

Abstract
Down-regulation of the forkhead transcription factor Foxp1 by integrin engagement controls monocyte differentiation in vitro. To determine whether Foxp1 plays a critical role in monocyte differentiation and macrophage functions in vivo, we generated transgenic mice (macFoxp1tg) overexpressing human FOXP1 in monocyte/macrophage lineage cells using the CD68 promoter. Circulating blood monocytes from macFoxp1tg mice have reduced expression of the receptor for macrophage colony-stimulating factor (c-Fms/M-CSFR), impaired migratory capacity, and diminished accumulation as splenic macrophages. Macrophage functions, including cytokine production, phagocytosis, and respiratory burst were globally impaired in macFoxp1tg compared with wild-type cells. Osteoclastogenesis and bone resorption activity were also attenuated in macFoxp1tg mice. In models of chemical and bacterial peritonitis, macFoxp1tg mice exhibited reduced macrophage accumulation, bacterial clearance, and survival. Enforced overexpression of c-Fms/M-CSFR reversed the cytokine production and phagocytosis defects in macFoxp1tg macrophages, indicating that repression of c-fms/M-CSFR is likely the dominant mechanism responsible for Foxp1 action in monocyte differentiation and macrophage function. Taken together, these observations identify down-regulation of Foxp1 as critical for monocyte differentiation and macrophage functions in vivo.
Introduction
Pluripotent hematopoietic stem cells undergo progressive restriction in their lineage potential to give rise to mature terminally differentiated cells. The process of hematopoietic differentiation is thought to follow a developmentally ordered pattern of gene expression. Transcription factors play a key role in the lineage determination and maturation of hematopoietic cells.1 Identification of these regulators and determining the mechanism of how they activate their target genes are important for understanding the development of monocytes and macrophages. In the case of monocyte differentiation, several transcription factors, including PU.1, C/EBPβ, AML1, EGR-1, MAFB, interferon regulatory factor-8/interferon consensus sequence-binding protein (IRF-8/ICSBP), and others, have been implicated based on experimental evidence obtained from knockdown and gain-in-function strategies both in vitro and in vivo.2 The transcriptional regulation of the c-Fms gene, which encodes for the macrophage colony-stimulating factor receptor (M-CSFR), is a focal point of investigation because it is required for the differentiation, proliferation, and survival of monocytic phagocytes.3,4
However, the precise external signals that control differentiation of peripheral blood monocytes to tissue macrophages are incompletely defined. Monocytes leave the bone marrow and travel through peripheral blood vessels. Once they reach a tissue, possibly in response to M-CSF, GM-CSF, monocyte chemoattractant protein-1 (MCP-1), and/or interleukin-3 (IL-3), they differentiate into macrophages by growing in size and increasing their lysosomal compartment, the amount of hydrolytic enzymes and the number and size of mitochondria, and the extent of their energy metabolism.5
We were intrigued by the possibility that cell adhesion molecules participating in the firm arrest and transmigration of blood-borne monocytes across endothelial and extracellular matrix barriers, could provide these signals. Integrins mediate adhesion of cells to extracellular matrices as well as intercellular interactions that are central to inflammation, immunity, hemostasis, and tumor metastasis.6 These adhesive interactions transduce “outside-in” signals that control complex cell functions, such as proliferation, differentiation, and survival, and require the regulation of gene expression.7 Neutrophil and monocyte recruitment in acute inflammation are mediated in part by the β2-integrin family of receptors, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), p150,95 (αXβ2, CD11c/CD18), and CD11d/CD18 (αDβ2). Engagement of β2-integrins by a broad repertoire of ligands generates “outside-in” signals leading to inflammatory cell activation and induction of genes encoding for IL-1β, TNF-α, and tissue factor.8,9 The cytoplasmic tail of LFA-1 interacts with the transcriptional coactivator JAB1 and modulates AP-1 activity by regulating JAB1 nuclear localization.10 Mac-1 associates with IL-1 receptor–associated kinase (IRAK1) and promotes activation of NF-κB activity in a cascade involving TNF receptor–associated factor 6 (TRAF6) and TGF-β–activated kinase 1 (TAK1).11
We previously described a new mechanism by which integrin engagement orchestrates monocyte differentiation signals through the forkhead transcription factor FOXP1.12 Based on prior observations from our laboratory that antibody13 and gene14 targeting of Mac-1 attenuate the biologic inflammatory and neointimal thickening responses to vascular injury, we hypothesized that clustering and activation of Mac-1 may initiate a novel gene program that promotes vascular inflammation. We cloned an 85 kDa forkhead transcription factor (originally termed Mac-1–regulated forkhead, MFH, found subsequently to be identical to FOXP1) using differential display polymerase chain reaction (PCR) that is down-regulated in Mac-1–clustered compared with -nonclustered monocytic THP-1 cells.12 FOXP1 is expressed in untreated HL-60 cells and its expression was markedly reduced during phorbol ester–induced monocyte differentiation. Overexpression of FOXP1 markedly attenuated phorbol ester–induced expression of c-FMS and was accompanied by decreased CD11b expression, cell adhesiveness, and phagocytosis. Using electromobility shift and reporter assays, we established that Foxp1 binds to forkhead binding sites within the c-FMS promoter and functions as a transcriptional repressor. Importantly, deficiency of Mac-1 is associated with altered regulation of FOXP1 and monocyte maturation in vivo. Taken together, these observations suggest that down-regulation of the forkhead transcription factor FOXP1 by integrin engagement is essential for the control of monocyte differentiation.
In this work, we directly tested whether Foxp1 plays a critical role in monocyte differentiation and macrophage functions in vivo by generating transgenic mice expressing human Foxp1 in monocyte/macrophage lineage cells using the CD68 promoter (macFoxp1tg). We found that macrophage functions were globally impaired in macFoxp1tg compared with wild-type cells. Osteoclastogenesis and bone resorption activity were also attenuated in macFoxp1tg mice. In models of chemical and bacterial peritonitis, macFoxp1tg mice exhibited reduced macrophage accumulation, bacterial clearance, and survival. These data delineate important physiologic roles for Foxp1 in monocyte differentiation and macrophage function.
Methods
Construction of human FOXP1 transgene vector
The strategy used for the construction of the transgenic vector is depicted in Figure 1. A 2.1-kb FOXP1 cDNA encoding the 677 amino acids of human FOXP1 with a C-terminal flag tag was obtained by reverse transcription–polymerase chain reaction (RT-PCR) of human peripheral monocyte mRNA with 5′ primer, ctt gcg gcc gct acc atg gga caa gaa tct ggg act gag aca aag and 3′ primer, tta tca ctt gtc gtc atc gtc ctt gta gtc ctc cat gtc ctc gtt tac tgg ttc. The PCR product was first cloned into pGEM-T-Easy (Promega), sequenced, and authentic in-frame expression of FOXP1-flag protein was confirmed by both TnT Quick Coupled Transcription/Translation Systems (Promega, Madison, WI) in the presence of S35 and by immunoblotting using anti-flag monoclonal (Sigma-Aldrich, St Louis, MO) and anti-Foxp1 polyclonal12 antibodies. To construct a transgene vector, the above 2.1-kb human FOXP1-flag fragment was then cloned into a plasmid containing 2.9-kb CD68 promoter with first intron enhancer IVS.15 Authentic FOXP1 expression directed by the CD68 promoter was detected by immunobloting with lysates of cells transfected by either the whole plasmid or the MfeI/DrdI double-excised 5.9-kb expression fragment. This 5.9-kb MfeI/DrdI fragment containing complete CD68 promoter-IVS-FOXP1-flag-bGHpA (bovine growth hormone polyadenylation sequence) cassette was then purified for pronuclear microinjection of C57BL/6J oocytes and the generation of Foxp1 transgenic mice (Transgenic Mouse Facility, Brigham and Women's Hospital, Harvard Medical School, Boston, MA).
