

Abstract
According to the current hypothesis, in fibrinogen, the COOH-terminal portions of two Aα chains are folded into compact αC-domains that interact intramolecularly with each other and with the central region of the molecule; in fibrin, the αC-domains switch to an intermolecular interaction resulting in αC polymers. In agreement, our recent NMR study identified within the bovine fibrinogen Aα374-538 αC-domain fragment an ordered compact structure including a β-hairpin restricted at the base by a 423–453 disulfide linkage. To establish the complete structure of the αC-domain and to further test the hypothesis, we expressed a shorter αC-fragment, Aα406-483, and performed detailed analysis of its structure, stability, and interactions. NMR experiments on the Aα406-483 fragment identified a second loose β-hairpin formed by residues 459–476, yielding a structure consisting of an intrinsically unstable mixed parallel/anti-parallel β-sheet. Size-exclusion chromatography and sedimentation velocity experiments revealed that the Aα406-483 fragment forms soluble oligomers whose fraction increases with increasing concentration. This was confirmed by sedimentation equilibrium analysis, which also revealed that the addition of each monomer to an assembling αC oligomer substantially increases its stabilizing free energy. In agreement, unfolding experiments monitored by CD established that oligomerization of Aα406-483 results in increased thermal stability. Altogether, these experiments establish the complete NMR solution structure of the Aα406-483 αC-domain fragment, provide direct evidence for the intra- and intermolecular interactions between the αC-domains, and confirm that these interactions are thermodynamically driven.
Fibrinogen is a multidomain plasma protein whose major function is to form fibrin clots that prevent the loss of blood upon vascular injury. In addition to its prominent role in haemostasis, fibrinogen also contributes to wound healing and participates in a number of other physiological and pathological processes through the interaction of its multiple regions/domains with various proteins and cell types. The three-dimensional structure of most of these regions has been established by crystallographic studies of proteolytically derived and recombinant fragments of human and bovine fibrinogen (1–4). The crystal structures of a proteolytically truncated bovine fibrinogen and intact chicken fibrinogen have also been solved (5, 6). However, the COOH-terminal regions of two fibrin(ogen) Aα chains including residues Aα221-610 (αC regions), as well as the NH2-terminal regions of the Bβ chains (βN-domains), that together account for about one-fourth of the molecule, were not identified in these structures.
The structure of the αC regions has been a controversial issue for a long time. It was originally proposed, based mainly on the sequence analysis and the results of limited proteolysis, that these regions represent “free swimming appendages” devoid of any ordered structure (7, 8). In contrast, microcalorimetry and electron microscopy data suggested the presence of compact structures in these regions (9–12). This suggestion was directly confirmed by the unfolding study of the recombinant human and bovine fibrinogen αC regions and their truncated variants (13). This study also revealed that the αC region consists of two structurally distinct portions, a compact αC-domain formed by its COOH-terminal half (residues Aα392-610 in human fibrinogen) and a flexible αC-connector formed by the remaining residues Aα221-391, which attaches the αC-domain to the bulk of the molecule. The presence of an ordered structure in the αC-domain has been confirmed by a recent NMR study of the recombinant bovine αC-domain fragment including residues Aα374-538 (corresponds to the human fibrinogen sequence Aα392-568) (14). Furthermore, it was shown that this fragment contains a disulfide-linked β-hairpin and a region of slower concerted motion termed a collapsed hydrophobic region (14). Although resonance ambiguity arising from the disordered portions of the fragment precluded further structural definition of this region, the slower cooperative motion suggested the presence of an ordered conformation. One of the goals of this study was to establish the NMR structure of this region.
The αC-domains contain a number of binding sites that mediate the interaction of fibrin(ogen) with several components of the coagulation and fibrinolytic systems (factor XIII, plasminogen, tPA, α2-antiplasmin) (15–18), and other proteins and cellular receptors [apolipoprotein(a), integrins αIIbβ3, αVβ3, αVβ5, and α5β1] (19–21). These interactions play an important role in the regulation of fibrin-dependent processes such as fibrinolysis, atherogenesis, angiogenesis, and tumorigenesis. The importance of the αC-domains is highlighted by the fact that congenital defects in them cause severe pathological consequences including familial recurrent thrombosis, pulmonary embolism, and renal amyloidosis (22–27). It should be noted that in fibrinogen, which is rather inert in the circulation, the αC-domain binding sites seem to be cryptic, while in fibrin they are highly reactive. Because in fibrin the αC-domains form high molecular mass polymers covalently cross-linked by factor XIIIa (28, 29), their activity appears to be connected with polymerization.
According to the current hypothesis, in fibrinogen the αC-domains interact intramolecularly with each other and with the central region containing fibrinopeptides B, while upon fibrin assembly they dissociate and switch from intra- to intermolecular interaction to form αC polymers in fibrin (29). This hypothesis is based on several observations. First, near the central region of fibrinogen, electron microscopy studies revealed an extra nodule, which was suggested to be formed by two COOH-terminal portions of the Aα chains (11, 12). Second, thermodynamic analysis of the thermal denaturation of fibrinogen and its fragments revealed that this nodule consists of two independently folded entities, denoted as αC-domains, and suggested that these domains may interact with each other (9, 10). Because the αC-domains may also interact in fibrin, in which they form cross-linked αC polymers (28), it was hypothesized that upon fibrin assembly they switch from intra- to intermolecular interactions (30). Third, another electron microscopy study revealed that the αC-domains may interact with the central region of fibrinogen and may be released upon removal of fibrinopeptides B (31, 32). Although this “intra- to intermolecular switch” hypothesis coherently explains the location of the αC-domains in fibrinogen and fibrin and suggests a possible mechanism for the exposure of their multiple binding sites upon conversion of fibrinogen into fibrin, it is not widely accepted. The major reason for the lack of consensus is that this hypothesis is based mainly on low resolution data obtained by electron microscopy. Direct evidence for the interactions between the αC-domains and between the αC-domains and the central region of fibrinogen are absent, as well as the understanding of the nature of driving forces for the “switch”. Another goal of this study was to confirm the interaction between the αC-domains.
