGCTTAAAAAGATTCAGGGAAGCTCTACATCTCTTGAATGTAGAGCTTCCCTGAATCGGG-3′) into pSUPER.retro-puro (OligoEngine) according to the manufacturer¹s instructions. Virus was isolated from 293FT cells and used to infect C4-2B PCa cells. Infected cells were selected with puromycin.
Protein blots
Cells were lysed in a buffer containing 20 mM Tris·HCl, pH 8.0, 137 mM NaCl, 1% NP-40, 10% glycerol, 1 mM Na3VO4, 1 mM PMSF, 1% aprotinin, and 20 μg/ml leupeptin. Protein concentrations are equalized using the Løwry method. Equal amounts of protein are separated by SDS-PAGE. The proteins are blotted to PVDF membranes and probed with antibodies for RKIP (Cat #37–3300, Zymed Laboratories, Invitrogen Corporation, Carlsbad, CA; or PARP (Cat#9542, Cell Signaling, Inc., Danvers, MA). Densitometry was performed using Scion Image (Scion Corporation, Frederick, MD). All blots were performed at least 3 times.
Radiation treatment
In vivo and in vitro irradiation was carried out using a Philips 250 kV orthovoltage unite at a dose rate of approximately 2 Gy/min. Dosimetry was carried out using an ionization chamber connected to an electrometer system that is directly traceable to a National Institute of Standards and Technology calibration.
Survival curves
Survival assays were performed as previously described (18). Briefly, cells were irradiated then trypsinized, counted with the Coulter counter (Hialeah, FL), and plated at clonal densities in 60mm tissue culture plates and fed once a week for 2 weeks. On the 14th day of culture, the cells were fixed in 7:1 methanol:acetic acid and stained with 0.5% crystal violet. Colonies were scored if they contained 50 or more cells. The colony forming efficiency (CFE) was calculated from control plates as the number of colonies divided by the number of cells plated. The surviving fractions of treated plates were calculated as the CFEs of plates from treated cells divided by the CFE of control cells. Survival curves were performed in triplicate at least 3 times.
Apoptosis assays
Apoptosis assays were performed according to manufacturer’s instructions (CellTrace™ and Caltag™ Invitrogen, Corp, Carlsbad, CA). Briefly, cells were plated onto chamber slides, fixed, and stained with either Calcein AM, Annexin V, or propidium iodide (PI) and viewed under fluorescence. These compounds indicate apoptosis. All immunofluorescence were performed at least 3 times in duplicate.
Animal studies
All animal protocols were approved by the University of Michigan Institutional Animal Use Committee. 1×106 cells were injected subcutaneously into each of the flanks of CB17 SCID mice. Ten animals were injected with C4-2B pBP cells, 10 with C4-2B fRKIP, and 10 with C4-2B shRKIP. When tumors were palpable (150–200 mm3), fifteen 2 GY fractions of ionizing radiation were given on days 1–5, 8–12 and 15–19. Tumor sizes were measured throughout the experiment. Mice were anesthetized with IP injections of ketamine (80 mg/kg) and xylazine (4mg/kg), and were placed prone in custom made holders. Cerrobend shielding was placed over the entire mouse minus the tumor creating a conformal treatment field. After radiation, mice were kept warm with heating pads until fully recovered from anesthesia. The shRKIP group lost one treated and one untreated animal 4 weeks after start of treatment due to extremely large tumors. The other groups lost one animal each after 88 days. The experiments were stopped around 100 days due to atrtion and large tumor growth in the shRKIP group.
RNA isolation and microarray analysis
Total RNA from C4-2B cells harboring pBP (vector), fRKIP, and shRKIP were isolated using TRIZOL reagent following the manufacturer’s instructions (Invitrogen). Oligonucleotide microarray was performed on Affymetrix Genechip (Human genome U133 Plus 2.0) array. Expression values were calculated after normalization of the data based on previously published procedure (19). Gene expression fold changes for fRKIP over pBP (fRKIP vs vector), and fRKIP over shRKIP (fRKIP vs shRKIP), shRKIP over pBP (shRKIP vs vector), and shRKIP over fRKIP (shRKIP vs. fRKIP) were calculated from two independent biological replicate array experiments. Expression values for each gene were calculated using a robust multi-array average (RMA) (19). This is a modeling strategy that converts the PM probe values into an expression value for each gene. The expression values are log2 transformed data. Overlap of genes that showed greater than or equal to 1.5 fold changes (data not shown) were used for Molecular Concepts Map (MCM) analysis.