Figure 1.

Generation of macFoxp1tg mice and expression of human FOXP1-flag transgene. (A) Construct used for pronuclear injection of C57 oocytes. Restriction enzymes used to release the CD68-FOXP1-flag sequence from the plasmid are indicated. (B) Murine (90 kDa) and human (85 kDa) FOXP1 expression was examined in bone marrow–derived monocytes/macrophages (Mono/Mac), neutrophils (PMN), and lymphocytes isolated from line 41 macFOXP1tg mice. B- and T-cell lymphocyte subpopulations were isolated by flow sorting of splenocytes. Cells were lysed with SDS-PAGE reduced sample buffer, then immunoblotted with an affinity purified anti-FOXP1 polyclonal antibody.12 Mouse Foxp1 and human FOXP1-flag expression during monocyte differentiation. Semiquantitative PCR (C) and corresponding quantitative PCR (D) of mouse Foxp1 and human FOXP1-flag from RNA isolated for peripheral blood monocytes (Mono) and thioglyoclate-elicited macrophages (Mac) isolated at day 3 from wild-type and line 41 and line 57 macFoxp1tg mice. Mean relative expression plus or minus SD; n = 2-3 separate experiments; *P < .04 (P value represents comparison of day 3 macrophage versus monocyte for each group). (E) Murine and human Foxp1 protein expression was examined in bone marrow–derived blood monocytes and thioglycolate-elicited macrophages isolated from macFoxp1tg41 mice at 1 day and 2.5 days. Cells were lysed with SDS-PAGE reduced sample buffer and then immunoblotted sequentially with antiFoxp1 and antitubulin antibodies.
Generation of macFoxp1-transgenic mice
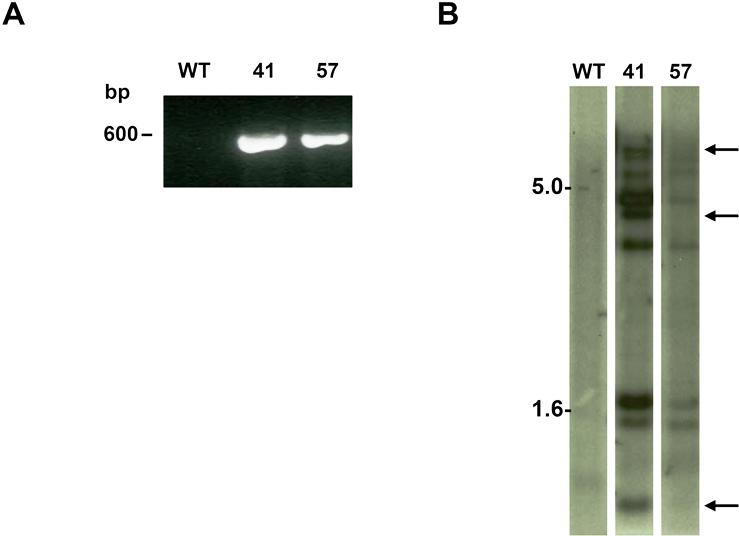
Microinjected embryos were transferred to uteri of pseudopregnant C57BL/6J foster mothers. Founders of FOXP1-transgenic mice (macFoxp1tg) were identified by DNA genotyping of mouse tail biopsies and PCR using the 5′ primer, ctg agt cag gct gtg ggt ggg atc atc t residing in the CD68 promoter, the 3′ primer, gaa gag ctg gtt gtt tgt cat tcc tct tgg ga within sequence of Foxp1. Founder lines (macFoxp1tg41 and macFoxp1tg57) were bred with C57BL/6 mice to establish F1 animals, and F1 progeny within each line were then intrabred. Integration of FOXP1 transgene into genome was characterized by Southern blot analysis using the 572-bp PCR product generated by the above genotyping primers as probe. Wild-type C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
Mice were maintained in animal facilities at Case Western Reserve University School of Medicine. Animal care and procedures were reviewed and approved by the Institutional Animal Care and Use Committee and performed in accordance with the guidelines of the American Association for Accreditation of Laboratory Animal Care and the National Institutes of Health.
Thioglycolate-induced peritonitis and isolation of macrophages
Peritonitis was induced in 12- to 20-week-old mice by intraperitoneal injection of 1 mL sterile 3% (wt/vol) thioglycolate broth.16 At various time points, peritoneal cavities were lavaged by injecting 10 mL sterile PBS buffer twice with gentle abdominal massage. Leukocytes in the lavaged fluid were counted and the proportion of neutrophils, monocytes, macrophages, and other leukocytes determined by flow cytometry. Macrophages were enriched after brief adhesion to tissue culture plastic to remove nonadherent lymphocytes, resulting in more than 95% macrophages.
Isolation of peripheral blood monocytes
Mouse monocytes were isolated from peripheral blood (∼1.5 mL per mouse via direct inferior vena caval puncture) by Ficoll-Hypaque centrifugation17 followed by adhesion to tissue culture plastic to remove nonadherent lymphocytes.
Isolation of bone marrow–derived neutrophils and monocytes/macrophages
Neutrophils and monocyte/macrophages were isolated from the bone marrow by Percoll gradient centrifugation.18
Splenocyte isolation
See Document S1, “Splenocyte isolation” (available on the Blood website; see the Supplemental Materials link at the top of the online article).
RNA purification, reverse-transcription PCR, and quantitative PCR
See Document S1, “RNA purification.”
Immunoblotting
Protein samples were denatured by boiling in sodium dodecyl sulfate (SDS) sample buffer, run on 6% to 12% reducing SDS–polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose. The membrane was then blotted with indicated antibody, and the bands visualized with horseradish peroxidase–conjugated secondary antibody followed by the enhanced chemiluminescence Western blotting detection system (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Immunohistochemistry
Lung, liver, and spleen were harvested, fixed with 10% formalin, and embedded. Standard avidin-biotin immunohistochemistry procedures were used for mouse Mac-3 (mAb M3/84; BD Biosciences) and mouse F4/80 (AbD Serotec). Positive staining area was quantified using computer-assisted imaging analysis (Scion Image) by a histologist blinded to genotype. Photomicrographs were imaged with a Leica DM 2000 microscope (Leica Microsystems, Bannockburn, IL) using AxioCam MRc5 camera with Axio Vision Release 4.5 imaging system software (Carl Zeiss Light Microscopy, Gottingen, Germany).
Flow cytometry
See Document S1, “Flow cytometry.”
Phagocytosis assay
Phagocytic capability of macrophages was measured using the Vybrant Phagocytosis Assay Kit (Molecular Probes, Eugene, OR). Macrophages (105/well) were incubated with fluorescein-labeled Escherichia coli K-12 bioparticles and intracellular uptake was quantified by measuring fluorescence emitted by engulfed particles. Extracellular fluorescence was quenched by trypan blue. For retrovirus infected phagocytosis experiments, we utilized Alexa-conjugated Staphyloccocus aureus bioparticles (Molecular Probes).
Cytokine production in vitro and in vivo
Thioglycolate-elicited peritoneal macrophages were incubated in OPTI MEM I (Invitrogen, Carlsbad, CA) and stimulated with 10 μg/mL E coli LPS overnight at 37°C. IL-1β, TNF-α, IL-12, IFN-γ, MCP-1, and M-CSF in cultured supernatants were then quantified by immunoassay with Quantikine ELISA kits (R&D Systems, Minneapolis, MN). Mice were also challenged with LPS 200 μg intraperitoneally, killed after 6 hours, and inferior vena caval venipuncture performed for the quantification of the plasma cytokine levels.
