

Abstract
Phage display of protein and peptide libraries offers a powerful technology for the selection and isolation of ligands and receptors. To date, the technique has been considered limited to soluble, non-membrane proteins. We report two examples of phage display of full-length, folded and functional membrane proteins. Consistent display required the recently reported KO7+ helper phage. The two proteins, full length caveolin-1 and HIV gp41, display well on the surface of the phage, and maintain their binding activities as shown by in vitro assays.
As cellular gatekeepers, membrane proteins regulate the bulk of information and molecules flowing across the plasma membrane. Reflecting the importance of these functions, roughly 30% of eukaryotic genomes encode membrane proteins,1 and the transmembrane G-protein coupled receptors (GPCRs) represent the largest family of drug targets.2 Yet despite the physiological importance of membrane proteins, the paucity of high resolution structural information and high-throughput mutagenesis assays limits insight into this class of proteins. A major rate-limiting step in studying membrane-associated proteins remains purification of sufficient quantities of folded and homogenous proteins.3 Phage display could offer abundant quantities of soluble, purified membrane-associated proteins.
Anecdotal reports of failures resulting from attempts to display membrane proteins on the phage surface buttress the conventional dogma that the technique only works for soluble proteins.4 However, our laboratory reported phage display of full-length HIV-1 Nef for inhibitor discovery and library versus library applications.5, 6 After our publications appeared, the Harris laboratory reported that Nef even without post-translational myristoylation can associate with membranes.7 The experience with Nef suggests an important exception to the requirement for protein solubility in phage display. Thus, we chose two proteins exemplifying this class to further demonstrate this capability–caveolin-1 and HIV-1 gp41.
The three isoforms of caveolin exhibit distinct tissue profiles with human caveolin-1 (featured here, and hereafter referred to as caveolin) found most typically in adipocytes, endothelial cells and smooth muscles.8 At the cell surface, caveolin binds cholesterol, which triggers assembly of homo-oligomers of 12–18 U.9 The caveolin oligomers associate with the Polymerase Transcript and Release Factor (PTRF aka Cavin), various cytoskeletal proteins and lipid rafts, which are detergent-resistant glycosphingolipid-rich membrane microdomains. The resultant complexes generate flask-shaped caveolae,10 and are involved in dynamic cellular processes including signal transduction and endocytosis (Figure 1).11, 12 Residues 102– 134 of caveolin embed in the membrane without crossing the lipid bilayer. The N- and C-termini remain inside the cytosol.9 Also in contact with the membrane, the caveolin scaffolding domain (CSD residues 82-101)13 is responsible for caveolin oligomerization14, 15 and for binding to and inhibiting PKA (Protein Kinase A), eNOS (endothelial Nitric Oxide Synthase) and adenylyl cyclase, cellular proteins involved in signal transduction and endocytosis.16 A number of pathogens including viruses, also use caveolin and other lipid raft-associated proteins to enter the cell.17, 18
Figure 1.
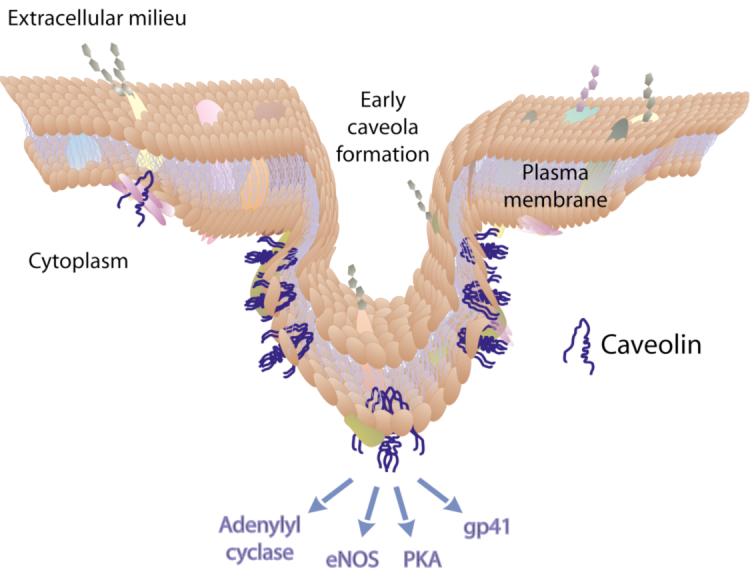
Schematic representation of caveolin membrane topology and function.