Based on the knowledge gained from our previous study with the bovine Aα374-538 αC-domain fragment (14), we constructed, expressed, and characterized a shorter fragment, Aα406-483, which is devoid of most of the previously observed disordered portions. NMR experiments on this fragment revealed in addition to the previously identified disulfide-linked β-hairpin (14) a second loose β-hairpin in the collapsed hydrophobic region yielding an intrinsically unstable parallel/anti-parallel β-sheet. We also observed a concentration-dependent oligomerization of this fragment, in agreement with the “intra- to intermolecular switch” hypothesis, and found that such oligomerization results in a substantial increase of its stability.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Bovine Fibrinogen αC-Fragments
The Recombinant bovine fibrinogen Aα374-538 fragment was expressed in E. coli and subsequently purified and refolded by the procedures described previously (13). Recombinant bovine fibrinogen Aα406-503 and Aα406-483 fragments were expressed in E. coli using the pET-20b expression vector (Novagen Inc.). The cDNAs encoding the Aα406-503 and Aα406-483 fragments were amplified by polymerase chain reaction using a plasmid carrying the full-length bovine αC region sequence (13). The following oligonucleotides were used as primers: 5′-AGAGACATATGATTGATAATGAGAAGGTC-3′ and 5′-AGAGAAAGCTTTTACCCTAAGGCTGCAAATTC-3′ for the Aα406-503 fragment, and 5′-AGAGACATATGATTGATAATGAGAAGGTC-3′ and 5′-AGAGAAAGCTTTTAGGTGAAGAAATCATCCTT-3′ for the Aα406-483 fragment. The forward primers, which were the same for both constructs, incorporated the NdeI restriction site immediately before the coding region; the final three bases of the NdeI site, ATG, code for the fMet residue that initiates translation. The reverse primers included a TAA stop codon immediately after the coding segment, followed by a HindIII site. The amplified cDNA fragments were purified by electrophoresis in agarose gel, digested with NdeI and HindIII restriction enzymes, and ligated into the pET-20b expression vector. The resulting plasmids were used for transformation of DH5α and then B834(DE3) pLysS E. coli host cells. The cDNA fragments were sequenced in both directions to confirm the integrity of the coding sequences.
Both αC-fragments were found in inclusion bodies, from which they were purified by the procedure described earlier (18). The purified fragments were refolded at 4 °C by slow dialysis from urea without any reducing agent using the protocol described in (13). The fractions of unfolded fragments were removed by size-exclusion chromatography on a Superdex S-75 column equilibrated with TBS1 (20 mM Tris buffer, pH 7.4, with 150 mM NaCl) and 0.2 mM PMSF. All chromatography experiments were performed at 4 °C. The refolded fragments were concentrated to 1–2 mg/mL with a Centriprep 10 concentrator (Millipore), filtered through 0.2 μm filter unit and stored at 4 °C.
15N- and 15N/13C-labeled Aα406-483 αC fragments were expressed in E. coli in minimal media supplemented with either 15NH4Cl or 15NH4Cl and 13C6-glucose and subsequently purified and refolded from inclusion bodies as described above. The refolded fragments were concentrated to 3–10 mg/mL and dialyzed against 10 mM KPO4 buffer, pH 6.5, containing 150 mM NaCl and 10% D2O.
Protein Concentration Determination
Concentrations of the recombinant Aα406-483 and Aα406-503 fragments were determined spectrophotometrically using extinction coefficients (E280, 1%) calculated from the amino acid composition with the equation: E280, 1% = (5,690W + 1,280Y + 120S-S)/(0.1 M), where W, Y and S-S represent the number of Trp and Tyr residues and disulfide bonds, respectively, and M represents the molecular mass (33). Molecular masses of these fragments were calculated based on their amino acid composition. The following values of molecular masses and E280, 1% were obtained: 8,960 Da and 6.1 for the Aα406-483 fragment; 11,060 Da and 5.0 for the Aα406-503 fragment. The molecular mass and E280, 1% for the recombinant Aα374-538 fragment (18,123 Da and 3.2, respectively) were determined previously (13). Note that these values take into account the NH2-terminal fMet residue present in all recombinant fragments (see above) while the numbering of the fragments does not.
NMR Data Collection and Structure Elucidation
NMR data were recorded using the 15N- or 15N/13C-labeled Aα406-483 αC fragment in 20 mM KPO4, pH 6.5, with 150 mM NaCl and 10% D2O. The majority of NMR experiments were performed at the fragment concentration of <3 mg/mL, at which most of the peaks were sharp and well resolved. Aligned samples to determine the residual dipolar couplings were prepared as above with ~15 mg/mL Pf1 phage (ASLA biotec). All NMR spectra were recorded at 282 K on a Bruker DRX-600 MHz spectrometer with a triple-resonance cryo-probe and gradients in the Z-axis. Backbone and side-chain resonances and NOE cross peaks were determined and assigned as described before (14) resulting in approximately 90% assignment of N, HN, Cα, Cβ, Hα, and Hβ resonances. All spectra were processed with NMRPipe (34) and analyzed with Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Backbone 15N relaxation rates, T1, T1ρ, and heteronuclear NOE, were recorded and determined as described previously (14). NOE distance restraints, backbone dihedral angle restraints from TALOS (35), and RDC alignment restraints were incorporated into Xplor-NIH (36), as also described previously (14). RDC values were determined by calculating the difference between the observed JN-HN of the isotropic and anisotropic data sets obtained from the 1H-15N HSQC/IPAP experiments. The original alignment tensor was determined from histogram analysis of the RDC values and refined by energy minimization of the structure while scanning the magnitude and rhombicity of the alignment tensor. Hydrogen bond restraints were only included for the already established hairpin (Aα425-445) based on NOE and CSI data. Of the 100 structures initially calculated, the 20 lowest energy representative structures are discussed.