Migration assay
Cell migration was evaluated using 24-well transwell plates (Costar) with polycarbonate membrane containing 5-μm pores.19 Peripheral blood monocytes (2 × 105) were added to the upper chamber of each well. The lower chamber contained 10 nM MCP-1 (R&D Systems). Transwell plates were then incubated at 37°C for 90 minutes, polycarbonate membranes removed, and the under surface of the membrane rinsed with trypsin/EDTA for the determination of the number of migrated cells in the lower chamber.
Reactive oxygen species assay
Superoxide production from peritoneal macrophages was estimated using reduction of cytochrome c (AbD Serotec, Raleigh, NC) with or without superoxide dismutase (AbD Serotec), as described previously.20
Retroviral transduction of bone marrow–derived monocytes/macrophages
See Document S1, “Retroviral transduction.”
In vivo bacterial challenge
Wild-type S aureus strain Newman (Dr T. Foster, Trinity College, Dublin, Ireland) was cultured on tryptic soy broth (TSB). Bacteria (3 × 107 for phagocytosis or 8 × 107 for survival experiments) were diluted in 0.5 mL PBS and injected intraperitoneally. After 48 hours, peritoneal lavage was performed by injecting 10 mL PBS buffer twice. Collected fluid was serially diluted in TSB and spread on TSB agar plates. Colonies were counted after overnight incubation at 37°C.
Osteoclast differentiation and enzymatic assay
See Document S1, “Osteoclast differentiation.”
Statistical analysis
Data are presented as the mean plus or minus standard deviation (SD). Comparisons between groups used a nonpaired t test. P values less than .05 were considered significant. Comparisons between 2 groups for graft survival used Kaplan-Meier and log rank (Mantel-Cox) test.
Results
Generation of monocyte/macrophage-specific FOXP1-overexpressing transgenic mice
To determine whether Foxp1 plays a critical role in monocyte differentiation and macrophage functions in vivo, we generated transgenic mice (macFoxp1tg) overexpressing human FOXP1 in monocyte/macrophage lineage cells using the CD68 promoter. Overexpression of human rather than mouse Foxp1 was selected intentionally to take advantage of the molecular weight size difference between species, thereby allowing for easy detection of the transgene in addition to the flag tag. Mouse Foxp1 contains an expanded polyglutamine tract (amino acid resides 78-108), resulting in a larger mouse (90 kDa) compared with human (85 kDa) protein. We used an expression cassette that combines 2.9 kb of the CD68 promoter region with 83-bp intron of the CD68 gene (Figure 1A), which has been shown to contain a macrophage-specific enhancer.15 This combination of human CD68 gene sequences directed macrophage-specific gene expression of a type III human SR-A21 and murine IL-10 in transgene mouse lines.22 Constructs for expression of human FOXP1-flag under the control of the CD68 promoter were microinjected into fertilized C57BL/6J oocytes. Potential transgenic founders were screened by PCR using genomic DNA and primers spanning the CD68 promoter and downstream human Foxp1-flag region. Two founder lines, macFOXP1tg41 and macFoxp1tg57, were bred with C57BL/6 mice to establish F1 animals (Figure S1A). Both lines of mice showed germ line transmission to F1 offspring in an approximately Mendelian pattern. Southern blot analysis of restriction-digested genomic DNA confirmed unique integration of the human FOXP1-flag transgene into the host genomes of each of the 2 lines, respectively (Figure S1B). Despite unique integration, we observed equivalent functional results with macFoxp1tg41 and macFoxp1tg57 mice in experimental assays outlined below, and therefore, for purposes of clarity, designate only macFoxp1tg in all experimental results.
Expression of human Foxp1 is constitutive and restricted to monocytes/macrophages in macFoxp1tg mice
We first set out to verify that the CD68 promoter directed the constitutive expression of the human FOXP1 transgene in vivo specifically in monocytes/macrophages. The expression of human FOXP1 in hematopoietic lineage cells from macFOXP1tg mice was examined by Western blotting. Monocytes/macrophages, neutrophils, and lymphocytes were isolated from the bone marrow by Percoll gradient centrifugation. Flow cytometry using lineage- specific markers verified the isolation of monocyte-enriched (percentage of MOMA-2 positive cells = 88), neutrophil-enriched (% mAb 7/4-positive cells = 85), and lymphocyte-enriched (percentage of CD3e-positive and B220-positive cells = 62) populations. Flow sorting of splenocytes was used to isolate B-lymphocyte (anti-B220) and T-lymphocyte (anti-CD3) cell populations. Immunoblotting was performed with an affinity purified anti-FOXP1 polyclonal antibody12 raised against the carboxyl-terminal sequence (CDHDRDYEDEPVNEDME) that is conserved in both human and murine FOXP1. Expression of human FOXP1 was detected only in monocytes/macrophages from macFoxp1tg mice (Figure 1B). Notably, there was no detectable transgene expression in neutrophil- and lymphocyte-enriched or lymphocyte-sorted populations.
We have previously shown that differentiation of human monocytes to macrophages is accompanied by down-regulation of the expression of Foxp1.12 Therefore, we next investigated whether the CD68 promoter is capable of directing the constitutive overexpression of the human FOXP1 transgene during mouse monocyte differentiation. Using unique human FOXP1 and flag tag specific primers that are able to distinguish murine and human Foxp1, semiquantitative (Figure 1C) and quantitative (Figure 1D) PCR showed that murine Foxp1 is rapidly down-regulated during monocyte to macrophage differentiation in wild-type (relative expression 3-day macrophage vs monocyte = 0.17 ± 0.03, P = .037), macFOXP1tg41 (relative expression = 0.18 ± 0.07, P = .017), and macFOXP1tg57 (relative expression = 0.10 ± 0.04, P < .001) mice. In contrast, human FOXP1 is expressed only in transgenic mice with higher transgene levels in macFOXP1tg41 compared with macFoxp1tg57 cells. We also determined copy numbers of mouse and human FOXP1 transcripts to quantify the magnitude of overexpression of Foxp1 in macFoxp1tg mice. In macrophages harvested from macFoxp1tg41 mice, human FOXP1 was expressed 13.1-fold higher than mouse Foxp1 (44 033 vs 3361 copies per ng RNA). In macrophages harvested from macFoxp1tg57 mice, human FOXP1 was expressed 1.2-fold higher than mouse Foxp1 (4374 vs 3773 copies/ng RNA). These results indicate that human FOXP1 is overexpressed more efficiently in line 41 compared with line 57 mice. Overexpression of human FOXP1 was also confirmed at the protein level in monocytes and macrophages isolated from macFoxp1tg41 mice (Figure 1E). Taken together, these results indicate that the CD68 promoter is capable of directing the overexpression of the human FOXP1 transgene in vivo specifically in monocytes/macrophages.
macFoxp1tg mice have normal monocyte numbers in the peripheral blood and reduced macrophage numbers in the spleen
Compared with wild-type mice, macFoxp1tg mice showed no major differences in complete blood count of peripheral blood (Table 1). Total white blood cell number (wild-type: 4.66 ± 1.74 × 103/μL vs macFoxp1tg: 4.26 ± 0.74, P = .66) and monocyte percentage (wild-type: 3.2 ± 1.10 vs macFoxp1tg: 3.0 ± 1.87, P = .84) were similar in wild-type and transgenic mice.
Table 1.