The HIV-1 transmembrane protein gp41 is thought to bind caveolin and lipid rafts during viral infection.19 HIV gp120 initiates viral entry into CD4+ T-cells, macrophages and dendritic cells, through cell specific binding receptors. Next, gp41 undergoes a conformational change triggered by gp120-chemokine receptor binding and decreased pH, which leads to the insertion of the glycine rich, hydrophobic ‘fusion’ peptide in the membrane of target cells.20-22 Two hydrophobic heptad repeat domains characteristic of coiled-coil motifs at the N- and C-termini of the gp41 ectodomain play an essential role in membrane fusion.23 In close proximity to the membrane, the CSD binds to the gp41 ectodomain, putatively as part of viral entry.18, 19
As an integral, monotopic membrane protein associated with detergent insoluble lipid rafts, both full-length caveolin and gp41 fail to express in E. coli in the absence of fusion to the phage coat protein (data not shown). Prokaryotes have different lipid bilayer compositions and lack the protein processing machinery of eukaryotes. As a result, bacteria often have difficulty with the over-expression, folding, assembly and stabilization of eukaryotic membrane proteins. Although some success has been reported in bacterial systems,24 the success rate of membrane protein expression in prokaryotes is around 10-30% compared to 45-76% in eukaryotes.25 The results described here demonstrate simply improving protein solubility by tethering caveolin and gp41 to the large solubilizing phage coat can solve the membrane protein expression problem in prokaryotes.
To demonstrate successful phage display of membrane proteins, we assayed display (Figure 2) of the monotopic, integral membrane protein caveolin (Figure 3A) and the transmembrane protein gp41 (Figure 3B) on M13 phage, through fusion to the major coat protein (P8) of M13 phage. To further verify the membrane protein functionality, we demonstrate the binding of caveolin with gp41 ectodomain (Figure 4).
Figure 2.
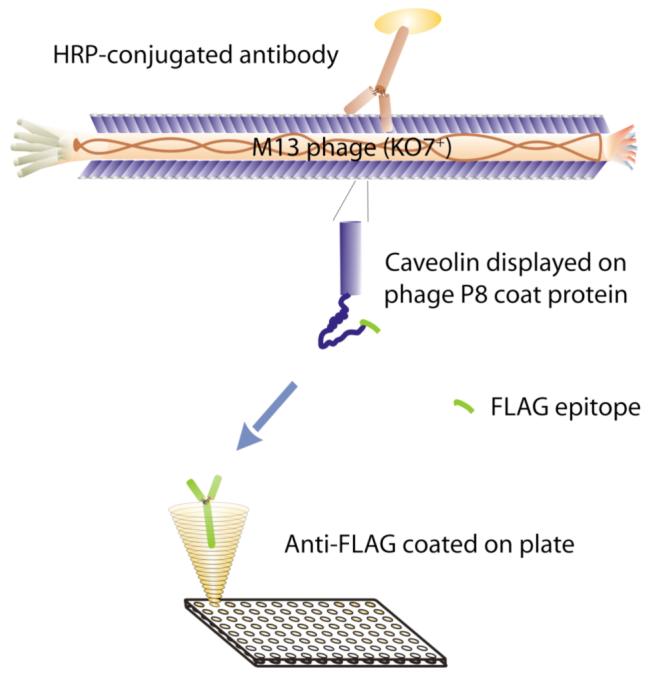
Diagram of the phage ELISA experiment. Anti-FLAG antibody coated on the plate binds to a FLAG epitope on the N-terminus of the phage-displayed protein. An anti-P8, HRP-conjugated antibody binds specifically to the phage.