Fluorescence Study
Fluorescence measurements of thermally-induced unfolding of the recombinant αC-fragments were performed in an SLM 8000-C fluorometer by monitoring the ratio of the intensity at 370 nm to that at 330 nm with excitation at 280 nm. The temperature was controlled with a circulating water bath programmed to raise the temperature at 1 °C/min. Fragment concentrations determined spectrophotometrically were 0.04–0.1 mg/mL. All experiments were performed in TBS.
Circular Dichroism Study
Circular dichroism (CD) measurements were made with a Jasco-810 spectropolarimeter. CD spectra of the Aα406-483 fragment in PBS (20 mM potassium phosphate buffer, pH 6.5, 150 mM NaCl) at 1.5, 3.0 and 6.2 mg/mL were recorded using a 0.01-cm path-length quartz cuvette. Thermally-induced unfolding curves were obtained by monitoring the ellipticity at 225 nm while increasing the temperature at a rate of 1 °C/min with a Peltier type FDCD attachment. Unfolding experiments were performed in TBS using a 0.1-cm path-length quartz cuvette. All CD data were expressed as the mean residue ellipticity, [θ], in units of degrees centimeter squared per decimole.
Size-Exclusion Chromatography
Analytical size-exclusion chromatography was used to analyze the aggregation state of the Aα406-483 αC-domain fragment. The experiments were performed with a fast protein liquid chromatography system (FPLC, Pharmacia) on a Superdex 75 column at flow rate of 0.5 mL/min and 4 °C. Typically, 50 μl of the fragment at different concentrations were loaded onto the column equilibrated with TBS and followed by elution with the same buffer. Protein elution was monitored by measuring absorbance at 280 nm. To determine the molecular masses of monomeric and oligomeric forms of the Aα406-483 fragment, the column was calibrated with the gel filtration LMW calibration kit (Amersham Biosciences).
Analytical Ultracentrifugation
Samples for analytical ultracentrifugation were prepared by overnight dialysis of the Aα406-483 fragment at 1.5, 3.0, and 6.2 mg/mL versus TBS. Sedimentation velocity experiments at all three concentrations of Aα406-483 were performed in a Beckman Optima XL-A analytical ultracentrifuge (Beckman Instruments, Palo Alto, CA) equipped with absorbance optics and an An60 Ti rotor, as previously described (37, 38). Due to their increased absorbance, samples of Aα406-483 at 3.0 and 6.2 mg/mL were monitored in double-sector cells with 3 mm centerpieces; standard 12 mm centerpiece cells were used for samples at 1.5 mg/mL.
Sedimentation velocity data were collected at 20 °C at a rotor speed of 45,000 rpm over a 10-hour period. The data were analyzed using both SVEDBERG [J. Philo, version 6.39 (39)] and DCDT+ [J. Philo, version 2.07 (40)] software to obtain the weight-average sedimentation coefficient (Sw) and distribution of sedimenting species, g (s*), respectively (41–43). All sedimentation coefficients have been corrected for solvent density and viscosity to obtain S20, w values.
Sedimentation equilibrium experiments were performed at the same concentrations of the Aα406-483 fragment, again using 3 mm centerpieces for the 3.0 and 6.3 mg/mL samples. Data were collected at 6,000 rpm and 8,000 rpm, as 3 sequential scans at 3 hour intervals following a 24 hour equilibration period at 4°C, and at 2 hour intervals following an 18 hour period at 20 °C. Sedimentation equilibrium data were analyzed with HeteroAnalysis (version 1.1.28, J.W. Cole and J.W. Lary, Analytical Ultracentrifugation Facility, Biotechnology/Bioservices Center, University of Connecticut, Storrs, CT) to obtain weight-average molecular weights (Mw) and to characterize the self-association of the Aα406-483 fragment with an isodesmic model.
RESULTS
Preparation and Characterization of Recombinant Truncated Variants of the αC-Domain
Our previous NMR study revealed that ~30 amino acid residues of the bovine fibrinogen Aα374-538 αC-domain fragment next to the identified β-hairpin may form a compact structure, described as a hydrophobic collapsed region (14). However, resonance ambiguity arising from the disordered portions of Aα454-483 precluded further structural definition of this region. To address this problem, we recombinantly removed these portions and constructed two truncated αC-domain fragment variants, Aα406-503 and Aα406-483, with the intent to preserve the compact structure of the originally studied Aα374-538 fragment (14) while limiting the disordered portions of the molecule. Both shorter fragments lack 32 residues at the NH2-termini and 35 or 55 residues at the COOH-termini (Figure 1A). Of these excluded residues, 33% are Gly or Ser, which are known to have a relatively high propensity for flexibility and lower propensity for structure (44–46). Comparatively, the hydrophobic collapsed region contains only 15% Gly/Ser. Furthermore, all deleted regions of the constructs were shown previously to have slower transverse NMR backbone relaxation rates and narrow 15N HSQC peak dispersion indicative of disorder and non-cooperative motion (14).
Figure 1.