Complete blood count analysis
| Wild-type | macFOXP1tg | P | |
|---|---|---|---|
| WBC, × 103/μL) | 4.66 ± 1.74 | 4.26 ± 0.74 | .66 |
| Neutrophils | 10.6% ± 4.83% | 10.2% ± 4.55% | .90 |
| Lymphocytes | 86.0% ± 5.79% | 86.6% ± 5.41% | .87 |
| Monocytes | 3.2% ± 1.10% | 3.0% ± 1.87% | .84 |
| Eosinophils | 0.2% ± 0.45% | 0.2% ± 0.45% | 1 |
| Basophils | 0 | 0 | |
| Hematocrit | 52.12% ± 3.38% | 49.26% ± 3.03% | .20 |
Although we observed no significant changes in peripheral blood monocyte number, we examined bone marrow monocytes to rule out significant effects of FOXP1 expression on monocyte maturation. We performed flow cytometric analysis of bone marrow in wild-type and macFoxp1tg mice using monoclonal antibodies, including ER-MP12 and Gr-1, that recognize antigens associated with distinct stages of increasing monocyte maturation.23 Monocyte maturation is accompanied by a shift from ER-MP12high to ER-MP12low populations of cells.24 Monocyte maturation was also investigated by staining for the myeloid marker Gr-1.25 Although ER-MP12 and Gr-1 recognize other hematopoietic cells, double staining with MOMA-2, which recognizes both bone marrow monocyte/macrophage precursors and mature macrophages,26 permits the definitive identification of cells of the monocytic lineage. Bone marrow cells were isolated and double-stained with MOMA-2 and either ER-MP12 or Gr-1 and then analyzed by flow cytometry. ER-MP12 (percentage of MOMA-2+ ER-MP12+ cells: 11.9 ± 0.7 vs 11.5 ± 1.7, respectively; P = .74) and Gr-1 (percentage of MOMA-2+ Gr-1+ cells: 6.1 ± 0.4 vs 6.0 ± 1.3, respectively; P = .9) staining were similar in MOMA-2+ wild-type and macFoxp1tg cells. Taken together, these observations suggest that the number of bone marrow or peripheral blood monocytes is not affected in macFOXP1tg mice.
We evaluated tissue macrophage content in wild-type and macFOXP1tg mice by staining lung, liver, and spleen with macrophage-specific antibodies, including F4/80 and Mac-3. Figure 2A and Table 2 show that there is no significant difference in the accumulation of F4/80+ macrophages in lung, liver, and spleen of macFoxp1 transgenic compared with wild-type mice. Mac-3–positive macrophages were reduced in macFoxp1tg mice, but this reduction was of borderline statistical significance (Figure 2B and Table 2: percentage of area wild-type 34.4 ± 8.6 vs macFoxp1tg 23.9 ± 3.0, P = .051). This reduction in macrophage content was further investigated using 4-color flow cytometry that permits the cellular identification of splenocytes as lymphocytes, neutrophils, monocytes, or macrophages/dendritic cells.27 Splenic CD11bhiCD90midB220midCD49bmidNK1.1midLy6Gmid cells were divided into F4/80hi CD11chiI-Abhi macrophages/dendritic cells and F4/80loCD11cloI-Ablo monocytes (Figure 2C). Macrophages/dendritic cells were more abundant in splenocytes harvested from wild-type compared with macFoxp1tg mice (13.8% ± 0.7% vs 8.1% ± 2.5%, P = .005). These observations are consistent with alteration in monocyte to macrophage maturation in the spleen of macFoxp1tg compared with wild-type mice.
Figure 2.

Tissue macrophage content in wild-type and macFoxp1tg mice was assessed using immunostaining and 4-color flow cytometry. Photomicrographs of lung (original magnification 40×/0.65), liver (×40), and spleen (10×/0.25) stained with the macrophage-specific antibodies F4/80 (A) and Mac-3 (B). (C) Four-color flow cytometry was performed to evaluate leukocyte subpopulations within the spleen. Splenic monocytes/macrophages (CD11bhiCD90midB220midCD49bmidNK1.1midLy6Gmid cells) were divided into F4/80hi CD11chiI-Abhi macrophages/dendritic cells and F4/80lo CD11cloI-Ab1o monocytes. Representative flow cytometry and percentage gate quantification of F4/80hi CD11chiI-Abhi macrophage/dendritic cells (mean ± SD; n = 3 spleens per genotype).
Table 2.
Tissue macrophage content
| Macrophage marker | Wild-type | macFOXP1tg | P |
|---|---|---|---|
| F4/80 | |||
| Lung, cell number | 18.7 ± 7.4 | 17.3 ± 3.9 | .73 |
| Liver, % area | 4.2 ± 1.0 | 4.5 ± 1.3 | .72 |
| Spleen, % area | 30.3 ± 7.3 | 26.1 ± 4.9 | .24 |
| Mac-3 | |||
| Lung, cell number | 31.5 ± 8.2 | 26.2 ± 9.7 | .36 |
| Spleen, % area | 34.4 ± 8.6 | 23.9 ± 3.0 | .051 |
Percentage of F4/80-positive or Mac-3–positive area was quantified using computer-assisted imaging analysis. For lung tissue, the number of F4/80-positive or Mac-3–positive cells per field was counted. Data represent means plus or minus SD from 5 individual mice per genotype.
Macrophages from macFoxp1tg mice have impaired cytokine production when stimulated with LPS in vitro
The consequence of overexpression of FOXP1 on macrophage function was first evaluated in vitro using peritoneal macrophages. Activated macrophages secrete multiple cytokines and chemokines that act via autocrine and paracrine pathways to initiate, amplify, and ultimately terminate the inflammatory response. Peritoneal macrophages were isolated and stimulated overnight with LPS. Compared with wild-type macrophages, stimulation of macFOXP1tg macrophages with LPS resulted in significantly reduced concentrations of IL-1β (% inhibition = 47), TNF-α (25%), IL-12 (72%), IFN-γ (72%), M-CSF (68%), and MCP-1 (40%) in cultured supernatants (Table 3).
Table 3.
macFoxp1tg macrophages and mice have diminished response to LPS in vitro and in vivo
| Cytokine, pg/mL | Wild-type | macFoxp1tg | % reduction | P |
|---|---|---|---|---|
| In vitro macrophages | ||||
| Unstimulated IL-1β | 3.1 ± 4.2 | 9.6 ± 6.3 | .13 | |
| LPS-stimulated IL-1β | 374 ± 162 | 198 ± 34 | 47 | .018 |
| Unstimulated TNF-α | 590 ± 144 | 348 ± 256 | .16 | |
| LPS-stimulated TNF-α | 10 287 ± 2907 | 7752 ± 1775 | 25 | .057 |
| Unstimulated IL-12 | ND | ND | ||
| LPS-stimulated IL-12 | 34.5 ± 15.7 | 9.7 ± 12.9 | 72 | .044 |
| Unstimulated IFN-γ | 15.1 ± 4.4 | 13.8 ± 3.9 | .43 | |
| LPS-stimulated IFN-γ | 803 ± 415 | 226 ± 93 | 72 | <.001 |
| Unstimulated M-CSF | ND | ND | ||
| LPS-stimulated M-CSF | 14.1 ± 4.9 | 4.5 ± 1.6 | 68 | <.001 |
| Unstimulated MCP-1 | ND | ND | ||
| LPS-stimulated MCP-1 | 3588 ± 794 | 2163 ± 635 | 40 | <.001 |
| In vivo LPS challenge | ||||
| Baseline IFN-γ | 433 ± 180 | 474 ± 51 | .64 | |
| 6 h post-LPS IFN-γ | 4453 ± 1951 | 2291 ± 1050 | 48 | .034 |
| Baseline IL-1β | 30.1 ± 41.8 | 47.6 ± 42.3 | .58 | |
| 6 h post-LPS IL-1β | 1464 ± 201 | 1047 ± 58 | .52 | |
| Baseline MCP-1 | 120 ± 19 | 69 ± 55 | .25 | |
| 6 h post-LPS MCP-1 | 216 ± 28 | 177 ± 56 | .11 |
Thioglycolate-elicted peritoneal macrophages from wild-type and macFoxp1tg mice were stimulated with LPS (10 μg/mL) overnight and cultured supernatants assayed for indicated cytokines by ELISA. Data represent mean plus or minus SD from 5 individual mice per genotype. For in vivo experiments, serum cytokine levels were determined in wild-type and macFoxp1tg mice challenged with LPS. Groups of 6 mice were injected via intraperitoneal route with 200 μg LPS. After 6 hours, mice were sacrificed and serum analyzed for cytokines by ELISA. Data represent mean plus or minus SD. ND indicates not detected. Percent reduction indicated for significant values only.