Figure 3.
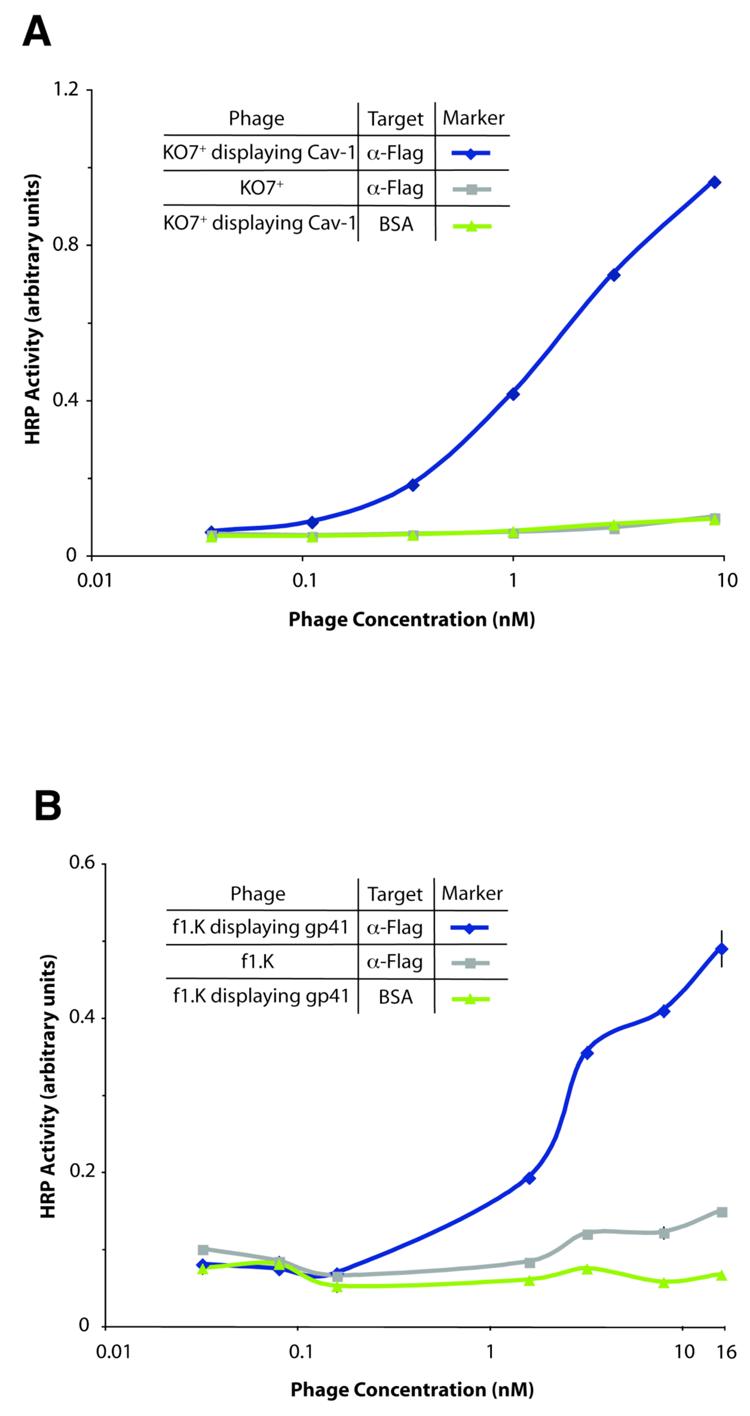
Phage display of two full-length membrane proteins. Using the format shown in Figure 2, display of caveolin or gp41 is assayed by binding to a FLAG epitope fused to the N-terminus of the protein. (A) Full-length human caveolin displayed on phage. Data points for caveolin phage binding to the negative control, BSA, overlay with data for the negative control KO7+ (no protein displayed on KO7+). All error bars indicate standard error (n=3 with the exception of 3B where n=2). (B) Full-length trimeric gp41 displayed on phage, assayed by an analogous method.