Molecular concepts analysis
Affymetrix probe ids that showed changes due to RKIP over expression (fRKIP vs pBP and fRKIP vs shRKIP) and RKIP knock-down (shRKIP vs pBP and shRKIP vs. fRKIP) consisted of 59 and 111 ids, respectively, and were uploaded to Oncomine database (20). The uploaded genes were subjected to biological concepts and pathway analysis using the integrated Oncomine Concepts MaP (OCM) tool (20, 21). As a result of the pairwise analysis between the uploaded set of genes and a total of about 13,364 concepts in the database, several concepts that showed significant association were obtained. However, only radiation related concepts are reported in this manuscript.
Results
To study the radiation affects on RKIP expression, we used the C4-2B PCa cells. C4-2B is a metastatic subline of LNCaP cells (reviewed in (22)). C4-2B cells have a low level of RKIP compared to it’s non-metastatic counterpart LNCaP (8). C4-2B cells were used to demonstrate that re-expression of RKIP could block PCa metastasis. Therefore, we chose to manipulate the expression of RKIP in C4-2B cells and use the resulting engineered cell lines for the studies presented here. Since chemical DNA damaging agents have been shown to increase RKIP protein levels and apoptosis (9), we tested whether or not ionizing radiation, another type of DNA damaging agent, could induce RKIP expression and apoptosis as well. We used a metastatic PCa cell line that expressed relatively small amounts of RKIP, C4-2B (8). We observed 28% and 85% increase in RKIP protein levels in C4-2B with 2 Gy of radiation at 1 hr after treatment and at 3 hr after treatment, respectively (Figure 1A). To assess for apoptosis we stained cells with calcein AM and propidium iodide (PI). We observed apoptosis in the irradiated samples and not in the un-irradiated samples as was evident by the bright/white appearance of the PI stain 48 hours after 2GY irradiation (arrows in Figure 1B). The background PI staining in the C4-2B cells is probably due to the fact that parental C4-2B cells have relatively low levels of RKIP (Figure 2) and were capable of baseline levels of apoptosis. Therefore, radiation-induced apoptosis correlated with radiation-induced RKIP expression.
Figure 1.

RKIP is induced by ionizing radiation. (A) C4-2B cells were irradiated with 0.2 Gy, 2.0 Gy, or 5.0 Gy. After 1 hour or 3 hours, proteins were harvested, separated on SDS-PAGE, blotted, and probed for RKIP. Protein blots were stripped and reprobed for actin as a loading control. Densitometry was performed using Scion Image. The amount of RKIP relative to the corresponding actin amounts and relative to untreated are given. These experiments were performed 3 times. A representative blot is shown. (B) Radiation induces apoptosis of C4-2B PCa cells. PCa cells were either untreated or treated with 2Gy radiation. After 24 hrs, cells were stained with Calcein Am and propidium iodide (PI). Bright staining in PI panels suggests apoptosis and is indicated by the arrows. These experiments were performed at least 3 times. A representative experiement is shown.
Figure 2.

Knockdown of RKIP expression confers radioresistance. (A) C4-2B cells contained vector (pBP), an over expression of a FLAG-tagged RKIP (fRKIP), or the shRNA knock-down for RKIP (shRKIP). The protein blot was probed with RKIP, stripped and probed for FLAG, and stripped again and probed for actin. Densitometry was performed using Scion Image. The amount of RKIP relative to the corresponding actin amounts and relative to the parental C4-2B cells are given. These blots have been performed numerous times with a representative one being shown here. (B) C4-2B cells containing vector (pBP), RKIP over expression (fRKIP), or RKIP knock-down (shRKIP) were subjected to a clonogenic survival assay after 2.0 Gy, 3.0 Gy, or 5 Gy. After 14 days, colonies of 50 cells or more were counted and the surviving fraction was determined by number of colonies divided by the number of cells plated. This experiment was performed at least 3 times.
To determine if increased RKIP expression promoted apoptosis, we used C4-2B PCa cells that were engineered to express varying levels of RKIP. In polyclonal populations of C4-2B cells RKIP expression was increased approximately 44% using a FLAG-tagged RKIP (fRKIP) or knocked down approximately 70% using a shRNA to RKIP (shRKIP) (Figure 2A) compared to the parental C4-2B cells. Again, there was a low level of apoptosis in the control cells probably due to the relatively low level of RKIP in those cells. The over expression of RKIP was confirmed by the presence of the FLAG tag (lane 3, Figure 2A).