macFoxp1tg mice are hyporesponsive to LPS challenge in vivo
We next studied whether macFoxp1tg mice produce fewer cytokines when challenged with LPS in vivo. After intraperitoneal injection of LPS, the serum levels of INF-γ, IL-1β, and MCP-1 were measured after 6 hours (Table 3). In macFoxp1tg mice, the response to LPS was dampened with a significant reduction in the level INF-γ and a trend for lower MCP-1.
Macrophages from macFoxp1tg mice have impaired phagocytic activity and superoxide production
We also tested the effect of overexpression of FOXP1 on phagocytic activity, the oldest known function of macrophages. Peritoneal macrophages were incubated with fluorescein-labeled E coli K-12 bioparticles. As shown in Figure 3A, macFOXP1tg macrophages engulfed fewer particles, indicating that they had significantly reduced phagocytic activity compared with wild-type macrophages.
Figure 3.

Phagocytosis and ROS production are reduced in macFOXP1tg macrophages. (A) Phagocytic capability of macrophages from wild-type and macFoxp1tg mice was measured using Vybrant Phagocytosis Assay Kit, as described in “Phagocytosis assay.” Phagocytosis of fluorescein-labeled E coli K-12 BioParticles was quantified by measuring intracellular fluorescence emitted by engulfed particles. Extracellular fluorescence was quenched by trypan blue. NIH 3T3-L1 fibroblasts were included as nonphagocytic control. Data represent mean plus or minus SD from 4 to 5 individual mice per genotype. (B) Superoxide production was detected using a standard assay involving the reduction of cytochrome c in the presence and absence of superoxide dismutase. Data represent the mean plus or minus SD from macrophages isolated from 5 mice per genotype. P value represents 2-way ANOVA.
Respiratory burst and the production of reactive oxygen species (ROS) are essential for the efficient killing of bacteria by phagocytes. Superoxide was detected using a standard assay involving the reduction of ferricytochrome c20 in the presence and absence of superoxide dismutase. Superoxide production was attenuated significantly over time (P < .001 by ANOVA) in macFoxp1tg compared with wild-type macrophages (Figure 3B).
Response of macFoxp1tg mice to inflammatory stimulus
The ability of macFoxp1tg monocytes to mount an acute inflammatory response was tested first in vivo with thioglycolate-induced peritonitis. Thioglycolate was injected intraperitoneally and, at selected time points, the elicited leukocytes were harvested, stained, and identified by flow cytometry. In the thioglycolate-induced peritonitis model, neutrophils are the most abundant cells up to 24 hours with monocytes predominating at 48 to 72 hours.28,29 We performed 2-color flow cytometry to distinguish neutrophil and monocyte/macrophage populations. Peritoneal neutrophils were defined as CD11bhiLy6GhiCD49hiCD90hiB220hiNK1.1hi cells and monocytes/macrophages as CD11bhiLy6GmidCD49midCD90midB220midNK1.1mid cells (Figure 4A). Twenty-four hours after thioglycolate injection, macFOXP1tg mice were noted to have reduced accumulation of peritoneal monocytes/macrophages compared with wild-type mice, as determined by percentage monocyte/macrophage gate (23.0 ± 4.1 vs 35.5 ± 3.8%, P = .001) (Figure 4B) or absolute monocyte/macrophage cell number (2.3 ± 0.4 × 106, n = 16 vs 3.1 ± 0.3 × 106, n = 17; P < .001). Double staining with MOMA-2 and either Gr-1 (% reduction = 29.7, P = .04), Mac-3 (36.6%, P = .003), F4/80 (38.1%, P = .002), or CD11b (33.1%, P = .016) showed a global reduction in the expression of macrophage markers consistent with a defect in macrophage accumulation (Figure 4C). At 72 hours after thioglycolate injection, when macrophages comprise more than 90% of cells based on MOMA-2 staining, we continued to observe a reduction in total peritoneal lavage fluid cell number in macFOXP1tg compared with wild-type mice (1.7 ± 1.0 × 107, n = 43 vs 2.5 ± 1.2 × 107, n = 43; P = .003; Figure 4D). To explore a possible mechanism for diminished accumulation of peritoneal monocytes/macrophages, we performed monocyte transmigration assays. Peripheral blood monocytes were harvested 6 hours after intraperitoneal injection of thioglycolate and incubated in Transwell inserts in presence or absence of murine MCP-1. Monocytes from macFoxp1tg mice showed defective migration compared with monocytes from wild-type mice (Figure 4E).
Figure 4.

Diminished ability of macFoxp1tg mice to mount an acute inflammatory response after chemical peritonitis. (A) Representative flow cytometry of peritoneal leukocytes. Thioglycolate was injected intraperitoneally, and leukocytes were harvested at 24 hours and stained by 2-color flow cytometry to distinguish neutrophil and monocyte/macrophage populations, as described in Document S1, “Flow cytometry.” CD11bhiLy6GhiCD49hiCD90hiB220hiNK1.1hi cells are designated as neutrophils (oval) and CD11bhiLy6GmidCD49midCD90midB220midNK1.1mid cells as monocytes/macrophages (rectangular box). (B) Neutrophil and monocyte/macrophage gates were quantified as mean plus or minus SD from peritoneal lavage obtained from 5 mice per genotype. (C) Double staining of peritoneal cells with MOMA-2 and either Gr-1, Mac-3, F4/80, or CD11b. Data represent percentage of gate (mean ± SD) from cells isolated from 3 mice per genotype. (D) Peritoneal lavage cell number (mean ± SD) in wild-type and macFoxp1tg mice 72 hours after intraperitoneal thioglycoate injection. (E) In vitro transmigration assay. Peripheral blood monocytes were harvested 6 hours after intraperitoneal injection of thioglycolate and incubated in Transwell inserts in presence or absence of murine MCP-1. Number of cells per well passing to the bottom chamber was counted (mean ± SD, n = 4-5 per genotype).
Defective bacterial clearance and reduced survival in macFoxp1tg mice
Activated macrophages play an essential role in host defense by clearing bacterial pathogens. In vitro experiments indicated that macFoxp1tg macrophages engulfed fewer fluorescein-labeled E coli K-12 bioparticles, indicating impaired phagocytic activity (Figure 3A). To investigate the integrated physiologic functions of monocyte/macrophage migration, phagocytosis, and bacterial killing, we inoculated S aureus bacteria into the peritoneal cavity of wild-type and macFOXP1tg mice. After 48 hours, peritoneal lavaged fluid was collected, diluted in TSB, and spread on TSB agar plates. The number of bacterial colonies was 52-fold higher (P = .023) in macFoxp1tg compared with wild-type mice (Figure 5A), indicating defective phagocytic capability and bacterial killing. Consistent with a defect in the activation of leukocyte antimicrobial functions in macFoxp1tg mice, microscopic analysis of lavage fluid collected 48 hours after S aureus inoculation revealed few appreciable bacteria in wild-type mice, whereas both free and inflammatory cell-associated bacteria were prevalent in macFoxp1tg mice. Many phagocytes appeared to be overwhelmed by bacteria and seemed physically disrupted (Figure 5B).