Figure 4.
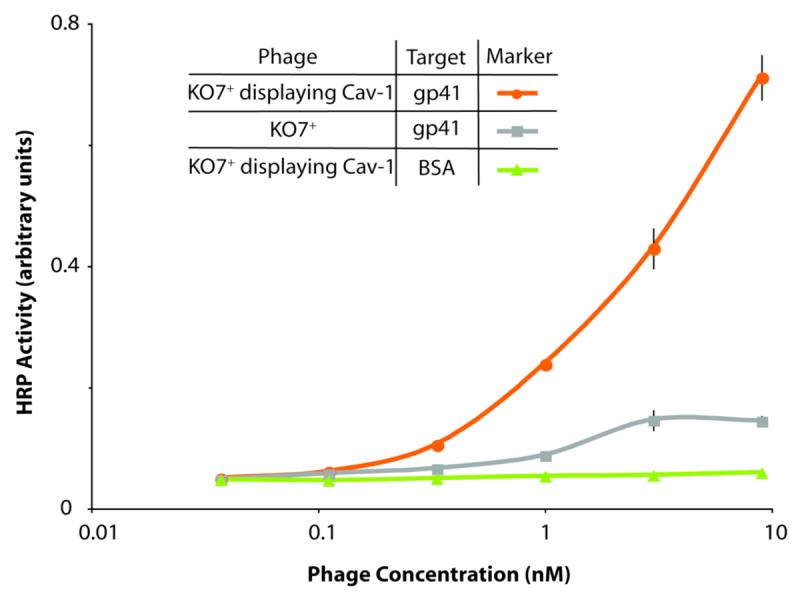
Phage-displayed, full-length caveolin binds to the gp41 ectodomain. In a format similar to the diagram in Figure 2, binding between phage-displayed caveolin and gp41 is assessed by ELISA targeting plate-bound gp41. High display levels of caveolin were demonstrated by anti-FLAG ELISA (data shown in graphical abstract).
First, the open reading frames (ORFs) encoding caveolin and gp41 were subcloned into a conventional phage display vector for display on the surface of the M13 bacteriophage. This plasmid encodes a fusion between the protein of interest and the major coat protein P8, which anchors the displayed protein to the surface of the phage. A linker (amino acid sequence of GGSG) separates the displayed protein from P8, and the FLAG peptide (amino acid sequence DYKDDDDK) fused to the N-terminus provides an antibody epitope for monitoring protein display levels. A conventional signal peptide encoded by the phagemid at N-terminal domain of the P8 fusion protein insures secretion into the periplasm. Although the phagemid also encodes an IPTG inducible promoter, the experiments reported here do not require induction. DNA sequencing verified construction of the phage display vector.
Figure 3A shows successful phage display of full-length caveolin (residues 2-178). In this ELISA, binding to the FLAG epitope on the N-terminus of phage-displayed caveolin demonstrates display of the full-length protein (Figure 2). Display levels were similar to those observed for phage display of soluble proteins. Some batches of anti-FLAG antibody resulted in high background binding between the control phage lacking a displayed protein and the antibody. However, consistently successful assays were obtained when the phage was packaged by a new type of helper phage, based upon f1.K first reported by Jamie Scott and co-workers.26 This variant of the M13 phage, referred to as KO7+, contains a short peptide (amino acid sequence AKAS) near the solvent-exposed N-terminus of the P8 coat protein; the additional lysine residue can disrupt non-specific binding between high pI target proteins (e.g., some anti-FLAG antibodies) and the negatively charged phage coat. Recently, our lab has shown that the use of KO7+ for phage display can remove or greatly reduce background binding in binding assays.27 For consistency, every figure in this paper uses KO7+ as the helper phage. Additionally, more consistent preparations of phage were obtained from phage-displayed caveolin, and gp41 produced using this helper phage. We cannot rule out the possibility that the extra positive charge on the surface of KO7+ virions assists in the display of caveolin and gp41.