We next tested whether radiation-induced apoptosis requires RKIP. C4-2B cells with modulated RKIP amounts were subjected to increasing amounts of radiation and used in a clonogenic survival assay. Colony growth indicated radiation-resistant cell survival. Knockdown of RKIP (shRKIP) resulted in cells exhibiting better survival than cells over expressing RKIP (fRKIP) or vector-only cells (pBP) (Figure 2B). These results indicate that radiation induces apoptosis through RKIP and are consistent with the concept that loss RKIP confers radiation resistance.
To determine the effect of RKIP expression on the extent of radiation-induced apoptosis, cells with modulated amounts of RKIP were subjected to ionizing radiation, stained with Annexin V, and analyzed by fluorescent immunocytochemistry staining for Annexin V and PI after 2Gy treatment (Figure 3A). Bright staining (at the arrows) indicates apoptosis. Only the cells over expressing RKIP were undergoing apoptosis following radiation treatment. These data indicate that RKIP promotes radiation-induced apoptosis.
Figure 3.

RKIP expression confers radiation-induced apoptosis. (A) C4-2B cells, C4-2B cells over expressing RKIP (fRKIP), and C4-2B cells with RKIP knocked-down (shRKIP) were stained with Annexin V and propidium iodide (PI) 48 hrs after 2 Gy of radiation and viewed in a fluorescent microscope. Bright staining suggests apoptosis and is indicated by the arrows. This experiment was performed 3 times. (B and C) C4-2B cells, vector-only (pBP), cells over expressing RKIP (fRKIP), and cells with RKIP knocked down (shRKIP) were treated with 0, 1, 5, or 10 Gy. After 3 hrs, protein was harvested, separated on SDS-PAGE, blotted, and probed for PARP. (B). Western blot for PARP. Densitometry was performed using Scion Imgae. The amounts of PARP cleavage (89 kd band) in treated cells relative to the untreated (0 Gy) are given for each cell type. (C) The amounts of PARP cleavage relative to the untreated (0 Gy) are shown in graphic form for the RKIP over expressors (fRKIP) (black bars) and under expressors (shRKIP) (white bars/black slashes). The amounts of PARP cleavage relative to the vector only cells (pBP) are shown for fRKIP (white bars/horizontal lines)and shRKIP (white bars/black diamonds).
As a further measure of the influence of RKIP on radiation-induced apoptosis, we measured PARP cleavage. PARP, a poly (ADP-ribose) polymerase, is involved in DNA repair in response to environmental stress (23) and is cleaved during apoptosis by caspases (24, 25). Over expression of RKIP (fRKIP) induced PARP cleavage products even at low levels of radiation (1 Gy); whereas, cells with knocked down RKIP (shRKIP) did not contain PARP cleavage fragments until they were treated with 5 Gy (Figure 3B). In cells over expressing RKIP (fRKIP), PARP cleavage was increased by greater than 20 fold with 5Gy of radiation compared to untreated cells and was not increased in cells that had RKIP knocked out (shRKIP). The amount of PARP cleavage relative to untreated is given for each cell type under each blot (Figure 3B) and in graphic form (Figure 3C). When comparing the amount of PARP cleavage to the vector only cells (pBP), cells over expressing RKIP (fRKIP) contained greater than 10-fold the amount of PARP cleavage product than pBP cells at 5 Gy, and the knockdown of RKIP (shRKIP) slightly decreased PARP cleavage. The amount of PARP cleavage relative to each treatment in the vector-only (pBP) cell blot is given for both the knockdown (shRKIP) and the over expressor (fRKIP) in both numerical (Figure 3B) and graphic (Figure 3C; fRKIP (rel) and shRKIP (rel)) form. These data indicate that RKIP deletion can protect cells against apoptosis and that RKIP over expression promotes apoptosis.
To determine if the in vitro results were relevant to the in vivo setting, mice were injected subcutaneously with C4-2B vector only cells (Figure 4A), C4-2B cells over expressing RKIP (Figure 4B), or C4-2B cells under expressing RKIP (Figure 4C). Radiation induced a strong anti-tumor response in control cells or cells expressing RKIP; whereas, the cells that had knockdown of RKIP did not respond to irradiation. These data are consistent with the in vitro data and indicate that loss of RKIP protects against radiation-induced apoptosis.