Figure 5.

Reduced bacterial clearance and survival in macFOXP1tg mice. (A) Bacterial colony counts (mean ± SD) per 100 μL peritoneal lavage fluid 2 days after intraperitoneal inoculation of S aureus bacteria (30 × 106) into the peritoneal cavity of wild-type (n = 11) and macFOXP1tg (n = 12) mice. (B) Gram stain of peritoneal lavaged fluid 2 days after intraperitoneal inoculation of S aureus (original magnification ×40/0.65). (C) Kaplan-Meier survival curves in wild-type (n = 6) and macFOXP1tg (n = 6) mice after intraperitoneal inoculation of S aureus (80 × 106) bacteria.
We examined the effect of impaired phagocytosis on survival in a separate cohort of mice. Figure 5C shows that the survival of macFoxp1tg mice (5 of 6 dead) was reduced significantly compared with wild-type (1 of 6 dead; P = .05 by log rank [Mantel-Cox] test) mice after the intraperitoneal inoculation of S aureus.
Peripheral blood monocytes in macFoxp1tg mice have diminished expression of M-CSF receptor
To gain insight into the possible mechanism accounting for diminished accumulation of splenic macrophages, we examined the expression of c-Fms, the gene encoding for the M-CSFR that is required for the differentiation, proliferation, and survival of monocytic phagocytes.3,4 We have shown previously that FOXP1 functions as a transcriptional repressor of c-FMS.12 Therefore, we reasoned that overexpression of Foxp1 would result in reduced expression of the c-Fms/M-CSFR in peripheral blood monocytes. Quantitative PCR indicated that the level of expression of c-Fms is reduced significantly (relative expression 0.31 ± 0.05, P < .003) in blood monocytes isolated from macFoxp1tg compared with wild-type mice (Figure 6A). Western blot analysis demonstrated reduced expression of both the 160-kDa and 135-kDa forms of M-CSFR in monocyte lysates from macFoxp1tg compared with wild-type mice (Figure 6B). Surface expression of M-CSFR was also investigated by flow cytometry using 2-color staining for monocytes (PE-conjugated MOMA-2) and M-CSFR using FITC enzymatic amplification staining30 (Figure 6C). The fraction of MOMA-2–positive cells expressing M-CSFR was significantly lower in macFOXP1tg compared with wild-type mice (percentage of double-positive cells: 4.9 ± 1.6 vs 10.1 ± 1.9%, n = 5 per genotype; P = .002).
Figure 6.

c-Fms/M-CSFR expression in macFoxp1tg peripheral blood monocytes. (A) The level of expression of c-fms was assessed by quantitative PCR using RNA harvested from wild-type and macFoxp1tg blood monocytes. Expression levels were normalized to β-actin and expressed relative to wild-type. (B) Expression of M-CSFR protein in wild-type and macFoxp1tg monocytes was investigated by Western blot analysis of cellular lysates immunoblotted sequentially with anti–M-CSFR and anti-tubulin antibodies. (C) Surface expression of M-CSFR was also investigated by flow cytometry using 2-color staining for monocytes (PE-conjugated MOMA-2) and M-CSFR using FITC enzymatic amplification staining (EAS), as described in Document S1, “Flow cytometry.” Representative flow cytometry for M-CSFR expression on MOMA-2–positive cells isolated from wild-type (WT) and macFoxp1tg mice. M-CSFR–positive cells were quantified by determining percentage gate of the M2 population defined by staining with an isotype-matched control antibody. (D) Overexpression of c-Fms reverses the cytokine production and phagocytosis defects in macFoxptg cells. LPS-stimulated IL-1β and TNF-α production in wild-type and macFoxp1tg bone marrow–dervied monocytes/macrophages retrovirally infected with GFP, GFP.c-Fms, or GFP.kinase-deficient c-Fms (GFP.c-Fms-mu). Values represent mean plus or minus SD (n = 3-5). Phagocytosis was assessed by culturing GFP-expressing cells in the presence of Alexa-conjugated S aureus bioparticles. Cells were viewed for internalization of the particles by fluorescence microscopy after quenching extracellular fluorescence with trypan blue (digital magnification 40×/0.55).
Enforced overexpression of c-fms rescues the functional impairment of macFoxp1tg macrophages
We next tested whether the repression of c-Fms is the dominant mechanism responsible for Foxp1 action in monocyte differentiation and macrophage function by enforcing overexpression of c-Fms/M-CSFR in macFoxp1tg cells. GFP, GFP.c-Fms, or GFP. K614A mutant c-Fms (GFP.c-Fms-mu) were introduced into wild-type or macFoxp1tg bone marrow–derived monocytes/macrophages by retroviral infection. Cytokine production (IL-1β and TNF-α) and phagocytosis were assayed as readouts of macrophage function. Enforced overexpression of c-Fms/M-CSFR reversed the IL-1β and TNF-α production defect and restored phagocytosis in macFoxp1tg macrophages (Figure 6D), indicating that repression of c-Fms/M-CSFR is likely the dominant mechanism responsible for Foxp1 action.
We also performed transcriptional profiling of wild-type and macFoxp1tg macrophages to identify candidate genes other than M-CSFR that are differentially expressed and that potentially participate in monocyte differentiation and/or macrophage activation. We identified 14 genes that are up-regulated and 20 genes that are down-regulated in macFoxp1tg compared with wild-type macrophages (Table S1). Several particularly interesting candidates known to be involved in cellular differentiation include inhibitor of DNA binding 4 (Idb4),31 insulin-like growth factor binding protein 5 (Igfbp5),32 DNA methyltransferase 3A (Dnmt3a),33 triggering receptor expressed on myeloid cells 2b (Trem2b),34 and B cell–stimulating factor 3 (Bsf3).35
Impairment of osteoclast differentiation and osteolytic activity in macFoxp1tg mice
Osteoclasts arise from the monocyte lineage and defects in monocyte differentiation are often accompanied by derangements in osteoclastogenesis. For example, deficiency of c-Fms and correspondingly M-CSFR is associated with large depletions of osteoclasts.3,4 Therefore, we hypothesized that down-regulation of Foxp1 may play an important role in osteoclast differentiation. Splenocytes were harvested from wild-type and macFoxp1tg mice and incubated with M-CSF and RANKL (receptor activator NF-κB ligand) to induce osteoclastogenesis. Osteoclast formation was monitored by staining for TRAP-positive cells (Figure 7A). We observed 21% fewer (P = .002) total TRAP-positive cells in macFoxp1tg compared with wild-type differentiated splenocytes (Figure 7B). Of note, large multinucleated giant cells were more prominent in wild-type compared with macFOXP1tg mice (Figure 7A). We also examined the bone resorptive capacity of these differentiated osteoclasts. Wild-type splenocytes incubated with M-CSF and RANKL exhibited a significant increase in osteolytic activity over 7 days. In contrast, osteolytic activity in macFoxp1tg cells was markedly attenuated (Figure 7C). To gain further insight into the potential mechanism by which FOXP1 regulates bone resorption, we examined the expression of endogenous murine Foxp1, human FOXP1 transgene under control of the CD68 promoter, and several key mediators of osteoclast formation by RT-PCR (Figure 7D). Similar to monocyte differentiation (Figure 1C,D), osteoclast differentiation induced by M-CSF and RANKL is accompanied by down-regulation of endogenous murine Foxp1. In contrast, expression of human FOXP1-flag transgene in macFoxp1tg splenocytes is constitutive and unaffected by the induction regimen. Reduced expression of Cathepsin K, TRAP, and c-Src was observed in macFoxp1tg compared with wild-type splenocytes treated with M-CSF and RANKL.