Thermally-induced unfolding of the recombinant bovine fibrinogen αC-domain fragments. Panel A represents schematic diagram of the Aα406-483, Aα406-503, and Aα374-538 fragments. The diagram shows location of the disulfide bond (S-S) linking the β-strands (two arrows) of the previously established β-hairpin (14) and the hydrophobic collapsed region (denoted by the bar with a question mark) with the tryptophan residue (Trp). Fluorescence- and CD-detected unfolding of all three fragments are shown in panels B and C, respectively. The unfolding curves in both panels have been arbitrary shifted along the vertical axis to improve visibility. The dashed straight lines in both panels represent the results of fitting of pre- and post-transition data; they provide the basis for estimating the fraction denatured for the Aα406-483, Aα406-503, and Aα374-538 fragments (curves 1, 2 and 3, respectively) presented in the insets. Note that the fraction denatured versus temperature profiles for Aα406-503 and Aα406-483 essentially coincide in both insets.
To test if the compact cooperative structure in the Aα406-503 and Aα406-483 fragments is preserved, we compared their folding status with that of Aα374-538 by fluorescence spectroscopy and circular dichroism (CD). When heated in the fluorometer while monitoring the ratio of fluorescence intensity at 370 nm to that at 330 nm as a measure of the spectral shift that accompanies unfolding, all three fragments exhibited sigmoidal unfolding transitions (Figure 1B). A similar result was obtained by CD, in which the ellipticity at 225 nM was monitored as a function of temperature (Figure 1C). These experiments confirmed that the Aα406-503 and Aα406-483 fragments contain compact structure. They also revealed that both fragments unfolded in the same temperature range. This becomes more obvious when the fluorescence and CD data are plotted as fraction denatured (insets in Figure 1B and C). This finding suggests that the presence of some extra residues in the longer Aα406-503 fragment do not contribute notably to its stability, i.e. these residues are disordered. Therefore, the shortest fragment, Aα406-483, was selected for further NMR studies. Another finding of these studies is that both the Aα406-503 and Aα406-483 fragments are slightly less stable than the Aα374-538 fragment because their heat-induced unfolding starts at a lower temperature. For that reason, all NMR experiments with the Aα406-483 fragment were performed at a low temperature, 282 K (9 °C), to minimize the fraction of unfolded molecules.
Relaxation and Dynamics of the Aα406-483 Fragment
15N NMR backbone T1ρ relaxation experiments conducted on the 15N-labeled Aα406-483 fragment revealed that slower concerted motion among its residues is comparable to that for the corresponding residues of the Aα374-538 fragment (Figure 2A). This implies that the ordered part of the Aα406-483 fragment covers similar residues as that of the Aα374-538 fragment and that its dynamics behavior is not influenced by the missing truncated residues. Further analysis of the relaxation data for the Aα406-483 fragment revealed several distinct regions of motion. The extreme NH2- and COOH-terminal residues, Aα405-408 and Aα481-483, exhibit the expected hallmark characteristics of flexibility with 1H-15N heteronuclear NOE values rapidly falling to 0 (Figure 2C) and T1ρ and T1 profiles increasing in disorder toward the ends of the fragment (Figures 2A and 2B, respectively). The rest of the fragment can be divided into three regions of slower concerted motions, Aα409-416, Aα420-453, and Aα454-477, based on similarity of the individual relaxation parameters whose averaging is represented by horizontal bars. One of them, Aα420-453, corresponds to the previously identified β-hairpin and bulge, while the other one, Aα454-477, includes most of the collapsed hydrophobic region. It should be noted that the corresponding regions of the Aα374-538 fragment (14) may have similar discrete motional characteristics, however, the higher acquisition temperature and fewer overall data points due to ambiguity and resonance overlap obfuscate the trends (Figure 2A).
Figure 2.

15N backbone relaxation data showing several areas of cooperative motion in the Aα406-483 fragment (filled circles). T1ρ (panel A), T1 (panel B) and NOE (panel C) relaxation data are shown. The T1ρ data for the Aα374-538 fragment (open circles in panel A) are shown for comparison. Horizontal lines show the average values for the region corresponding to the previously identified disulfide-linked β-hairpin (blue), and the NH2-terminal (red) and hydrophobic collapsed (green) regions shaded in gray. Vertical bars represent experimental errors.
NMR Solution Structure of the Aα406-483 Fragment
The NMR solution structure of the 15N-13C labeled Aα406-483 fragment was determined using the NMR structural restraints and statistics as shown in Table 1. The atomic coordinates and restraints for the 20 lowest energy conformations were deposited in the Protein Data Bank under accession code 2JOR, and the chemical shift assignments were deposited in the BioMagResBank (BMRB) under the accession number 15192. In addition to the already determined disulfide-linked β-hairpin and bulge formed by residues Aα425-445 and Aα446-452, respectively (14), we identified a second hairpin-like structure formed by residues Aα457-476 of the collapsed hydrophobic region (Figure 3). Due to instability and conformational heterogeneity in this region, consistent β-strand CSI values are lacking and only twelve unambiguous inter-strand NOE restraints are observed. Therefore, we were unable to unequivocally confirm hydrogen bonding patterns and define the register of the hairpin. However, since the potential for β-hairpin structure is evident, we defined this structure as a loose β-hairpin. Weak 15N-NOESY HN-HN cross-peaks observed between F458 and C423, and H461 and V426, and S466 and T432 suggest interaction between these residues that provides formation of a mixed parallel/anti-parallel 2↑1↓3↓4↑ β-sheet structure. What was classified previously as a bulge (14) is clearly seen in the structure as an extended loop formed by residues Aα448-457. This loop allows for parallel contact between strands 1 and 3 of the β-sheet.