Figure 4.
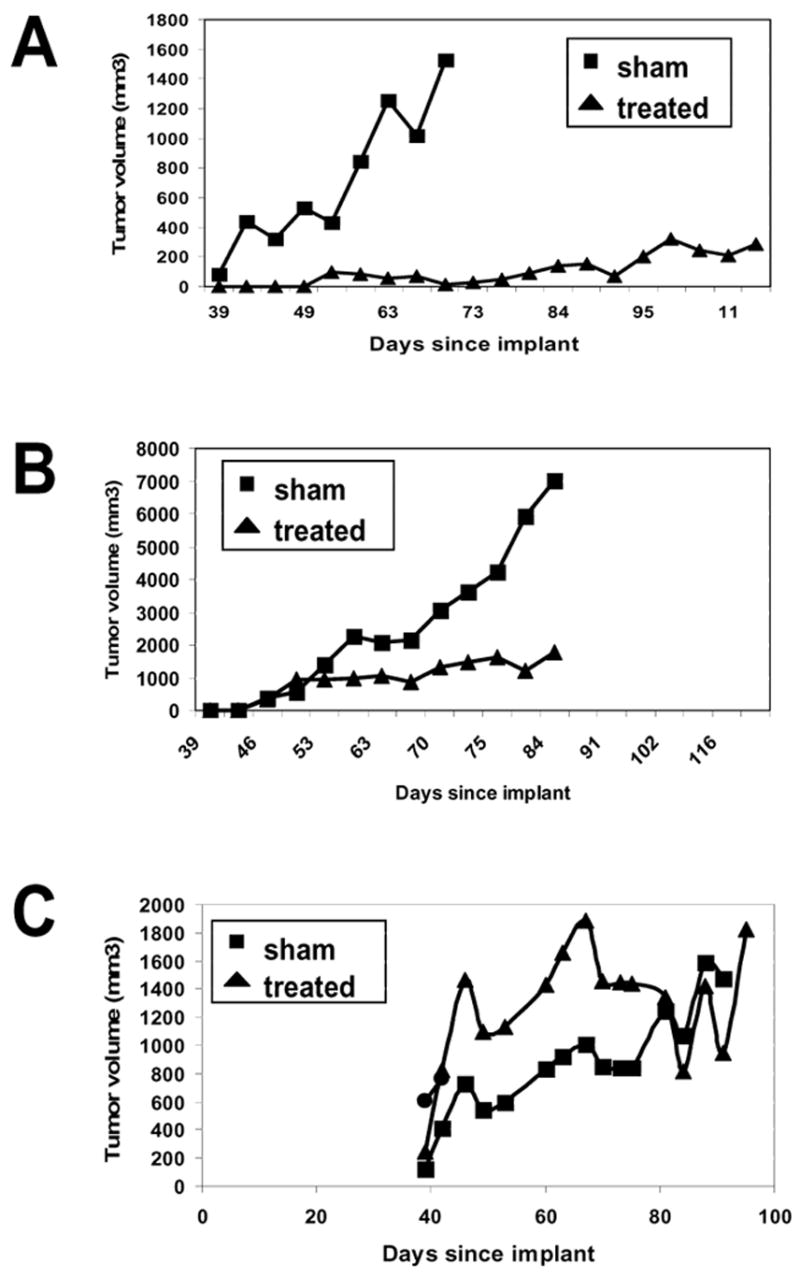
Knock-down of RKIP protects tumors from radiation. 1×106 C4-2B cells with (A) vector (pBP), (B) overexpression of RKIP (fRKIP), or (C) RKIP knockdown (shRKIP) were injected into the flanks of male CB17 SCID mice. When tumors were palpable (150–200 mm3), they were either treated with 2 Gy for 15 cycles or sham treated. Tumor volumes are graphed. Average surviving fractions are plotted versus radiation dose. Standard errors are given on the graph.