Phage display of full-length gp41 was also successful (Figure 3B). Full-length gp41, corresponding to residues 512-868 of the precursor protein gp160, was subcloned into the phagemid as described above. In the gp41-P8 ORF, an amber stop codon between the two genes ensured the production of both free and P8-fused gp41 monomers, when grown in an amber suppressor strain of E. coli (XL-1 Blue). The two gp41 constructs can form trimeric protein in the periplasm before display on the surface of the phage. A similar anti-FLAG ELISA format demonstrated efficient display of full-length gp41 on the phage. The nonlinearity of the data in Figure 3B could result from pipetting error. Previous reports suggest that C-terminal membrane-spanning helices anchor gp41 to the viral membrane. Notably, neither full-length gp41 nor caveolin – detached from the phage coat – express to any degree in E. coli, likely due to insolubility of such transmembrane regions.
After confirming phage display of caveolin, soluble gp41 ectodomain (residues 546-578 and 624-655 attached by glycine-rich linker) produced as described previously served as a target to demonstrate display of functional caveolin.28 As described above, the two proteins have been shown to interact both by immunoprecipitation and SPR binding assays with the 20-residue CSD and gp41 ectodomain.19 Various concentrations of phage displaying full-length caveolin were added to plate-adhered gp41 ectodomain, for a phage-based ELISA analogous to the assays with anti-Flag antibody (Figure 4). The results demonstrate a strong interaction between the two proteins at nanomolar phage concentrations. Thus, amongst its many functions, caveolin could also facilitate HIV entry.
In addition to the binding studies with membrane-associated proteins described here, the approach could allow high-throughput mutagenesis to link specific residues from membrane proteins to their function. For example, shotgun scanning could identify residues required for binding interactions and protein stability.29, 30 Other proteins too insoluble for conventional E. coli expression could also prove amenable to this strategy, which leverages the 16.5 mD size of the bacteriophage as a solubilizing fusion protein. Recently, phage-displayed proteins have been used directly in biosensors and screens for small molecule probes.5, 6, 31 With their wide range of important activities and binding specificities,32 membrane proteins displayed on phage could extend drug screening and virus electrodes into new areas.
Supplementary Material
Acknowledgement
We thank the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH for providing the gp41 cDNA (pHXB2-env) from Dr. Kathleen Page and Dr. Dan Littman. We thank Juan E. Diaz and Jorge A. Lamboy for helpful discussions. This work was supported financially by the the National Institutes of Health, National Institute of General Medical Sciences (R01 GM078528-01 to GAW and NIH-MARC training grant number GM-069337 to ASM).
Footnotes
Supplementary Data
Supplementary data associated with this article can be found in the online version at ….
References
- 1.Liu J, Rost B. Protein Sci. 2001;10:1970. doi: 10.1110/ps.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lagerström MC, Schioth HB. Nat. Rev. Drug Discov. 2008;7:339. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 3.Wiener MC. Methods. 2004;34:364. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 4.Weiss GA, Penner RM. Anal. Chem. 2008;80:3082. doi: 10.1021/ac086009h. [DOI] [PubMed] [Google Scholar]
- 5.Olszewski A, Sato K, Aron ZD, Cohen F, Harris A, McDougall BR, Robinson WE, Jr., Overman LE, Weiss GA. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14079. doi: 10.1073/pnas.0406040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olszewski A, Weiss GA. J. Am. Chem. Soc. 2005;127:12178. doi: 10.1021/ja053316l. [DOI] [PubMed] [Google Scholar]