Table 1.
Solution NMR restraints and structural statistics for the 20 lowest energy conformations of the 420-478 region of the Aα406-483 fragment
| Restraints | Number | Average rmsd |
|---|---|---|
| NOE distance restraints | 297 | 0.054 ± 0.004 Å |
| Long-range NOEs | 24 | |
| Ambiguous NOEs | 28 | |
| Violations > 0.5 Å | 0 | |
| Backbone dihedral angle restraints | 50 | 0.407 ± 0.026 ° |
| Residual dipolar coupling restraints | 71 | |
| Hairpin | 32 | 2.11 ± 0.197 Hz |
| Othera | 39 | 0.773 ± 0.139 Hz |
| Hydrogen bonds (hairpin) | 11 | 0.021 ± 0.003Å |
| Structural Statisticsb | Atomic Pairwise rmsd (Å) | |
| Backbone (420–478) | 2.40 | |
| Heavy (420–478) | 3.32 | |
| Backbone (hairpin; 423–447) | 0.78 | |
| Backbone (loose-hairpin; 455–478) | 2.60 | |
| Ramachandran Statistics | Percent of Total | |
| Residues in favored regions | 83.7% | |
| Generously allowed | 10.4% | |
| Disallowed | 5.9% |
Using a half-open potential (58).
Backbone rmsd was calculated based on backbone amide N, CA, C′, and O atoms.
Heavy atom rmsd was calculated based on all C, N, S, and O atoms.
Figure 3.
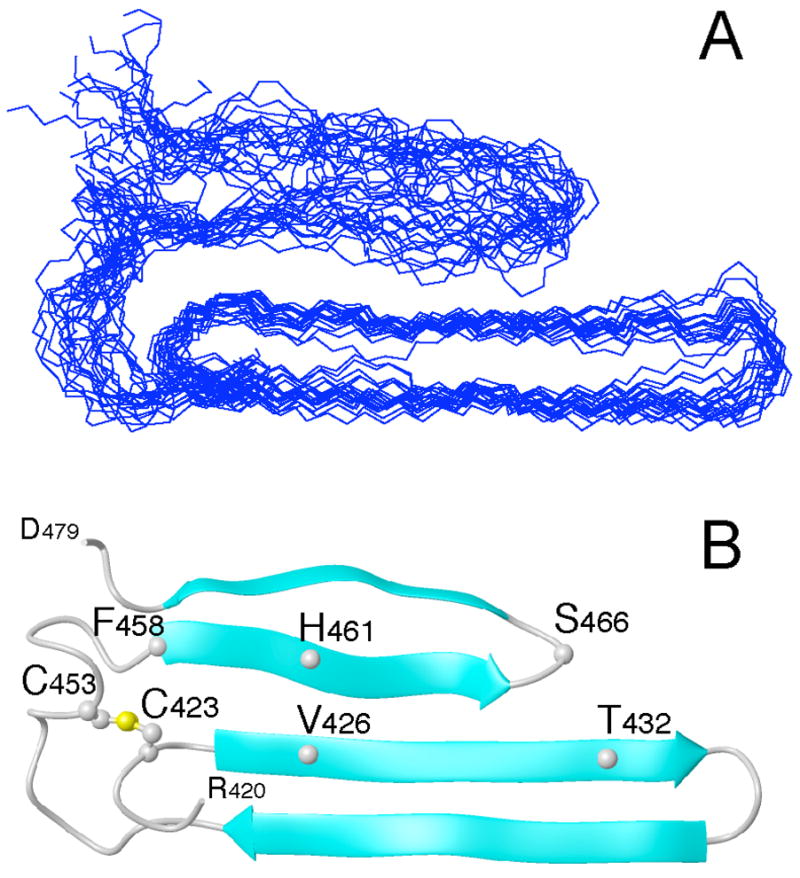
Overlay of the 20 lowest energy conformations of the bovine fibrinogen Aα406-483 αC-domain fragment (panel A). Molecules were superimposed over residues 420–478 using the backbone amide atoms C, CA, N, and O. Panel B displays the ribbon diagram of the average minimized conformation. For clarity, only the ordered residues, 420–478, are shown. The locations of some residues mentioned in the text, including C423 and C453 forming the disulfide bond, are indicated.
The lack of extensive NOE and CSI (chemical shift index) data and two 15N-HSQC peaks for residues S451, G454, W460, H461, and R476 in the region of the loose β-hairpin suggests the presence of alternative conformations. In order to ensure a representative ensemble of the structures presented in Figure 3, potential hydrogen bonds between residues W460 and D472, and H462 and L470 of the loose β-hairpin, which would restrict the structure and define the register, were not included in the structure calculation and refinement process. Despite the lack of an abundance of restraints, the heavy atom backbone rmsd for the 20 lowest energy conformations of the loose β-hairpin is 2.60 Å. It is readily apparent that the majority of uncertainty in the structure arises from the variability in conformation and positioning of this hairpin since the rmsd of the entire hydrophobic collapsed region is 2.40 Å and that of the disulfide-linked hairpin, including the extended loop, is 0.78 Å.