In an attempt to identify potential mechanisms through which RKIP sensitizes cells to radiation-induced apoptosis, we performed molecular concept mapping using the Oncomine Concepts Map function (OCM) analysis of genes that showed changes in response to either over-expression or knock-down of RKIP (please see methods for details). OCM is a tool designed to use microarray data to indicate phenotypes that are associated with gene expression patterns (20, 21) and is currently the largest database of cancer microarray data. Two of the concepts from literature that defined the concept type of radiation-induced expression signature are given in Table 1. Both of these concepts are comprised of genes down-regulated in fibroblasts due to UV irradiation (26, 27) and were associated with gene signatures over-expressed in RKIP knock-down (shRKIP over vector, and shRKIP over fRKIP). Essentially, we found that genes that are down-regulated in response to UV-irradiation were up-regulated in C4-2B cells with RKIP knocked down (shRKIP); therefore, genes that are down-regulated in the response to radiation were present in higher amounts in cells with a knockdown of RKIP. Functions of the genes that were identified in this screen that have a role in apoptosis or tumor progression are given in Table 2. Genes that were upregulated in response to loss of RKIP include functions such as increasing cell migration, increasing proliferation, binding death-domain containing proteins, and playing a role in regulating the balance between cell growth and cell death. These data are supportive evidence that loss of RKIP promotes radioresistance.
Table 1.
Genes over expressed in shRKIP expressing C4-2B cells
| Query concept: Genes over expressed in shRKIP compared to fRKIP molecular concept: down-regulated genes in WS1 human skin fibroblasts after exposure to low dose UVC radiation | Query concept: Genes over expressed in shRKIP compared to pBP molecular concept: Down-regulated genes in human fibroblast cells expressing cancer-prone XPB/TTD allele 8h after UVC-irradiation compared to unirradiated cells |
| Odds Ratio = 9.71
P-value = 0.005 |
Odds Ratio = 3.92
P-value = 0.003 |
|
| |
| Overlapped genes:
SEMA3C (semaphorin 3C) PFT K1 (pftaire protein kinase 1) UTRN (utrophin) |
Overlapped genes:
SEMA3C (semaphorin 3C) PDLIM5 (Pdz and lim domain 5) DAPK1 (death-associated protein 1) GNAI1 (G protein binding protein) ID2 (inhibitor of DNA binding 2) ATRX (rad54 homolog) BICD1 (bicaudal homolog 1) TCEA1 (transcription elongation factor a) |
Table 2.
Genes from Table 1 with links to apoptosis or cancer progression
| Gene | Activity |
|---|---|
| PDLIM5 | LIM domains bind to ID2, PDZ domain binds actin (39) |
| ID2 | Blocks proliferation and increases proliferation (40); affects the balance of cell growth and death (41) |
| DAPK1 | Positive mediator of apoptosis (42) |
| ATRX | In complex with death-domain protein DAXX (43) |
| SEMA3C | Influences cell migration and may contribute to tumorigenesis and tumor progression (44, 45) |
| GNAI1 | Mutations cause this gene to become an oncogene (46) |
Discussion
In this manuscript, we showed that ionizing radiation induced the expression of the metastasis suppressor RKIP in the metastatic PCa cell line C4-2B. We then engineered the C4-2B cells to express various amounts of RKIP. Cells with over expression of RKIP readily underwent apoptosis when treated with radiation in vitro; whereas, cells with an under expression of RKIP did not undergo apoptosis with radiation. Furthermore, we showed that C4-2B cells with over expression of RKIP were treatable with radiation when grown as a subcutaneous tumor in mice. However, tumors arising from C4-2B cells with an under expression of RKIP were resistant to radiation therapy. The data presented here indicate that the loss of RKIP as PCa progresses would favor a proliferative response, and that the presence of RKIP as seen in localized PCa mediates apoptosis in response to a stress signal, such as trying to grow at a distant site.
Patients with clinically-localized PCa can be cured with local therapy including surgery and radiation therapy. However, once PCa advances beyond the localized stage, localized treatments are no longer effective. Therefore, determining how regional PCa evades localized treatment and becomes metastatic is necessary to identify patients who may be cured with local therapy and to develop improved treatments for more advanced, metastatic PCa.
RKIP has been associated with metastasis suppressor activity both in vitro and in vivo (6). Loss of RKIP has also been demonstrated in the progression of several human cancers (28–30), including PCa (7). However, the mechanism of metastasis suppression by RKIP is unknown.
Metastasis suppression can occur at any number of steps along the metastatic cascade, including blocking motility, intravasation, endothelial cell binding, extravasation, and growth at the distant site. We have shown RKIP expression to be down-regulated in 50% of primary PCa and in the majority of metastatic PCa (7) and have identified RKIP as a metastasis suppressor (8).