Figure 7.

Osteoclastogenesis and bone resorptive capacity is impaired in splenocyte-dervied cells from macFoxp1tg mice. (A) Splenocytes, obtained by homogenization of whole mouse spleen, were incubated in 96-well plates with RANKL (200 ng/mL) and M-CSF (30 ng/mL) for up to 9 days to induce osteoclast differentiation. Osteoclasts were identified by TRAP-positive staining (original magnification 40×/0.65). (B) Total osteoclast number/well was quantified and expressed as mean plus or minus SD from splenocytes isolated from 5 individual spleens per genotype. (C) Osteolytic activity (mean ± SD) of splenocyte-differentiated osteoclasts was investigated using the OsteoLyse Assay Kit (Cambrex, East Rutherford, NJ). (D) Semiquantitative PCR of unstimulated splenocytes and splenocytes stimulated with RANKL and M-CSF for 7 days (2 individual spleens per genotype). (E) Photomicrograph of distal femur (original magnification 20×/0.40) stained for TRAP. (F) TRAP-positive area was quantified by computer-assisted imaging analysis and expressed as mean plus or minus SD from 8 individual femurs per genotype.
Finally, to verify the physiologic relevance of these observations, we performed TRAP staining on proximal femurs from 8-week-old wild-type and macFoxp1tg mice (Figure 7E). TRAP-positive area was reduced significantly by 59% (3734 ± 2882 vs 9143 ± 5951 μm2, P = .02) in macFoxp1tg compared with wild-type mice (Figure 7F).
Discussion
In this study, we have determined that down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. This conclusion is supported by the following data: (1) circulating blood monocytes from macFoxp1tg mice have reduced expression of c-Fms/M-CSFR, impaired migration, and diminished accumulation as splenic macrophages; (2) macrophage functions, including cytokine production, phagocytosis, and respiratory burst were globally impaired in macFoxp1tg compared with wild-type cells; (3) enforced overexpression of c-Fms/M-CSFR reversed the cytokine production and phagocytosis defects in macFoxp1tg macrophages; (4) osteoclastogenesis and bone resorption activity were attenuated in macFoxp1tg mice; and (5) in models of chemical and bacterial peritonitis, macFoxp1tg mice exhibited reduced macrophage accumulation, bacterial clearance, and survival.
Foxp1 is a member of the hepatic nuclear factor-3/forkhead domain family of winged-helix transcription factors.36 More than 100 distinct forkhead transcription factors take part in a wide range of normal development events, including cellular differentiation and proliferation, pattern formation, and signal transduction.37 Foxp1 is expressed ubiquitously in both hematopoietic and nonhematopoietic tissues. Foxp1 appears to be involved in development of the lung38 as well as the brain and central nervous system.39,40 Deficiency of FOXP1 results in embryonic lethality due to severe defects in cardiac morphogenesis.41 Whereas down-regulation of FOXP1 is required for monocyte differentiation, we have shown previously that FOXP1 expression level is unaffected in neutrophil differentiation12 and provided evidence that FOXP1 is down-regulated via signaling pathways involving NF-κB, but with little contribution from PI3-kinase, tyrosine kinases, or MAP kinase. Thus, down-regulation of FOXP1 is likely highly contextual. An additional level of tissue specificity of FOXP1 relates to our recent finding that a functional interaction between FOXP1 and the corepressor SMRT (silencing mediator for retinoid and thyroid receptor) is required for cardiac growth and regulation of macrophage differentiation.42
Altered regulation of c-Fms/M-CSFR provides one possible mechanism by which overexpression of FOXP1 regulates monocyte differentiation. Prior observations from gene-targeted mice indicate that c-Fms/M-CSFR plays an indispensable role in monocyte and osteoclast differentiation.3,4 We observed that M-CSFR expression on peripheral blood monocytes was lower in macFOXP1tg compared with wild-type mice (Figure 6). Furthermore, altered osteoclastogenesis in macFoxp1tg splenocytes stimulated with RANKL and M-CSF was accompanied by reduced expression of c-Fms (Figure 7D). Enforced overexpression of c-Fms/M-CSFR reversed the cytokine production and phagocytosis defects in macFoxp1tg macrophages (Figure 6D), indicating that repression of c-Fms/M-CSFR is likely the dominant mechanism responsible Foxp1 action in monocyte differentiation and macrophage function. Given the versatile and broad repertoire of M-CSFR functions, including cell adhesion, motility and phagocytosis via Rac/Rho signaling, differentiation involving PP2A, Gab3, and Blimp-1, and proliferation and survival via MAP kinase cascades,43 c-fms/M-CSFR is likely one of the most important targets of Foxp1 action regulating immune cell function and bone turnover.
Transcriptional profiling of wild-type and macFoxp1tg macrophages identified candidate genes other than c-Fms/M-CSFR that are differentially expressed and that potentially participate in monocyte differentiation and/or macrophage activation. Several particularly interesting candidates known to be involved in cellular differentiation include inhibitor of DNA binding 4 (Idb4),31 insulin-like growth factor binding protein 5 (Igfbp5),32 DNA methyltransferase 3A (Dnmt3a),33 triggering receptor expressed on myeloid cells 2b (Trem2b),34 and B cell–stimulating factor 3 (Bsf3).35 Interestingly, the promoters of Dnmt3a, Igfbp5, Aldh1a1, Idb4, and betacellulin all contain consensus forkhead-binding sites (ttgttt or ttattt). Whether Foxp1 binds to and regulates the expression of these additional target genes is the focus of ongoing studies.
IFN-γ and TGF-β1 are 2 potent immunomodulatory, pleiotropic factors that have antithetical effects on macrophage function and proinflammatory gene expression.44–46 Emerging studies suggest that the mutual antagonism between these 2 signaling pathways in macrophages is largely controlled by different combinatorial interactions between downstream transcription factors (eg, Stat1, KLF4, and Smad3) and competition for coactivators (eg, p300/CBP).47–49 Future studies will be important to examine whether Foxp1 alters macrophage responsiveness in response to these 2 mutually antagonistic pathways.
The precise external signals that control differentiation of peripheral blood monocytes to tissue macrophages have remained elusive. We were intrigued by the possibility that cell adhesion molecules participating in the firm arrest and transmigration of blood-borne monocytes across endothelial and extracellular matrix barriers, could provide these signals. Our prior report provided in vivo evidence that the leukocyte integrin Mac-1 regulates the expression of FOXP1 and monocyte maturation.12 The present study identifies down-regulation of Foxp1 as critical for monocyte differentiation and macrophage functions in vivo. Mac-1–dependent signals promoting down-regulation of Foxp1 and, in turn, monocyte differentiation, are therefore an obvious target for a new type of anti-inflammatory treatment.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 HL57506, HL085816, and HL073852 (D.I.S.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.S. and M.S. designed and performed research, analyzed data, and wrote the paper; T.M. and K.J.C. performed research and analyzed data; A.L., H.G., and A.S. performed research; D.K. designed research and analyzed data; D.R.G. contributed vital reagent; Y.M. analyzed data and wrote the paper; and D.I.S. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel I. Simon, MD, Director, Case Cardiovascular Center, Case Western Reserve University School of Medicine, 11100 Euclid Avenue, Cleveland, OH 44022; e-mail: daniel.simon@uhhospitals.org.