- 7.Bentham M, Mazaleyrat S, Harris M. J. Gen. Virol. 2006;87:563. doi: 10.1099/vir.0.81200-0. [DOI] [PubMed] [Google Scholar]
- 8.Razani B, Woodman SE, Lisanti MP. Pharmacol. Rev. 2002;54:431. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 9.Monier S, Parton RG, Vogel F, Behlke J, Henske A, Kurzchalia TV. Mol. Biol. Cell. 1995;6:911. doi: 10.1091/mbc.6.7.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, Hancock JF, Parton RG. Cell. 2008;132:113. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. J. Cell Biol. 1993;122:789. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran D, Carpentier JL, Sawano F, Gorden P, Orci L. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7957. doi: 10.1073/pnas.84.22.7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arbuzova A, Wang L, Wang J, Hangyas-Mihalyne G, Murray D, Honig B, McLaughlin S. Biochemistry. 2000;39:10330. doi: 10.1021/bi001039j. [DOI] [PubMed] [Google Scholar]
- 14.Sargiacomo M, Scherer PE, Tang Z, Kubler E, Song KS, Sanders MC, Lisanti MP. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9407. doi: 10.1073/pnas.92.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. J. Biol. Chem. 1997;272:6525. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. J. Biol. Chem. 1998;273:5419. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 17.Manes S, del Real G, Lacalle RA, Lucas P, Gomez-Mouton C, Sanchez-Palomino S, Delgado R, Alcami J, Mira E, Martinez AC. EMBO Rep. 2000;1:190. doi: 10.1093/embo-reports/kvd025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hovanessian AG, Briand JP, Said EA, Svab J, Ferris S, Dali H, Muller S, Desgranges C, Krust B. Immunity. 2004;21:617. doi: 10.1016/j.immuni.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Huang JH, Lu L, Lu H, Chen X, Jiang S, Chen YH. J. Biol. Chem. 2007;282:6143. doi: 10.1074/jbc.M607701200. [DOI] [PubMed] [Google Scholar]
- 20.Allan JS, Strauss J, Buck DW. Science. 1990;247:1084. doi: 10.1126/science.2309120. [DOI] [PubMed] [Google Scholar]
- 21.Chan DC, Fass D, Berger JM, Kim PS. Cell. 1997;89:263. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 22.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Nature. 1997;387:426. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 23.Eckert DM, Kim PS. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11187. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grisshammer R. Curr. Opin. Biotechnol. 2006;17:337. doi: 10.1016/j.copbio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Aricescu AR, Assenberg R, Bill RM, Busso D, Chang VT, Davis SJ, Dubrovsky A, Gustafsson L, Hedfalk K, Heinemann U, Jones IM, Ksiazek D, Lang C, Maskos K, Messerschmidt A, Macieira S, Peleg Y, Perrakis A, Poterszman A, Schneider G, Sixma TK, Sussman JL, Sutton G, Tarboureich N, Zeev-Ben-Mordehai T, Jones EY. Acta Crystallogr., Sect. D. 2006;62:1114. doi: 10.1107/S0907444906029805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Houten NE, Zwick MB, Menendez A, Scott JK. Vaccine. 2006;24:4188. doi: 10.1016/j.vaccine.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamboy JA, Tam PY, Lee LS, Jackson PJ, Avrantinis SK, Lee HJ, Corn RM, Weiss GA. 2008. submitted for publication. [DOI] [PMC free article] [PubMed]
- 28.Shu W, Liu J, Ji H, Radigen L, Jiang S, Lu M. Biochemistry. 2000;39:1634. doi: 10.1021/bi9921687. [DOI] [PubMed] [Google Scholar]
- 29.Morrison KL, Weiss GA. Curr. Opin. Chem. Biol. 2001;5:302. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 30.Kotz JD, Bond CJ, Cochran AG. Eur. J. Biochem. 2004;271:1623. doi: 10.1111/j.1432-1033.2004.04076.x. [DOI] [PubMed] [Google Scholar]
- 31.Yang LM, Tam PY, Murray BJ, McIntire TM, Overstreet CM, Weiss GA, Penner RM. Anal. Chem. 2006;78:3265. doi: 10.1021/ac052287u. [DOI] [PubMed] [Google Scholar]
- 32.Sidhu SS. Curr. Opin. Biotechnol. 2000;11:610. doi: 10.1016/s0958-1669(00)00152-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.