Gamma radiation has been shown for some time to be capable of inducing DNA damage in a variety of ways: inducing breakage of the thymine moiety of DNA (11, 12), inducing double-stranded breaks (13), and inducing incorporation of wrong bases into DNA (14). Extensive DNA damage can lead to apoptosis, presumably through a p53-mediated process. However, the immediate molecular consequences of radiation-induced DNA damage are not known. In the current study, we showed that radiation induced a metastasis suppressor molecule, RKIP. In turn, RKIP expression was associated with PARP cleavage and apoptosis. In concordance with these data, we have observed that another PCa cell line that expresses very low levels of RKIP, PC-3 cells, undergo apoptosis when forced to stably over express RKIP (data not shown).
Millions of cells from primary tumors are shed into the blood stream daily; however, only a few cells successfully develop into clinical metastases (31, 32). Interestingly, some of the cells that seed distant sites remain dormant for years (33, 34). However, work by the Uhr group (35, 36) has shown that these dormant cells can proliferate but do not form clinical metastases because ongoing apoptosis balances out the proliferation. Therefore, loss of the ability to undergo apoptosis in dormant cells may lead to the formation of clinical metastases. Accordingly, it is conceivable that loss of RKIP at distant sites confers a growth advantage upon PCa cells since loss of RKIP could decrease apoptosis and favor proliferative activity. We have observed that genes which need to be down-regulated for a successful radiation-induced apoptotic response are present in higher amounts in cells with a knockdown of RKIP (Table 2). It is possible that down-regulating these genes to a level needed to induce apoptosis may be impossible in cells where RKIP is knocked down. This hypothesis would explain how PCa cells that have lost RKIP escape treatment and seed a distant site
Taken together, our data indicate that cells expressing RKIP can readily undergo apoptosis under certain conditions. Therefore, we hypothesize that of the cells that disseminate in the body, those expressing RKIP will undergo apoptosis, and those that have lost RKIP will be able to survive.
Although radiation therapy is a highly effective local treatment for clinically localized PCa, some aggressive cases of PCa may be resistant to radiation therapy and subsequently develop metastatic potential. Our previous observation that approximately 50% of primary tumors have low RKIP expression (7) along with the data from the current study, indicating that RKIP is needed for radiation to induce apoptosis, suggest that these PCa tumors will be radioresistant. New therapies for radioresistant PCa are needed in order to cure men with aggressive disease. Based on the results presented here, we hypothesize that PCa may escape the initial radiation therapy because of a loss of RKIP. Therefore, increasing RKIP in vivo may be a viable treatment for advanced PCa or can be an effective adjuvant for radiation therapy.
Acknowledgments
These experiments were funded, in part, by National Institutes of Health grant CA098513.
Footnotes
Conflict of Interest
There are no conflicts of interest for any of the authors concerning this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banfield M, Barker J, Perry A, Brady R. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure. 1998;6:1245–54. doi: 10.1016/s0969-2126(98)00125-7. [DOI] [PubMed] [Google Scholar]
- 2.Yeung K, Janosch P, McFerran B, et al. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–85. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung K, Seitz T, Li S, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 4.Frayne J, Ingram C, Love S, Hall L. Localisation of phosphatidylethanolamine-binding protein in the brain and other tissues of the rat. Cell Tissue Res. 1999;298:415–23. doi: 10.1007/s004419900113. [DOI] [PubMed] [Google Scholar]
- 5.Bollengier F, Mahler A. Localization of the novel neuropolypeptide h3 in subsets of tissues from different species. J Neurochem. 1988;50:1210–4. doi: 10.1111/j.1471-4159.1988.tb10594.x. [DOI] [PubMed] [Google Scholar]
- 6.Keller ET, Fu Z, Brennan M. The biology of a prostate cancer metastasis suppressor protein: Raf kinase inhibitor protein. Journal of Cellular Biochemistry. 2005;94:273–8. doi: 10.1002/jcb.20169. [DOI] [PubMed] [Google Scholar]
- 7.Fu Z, Kitagawa Y, Shen R, et al. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66:248–56. doi: 10.1002/pros.20319. [DOI] [PubMed] [Google Scholar]
- 8.Fu Z, Smith P, Zhang L, et al. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–89. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee D, Bai Y, Wang Z, et al. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. Journal of Biological Chemistry. 2004;279:17515–23. doi: 10.1074/jbc.M313816200. [DOI] [PubMed] [Google Scholar]