Detection of Oligomers in Preparations of the Aa406-483 Fragment
To increase spectral quality, the initial NMR data of the 15N-labeled Aα406-483 fragment were obtained at 10 mg/mL. However, the 15N-HSQC spectrum revealed that most of the peaks were weak and broad due to a decrease in relaxation time, which is most probably connected with the formation of larger complexes (soluble oligomers). The presence of soluble oligomers at high concentrations of the Aα406-483 fragment was confirmed by size exclusion chromatography. When this fragment at 11.1 mg/mL was applied onto a column, it eluted in two well separated peaks (Figure 4A). The first peak, corresponding to oligomers, contained about 20% of the total applied material. When Aα406-483 was applied at lower concentrations, 5.8 and 3.0 mg/mL, the fraction of oligomers was lower, about 13 and 8%, respectively; this fraction was significantly reduced when the fragment was applied at 1.5 mg/mL (Fig. 4B-D). These results indicate that oligomerization of the Aα406-483 fragment is a concentration-dependent process. It should be noted that when the fragment at 5.8 mg/mL, containing 13% oligomers, was diluted to 3.0 mg/mL and analyzed by size-exclusion chromatography, the amount of oligomers decreased to 9% suggesting that the oligomerization process is reversible.
Figure 4.
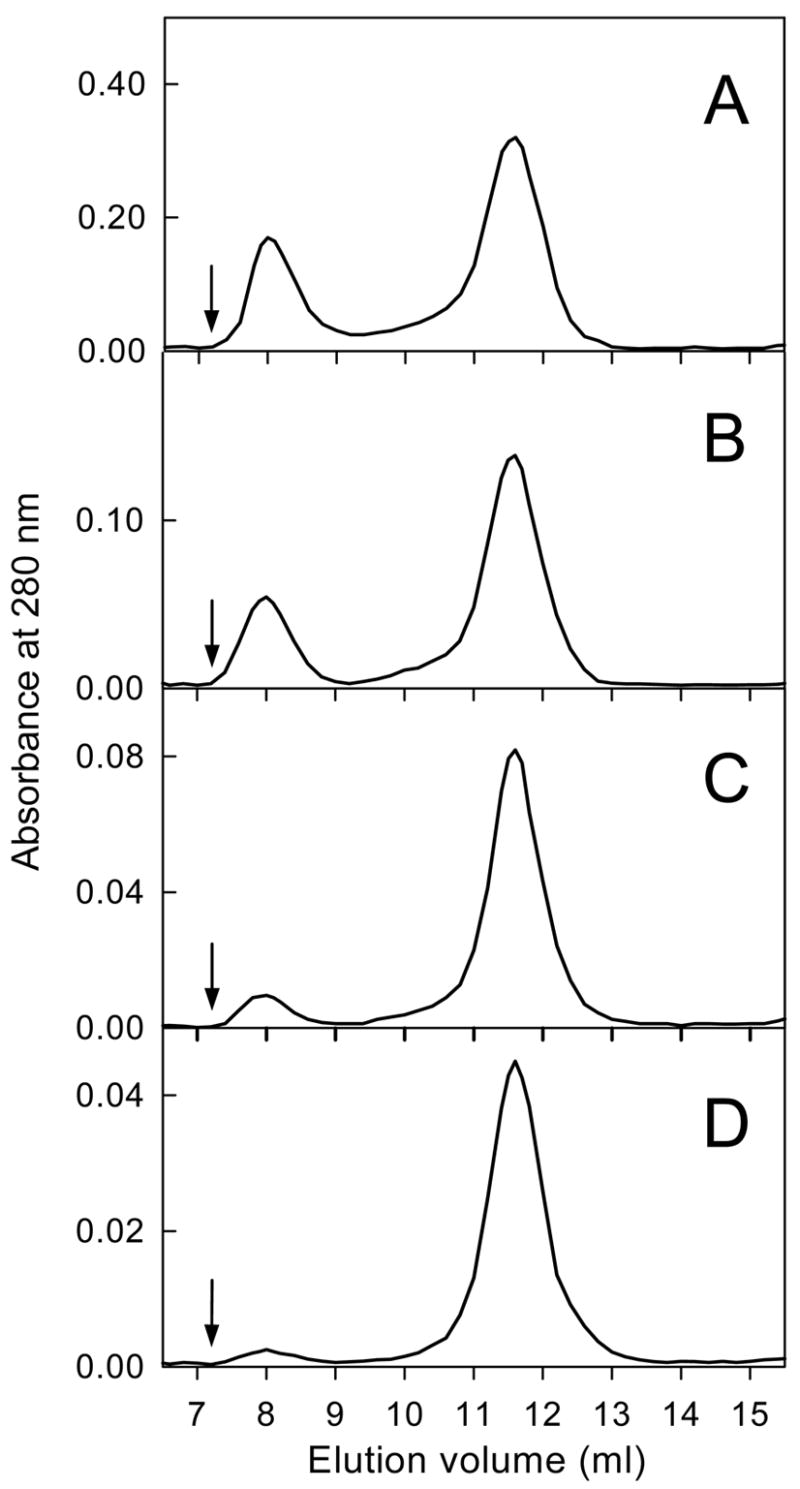
Size-exclusion chromatography profiles of the Aα406-483 fragment applied at different concentrations, 11.1 (A), 5.8 (B), 3.0 (C) and 1.5 mg/mL (D). Arrows indicate the free volume of the Superdex 75 column used in the experiments. The experiments were performed in TBS at 4 °C.
The apparent molecular masses of the material eluted in the first and second peaks were respectively about 10- and 2-fold higher than that expected for the monomeric fragment. This suggests that the oligomers may consist of ten monomers and the material eluted in the second peak may be represented by dimers. At the same time, taking into account the previously observed abnormal chromatographic mobility for the larger Aα374-568 fragment (47), one cannot exclude that the mobility of Aα406-483 is also abnormal and therefore this suggestion may not be correct. The fractions of oligomers determined at different concentrations may also be less accurate because the samples were diluted upon chromatography. Therefore, analytical ultracentrifugation was employed to further characterize the oligomerization of the Aα406-483 fragment.