References
- 1.Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. [PubMed] [Google Scholar]
- 2.Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene. 2002;21:3377–3390. doi: 10.1038/sj.onc.1205324. [DOI] [PubMed] [Google Scholar]
- 3.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Jr, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A. 1990;87:4828–4832. doi: 10.1073/pnas.87.12.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–120. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- 5.Valledor AF, Borras FE, Cullell-Young M, Celada A. Transcription factors that regulate monocyte/macrophage differentiation. J Leukoc Biol. 1998;63:405–417. doi: 10.1002/jlb.63.4.405. [DOI] [PubMed] [Google Scholar]
- 6.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 7.Giancotti FG. Integrin signaling: specificity and control of cell survival and cell cycle progression. Curr Opin Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- 8.Fan ST, Edgington TS. Coupling of the adhesive receptor CD11b/CD18 to functional enhancement of effector macrophage tissue factor response. J Clin Invest. 1991;87:50–57. doi: 10.1172/JCI115000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rezzonico R, Chicheportiche R, Imbert V, Dayer JM. Engagement of CD11b and CD11c beta2 integrin by antibodies or soluble CD23 induces IL-1beta production on primary human monocytes through mitogen-activated protein kinase-dependent pathways. Blood. 2000;95:3868–3877. [PubMed] [Google Scholar]
- 10.Bianchi E, Denti S, Granata A, et al. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 2000;404:617–621. doi: 10.1038/35007098. [DOI] [PubMed] [Google Scholar]
- 11.Shi C, Zhang X, Chen Z, Robinson MK, Simon DI. Leukocyte integrin Mac-1 recruits toll/interleukin-1 receptor superfamily signaling intermediates to modulate NF-kappaB activity. Circ Res. 2001;89:859–865. doi: 10.1161/hh2201.099166. [DOI] [PubMed] [Google Scholar]
- 12.Shi C, Zhang X, Chen Z, et al. Integrin engagement regulates monocyte differentiation through the forkhead transcription factor FOXP1. J Clin Invest. 2004;114:408–418. doi: 10.1172/JCI21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rogers C, Edelman ER, Simon DI. A monoclonal antibody to the β2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simon DI, Chen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in Mac-1(−/−) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greaves DR, Quinn CM, Seldin MF, Gordon S. Functional comparison of the murine macrosialin and human CD68 promoters in macrophage and nonmacrophage cell lines. Genomics. 1998;54:165–168. doi: 10.1006/geno.1998.5546. [DOI] [PubMed] [Google Scholar]
- 16.Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fogelman AM, Haberland ME, Seager J, Hokom M, Edwards PA. Factors regulating the activities of the low density lipoprotein receptor and the scavenger receptor on human monocyte-macrophages. J Lipid Res. 1981;22:1131–1141. [PubMed] [Google Scholar]
- 18.Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 19.Syrovets T, Tippler B, Rieks M, Simmet T. Plasmin is a potent and specific chemoattractant for human peripheral monocytes acting via a cyclic guanosine monophosphate-dependent pathway. Blood. 1997;89:4574–4583. [PubMed] [Google Scholar]
- 20.Crapo JD, McCord JM, Fridovich I. Preparation and assay of superoxide dismutases. Methods Enzymol. 1978;53:382–393. doi: 10.1016/s0076-6879(78)53044-9. [DOI] [PubMed] [Google Scholar]
- 21.Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001;103:351–361. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–3411. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 23.Henkel GW, McKercher SR, Leenen PJ, Maki RA. Commitment to the monocytic lineage occurs in the absence of the transcription factor PU. 1. Blood. 1999;93:2849–2858. [PubMed] [Google Scholar]
- 24.de Bruijn MF, Slieker WA, van der Loo JC, Voerman JS, van Ewijk W, Leenen PJ. Distinct mouse bone marrow macrophage precursors identified by differential expression of ER-MP12 and ER-MP20 antigens. Eur J Immunol. 1994;24:2279–2284. doi: 10.1002/eji.1830241003. [DOI] [PubMed] [Google Scholar]
- 25.Hestdal K, Ruscetti FW, Ihle JN, et al. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J Immunol. 1991;147:22–28. [PubMed] [Google Scholar]
- 26.Kraal G, Rep M, Janse M. Macrophages in T and B cell compartments and other tissue macrophages recognized by monoclonal antibody MOMA-2. An immunohistochemical study. Scand J Immunol. 1987;26:653–661. doi: 10.1111/j.1365-3083.1987.tb02301.x. [DOI] [PubMed] [Google Scholar]
- 27.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 29.Hobbs JA, May R, Tanousis K, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23:2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan D, Smith D. Enzymatic amplification staining for flow cytometric analysis of cell surface molecules. Cytometry. 2000;40:81–85. doi: 10.1002/(sici)1097-0320(20000501)40:1<81::aid-cyto11>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 31.Yokota Y. Id and development. Oncogene. 2001;20:8290–8298. doi: 10.1038/sj.onc.1205090. [DOI] [PubMed] [Google Scholar]
- 32.Lochrie JD, Phillips K, Tonner E, et al. Insulin-like growth factor binding protein (IGFBP)-5 is upregulated during both differentiation and apoptosis in primary cultures of mouse mammary epithelial cells. J Cell Physiol. 2006;207:471–479. doi: 10.1002/jcp.20587. [DOI] [PubMed] [Google Scholar]
- 33.Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10:631–647. [PubMed] [Google Scholar]
- 34.Humphrey MB, Daws MR, Spusta SC, et al. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21:237–245. doi: 10.1359/JBMR.051016. [DOI] [PubMed] [Google Scholar]
- 35.Vlotides G, Zitzmann K, Hengge S, Engelhardt D, Stalla GK, Auernhammer CJ. Expression of novel neurotrophin-1/B-cell stimulating factor-3 (NNT-1/BSF-3) in murine pituitary folliculostellate TtT/GF cells: pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal peptide-induced stimulation of NNT-1/BSF-3 is mediated by protein kinase A, protein kinase C, and extracellular-signal-regulated kinase1/2 pathways. Endocrinology. 2004;145:716–727. doi: 10.1210/en.2003-0813. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Tucker PW. DNA-binding properties and secondary structural model of the hepatocyte nuclear factor 3/fork head domain. Proc Natl Acad Sci U S A. 1993;90:11583–11587. doi: 10.1073/pnas.90.24.11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai E, Clark KL, Burley SK, Darnell JE., Jr Hepatocyte nuclear factor 3/fork head or “winged” proteins: a family of transcription factors of diverse biologic function. Proc Natl Acad Sci U S A. 1993;90:10421–10423. doi: 10.1073/pnas.90.22.10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- 39.Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E. Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. 2004;124:261–267. doi: 10.1016/j.neuroscience.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 40.Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- 41.Wang B, Weidenfeld J, Lu MM, et al. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- 42.Jepsen K, Gleiberman AS, Shi C, Simon DI, Rosenfeld MG. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008;22:740–745. doi: 10.1101/gad.1637108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bourette RP, Rohrschneider LR. Early events in M-CSF receptor signaling. Growth Factors. 2000;17:155–166. doi: 10.3109/08977190009001065. [DOI] [PubMed] [Google Scholar]
- 44.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2005 doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 45.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 46.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 47.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh AK, Yuan W, Mori Y, Chen S, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276:11041–11048. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 49.Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem. 2005;280:38247–38258. doi: 10.1074/jbc.M509378200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}