- 10.Dattoli M, Wallner K, True L, Cash J, Sorace R. Long-term outcomes after treatment with brachytherapy and supplemental conformal radiation for prostate cancer patients having intermediate and high-risk features. Cancer. 2007;110:551–5. doi: 10.1002/cncr.22810. [DOI] [PubMed] [Google Scholar]
- 11.Teoule R, Bonicel A, Bert C, Cadet J, Polverelli M. Identification of radioproducts resulting from the breakage of thymine moiety by gamma irradiation of E. coli DNA in an aerated aqueous solution. Radiat Res. 1974;57:46–58. [PubMed] [Google Scholar]
- 12.Swinehart J, Lin W, Cerutti P. Gamma-ray induced damage in thymine in mononucleotide mixtures, and in single- and double-stranded DNA. Radiat Res. 1974;58:166–75. [PubMed] [Google Scholar]
- 13.Corry P, Cole A. Radiation-induced double-strand scission of the DNA of mammalian metaphase chromosomes. Radiat Res. 1968;36:528–43. [PubMed] [Google Scholar]
- 14.Saffhill R. The incorporation of wrong bases by DNA polymerase I following gamma-irradiation of DNA-like templates. Biochim Biophys Acta. 1974;349:23–31. doi: 10.1016/0005-2787(74)90004-5. [DOI] [PubMed] [Google Scholar]
- 15.Wu HC, Hsieh JT, Gleave ME, et al. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–12. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 16.Thalmann GN, Anezinis PE, Chang SM, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Research. 1994;54:2577–81. [PubMed] [Google Scholar]
- 17.Woods Ignatoski KM, Maehama T, Markwart SM, et al. ERBB-2 overexpression confers PI 3′-kinase-dependent invasion capacity on human mammary epithelial cells. Br J Cancer. 2000;82:666–74. doi: 10.1054/bjoc.1999.0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao GS, Murray S, Ethier SP. Radiosensitization of human breast cancer cells by a novel ErbB family receptor tyrosine kinase inhibitor. Int J Radiat Oncol Biol Phys. 2000;48:1519–28. doi: 10.1016/s0360-3016(00)01358-4. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 19.Irizarry R, Hobbs B, Collin F, et al. Speed exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostat. 2003;4:149–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 20.Rhodes D, Kalyana-Sundaram S, Tomlins S, et al. Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia. 2007;9:443–54. doi: 10.1593/neo.07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomlins S, Mehra R, Rhodes D, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 22.Logothetis CJ, Lin S-H. Osteoblasts in prostate cancer metastasis to bone. Nature Reviews Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 23.Satoh M, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 24.Lazebnik Y, Kaufmann S, Desnoyers S, Poirier G, Earnshaw W. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–7. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 25.Cohen G. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentile M, Latonen L, Laiho M. Cell cycle arrest and apoptosis provoked by UV radiation-induced DNA damage are transcriptionally highly divergent responses. Nucleic Acids Research. 2003;31:4779–90. doi: 10.1093/nar/gkg675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Costa R, Riou L, Paquola A, Menck C, Sarasin A. Transcriptional profiles of unirradiated or UV-irradiated human cells expressing either the cancer-prone XPB/CS allele or the noncancer-prone XPB/TTD allele. Oncogene. 2005;24:1359–74. doi: 10.1038/sj.onc.1208288. [DOI] [PubMed] [Google Scholar]
- 28.Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005;24:3535–40. doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- 29.Hagan S, Al-Mulla F, Mallon E, et al. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clinical Cancer Research. 2005;11:7392–7. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 30.Al-Mulla F, Hagan S, Behbehani AI, et al. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. Journal of Clinical Oncology. 2006;24:5672–9. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 31.Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 32.Fidler I. The pathogenesis of cancer metastasis: the ‘seed and soil; hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 33.Allan A, Vantyghem S, Tuck A, Chambers A. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006–2007;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 34.Brackstone M, Townson J, Chambers A. Tumour dormancy in breast cancer: an update. Breast Cancer Res. 2007;9:208. doi: 10.1186/bcr1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yefenof E, Picker L, Scheuermann R, et al. Cancer dormancy: isolation and characterization of dormant lymphoma cells. Proc Natl Acad Sci U S A. 1993;90:1829–33. doi: 10.1073/pnas.90.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marches R, Scheuermann R, Uhr J. Cancer dormancy: from mice to man. Cell Cycle. 2006;5:1772–8. doi: 10.4161/cc.5.16.2995. [DOI] [PubMed] [Google Scholar]