Sedimentation Velocity Study of the Aα406-483 Fragment
To determine the oligomeric composition of the Aα406-483 fragment at different concentrations, we performed sedimentation velocity experiments (Figure 5A). Analysis of sequential scans obtained at 1.5 mg/mL with the DCDT+ algorithms (40) yielded a single species characterized by 1.04 ± 0.05 S and a molecular mass of 8.56 ± 0.90 kDa, which was very close to that of monomeric Aα406-483. At higher concentrations, the Aα406-483 fragment exhibited a second species with higher mobility, which most probably represented oligomers. Sedimentation coefficients of 3.6 ± 0.3 S and 3.8 ± 0.3 S were then obtained for Aα406-483 oligomers at 3.0 and 6.2 mg/mL, respectively. As illustrated in Figure 5C, inset, applying time-derivative analysis to sequential scans also enabled the measurement of the quantities of oligomeric species: at 3.0 mg/mL, 16.5 ± 5.5%, and at 6.2 mg/mL, 27.0 ± 3.7%.
Figure 5.

Distribution of Aα406-408 monomers and oligomers observed by analytical ultracentrifugation at 20 °C. Sedimentation velocity data obtained with Aα406-408 at 1.5 mg/mL revealed a single species sedimenting at 1.0 S (panel A, diamonds; the solid line depicts the weight fraction, g(S), vs. S profile obtained by time-derivative analysis with DCDT+). At higher concentrations, 3.0 mg/mL (panel B, circles) and 6.0 mg/mL (panel C, triangles), both a 1 S and a 4 S component were present; the contributions of the faster and slower species are shown by short-dashed and long-dashed lines, respectively, the solid lines depict the composite fit obtained with both terms. Concentration-dependent increases in the quantity of oligomeric species are presented in panel C, inset, with data obtained by DCDT+ shown as solid triangles; complementary data obtained by SVEDBERG are shown as open circles.
Analysis of the complete sedimentation velocity profile at 1.5 mg/mL with SVEDBERG (39) yielded one species with 1.4 S and a molecular mass of 8.1 kDa. Analysis of the data obtained at 3.0 and 6.2 mg/mL revealed 2 species with 1.2 and 3.7 S, and 1.3 and 3.6 S, respectively. The quantity of the ~ 4 S material increased from 13% at 3.0 mg/ml to 23% at 6.2 mg/mL (Figure 5, inset). These values are in agreement with those obtained by the time-derivative analysis. Altogether, these results indicate that the amount of oligomers formed by the Aα406-483 fragment increases with concentration.
Effect of Oligomerization of the Aα406-483 Fragment on its Thermal Stability
Since oligomerization of proteins is often accompanied by their stabilization, we tested the stability of the Aα406-483 fragment samples prepared for analytical ultracentrifugation by CD. When the samples at 1.5, 3.0 and 6.2 mg/mL were heated in the spectropolarimeter while monitoring the ellipticity at 225 nM, they all exhibited sigmoidal unfolding transitions (Figure 6). At 1.5 mg/mL, the all monomeric Aα406-483 fragment exhibited a transition with a midpoint (Tm) of 24.9 °C. At the concentration of 3 mg/mL, at which some oligomers were detected (Figure 4 and 5), the transition was slightly more stable, Tm = 26.8 °C. Increasing the fragment concentration to 6.2 mg/mL, at which oligomers accounted for 23–27% of all material (Figure 6C, inset A), resulted in further stabilization of its transition, Tm = 34.9 °C. These experiments indicate that oligomerization of the Aα406-483 fragment significantly increases its thermal stability. The CD spectra of this fragment obtained at all three concentrations are presented in Figure 6, inset B. The presence in all spectra of a negative band at 213–217 nm is in a good agreement with the identified β-sheet structure in Aα406-483. Moreover, the higher amplitudes of this band at 3.0 and 6.2 mg/mL, and its shift to 217 nm suggest that oligomerization of the Aα406-483 fragment results in increased amount of this structure.
Figure 6.
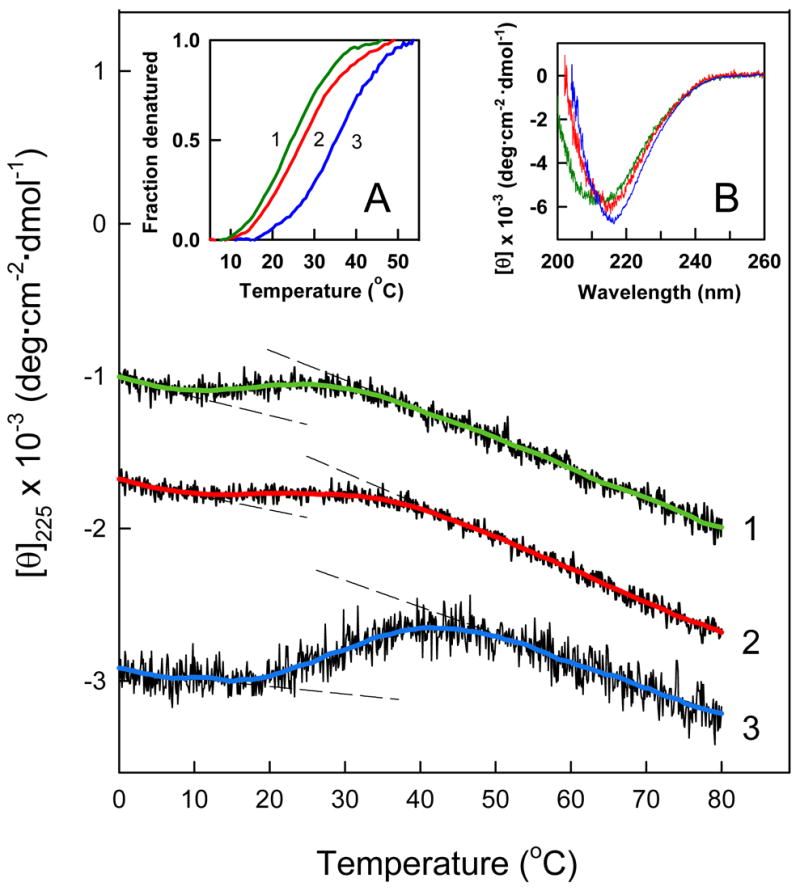
CD-detected thermal unfolding of the Aα406-483 fragment. The unfolding experiments were performed at 1.5, 3.0 and 6.2 mg/mL Aα406-483 (curves 1, 2 and 3, respectively) in TBS. The unfolding curves have been arbitrary shifted along the vertical axis to improve visibility and smoothed (colored curved lines) to reduce the noise. The dashed straight lines represent the results of fitting of pre- and post-transition data; they provide the basis for estimating the fraction denatured at 1.5 (green curve 1), 3.0 (red curve 2) and 6.2 mg/mL (blue curve 3) presented in the inset A. The CD spectra presented in inset B were obtained at the same three concentrations of Aα406-483, 1.5 (green), 3.0 (red) and 6.2 mg/mL (blue) in PBS at 4 °C.
The unfolding experiments presented in Figure 6 also revealed that at 20 °C, at which the sedimentation velocity studies had been performed, the Aα406-483 fragment at 1.5 and 3 mg/mL contained a fraction of unfolded molecules (~25%), which could potentially influence its oligomerization. Therefore, we also performed analytical ultracentrifugation at 4 °C, at which the Aα406-483 fragment did not contain any unfolded material. Because sedimentation velocity experiments at this temperature are challenging, we analyzed the Aα406-483 fragment by sedimentation equilibrium.
Sedimentation Equilibrium Study of the Aα406-483 Fragment
Sedimentation equilibrium experiments at 4 °C confirmed that the Aα406-483 fragment exhibits concentration-dependent oligomerization, as illustrated in Figure 7. This figure depicts the concentration gradients obtained during a sedimentation equilibrium experiment that examined 3 concentrations of Aα406-483, each at 2 rotor speeds. Molecular weight determinations with HeteroAnalysis using data obtained at 1.5 mg/mL (Figure 7A), yielded 5940 ± 580. This value is lower than the monomer’s calculated molecular weight of 8960 due to the shallow concentration gradients which hampered accurate analyses. At the same time, HeteroAnalysis of data obtained at 3.0 mg/mL (Figure 7B) and 6.2 mg/mL (Figure 7C) yielded Mw ~ 52290 ± 990 (Figure 7B and C). This result indicates that in this concentration range the Aα406-483 fragment forms oligomers consisting, on average, of 5–6 monomers.
Figure 7.

Analysis of Aα406-408 oligomerization by analytical ultracentrifugation performed at 4 °C, and at 6000 rpm (open symbols) and 8000 rpm (solid symbols). Sedimentation equilibrium data demonstrate that Aα406-408 remains monomeric at 1.5 mg/mL (panel A). HeteroAnalysis of data obtained at 4 °C yielded the solid lines corresponding to Mw ~ 5940 using data at 6000 rpm (open diamonds) and at 8000 rpm (solid diamonds, offset by 0.025 absorbance units for clarity). The data obtained at higher concentrations, 3.0 mg/mL (panel B) and 6.2 mg/mL (panel C) demonstrate oligomerization. HeteroAnalysis of these data yielded Mw ~ 52290; the solid lines describe an isodesmic self-association model with Ka = 5.28 × 103 M−1. This model accounts for both rotor speed and concentration-dependence as evidenced by the correspondence between data and fitted lines in panels B and C, as well as the narrow distribution of residuals presented in the inset to panel C. The data in panel A were obtained in a 12 mm path length cell, hence their absorbance values have been divided by 4 to facilitate direct comparison to the data obtained at higher concentrations in 3 mm path length cells (panels B and C).
The sedimentation equilibrium data obtained at 3.0 and 6.2 mg/ml were also utilized to determine the association constant (Ka) for self-association of the 8,96 kDa Aα406-483 fragment using an isodesmic association model: Mn−1 + M = Mn, where Ka is the same for all species with n>2 (48, 49). As illustrated by the correspondence between data and fitted lines in Figure 7, the resultant Ka = 5.28 ± 0.03 × 103 M−1 reliably describes both the concentration and rotor speed-dependence of these data. The quality of the fit is further evidenced by the tight, nearly random distribution of the residuals, as shown in Figure 7C, inset. The resultant Ka was utilized to calculate the free energy of association (ΔG) using the equation ΔG = −RT lnKa. The ΔG was found to be −4.98 ± 0.04 kcal/mol. This finding indicates that addition of each monomer to an assembling Aα406-483 oligomer may add as much as 5 kcal/mol of the stabilizing free energy. It should be noted that we also performed a similar experiment at 20 °C; however, only the concentrated Aα406-483 fragment (6.2 mg/ml), which at this temperature contained practically no unfolded material (Figure 6), was tested. Analysis of the sedimentation equilibrium data resulted in Ka = 14.4 ± 0.43 × 103 M−1 and ΔG = −5.56 ± 0.06 kcal/mol, further reinforcing the above conclusion. It should be noted that the increased Ka at this temperature may suggest that oligomerization is entropy-driven (50).
DISCUSSION
To establish the structure of the fibrinogen αC-domain, we prepared recombinant human and bovine αC-fragments (13); however, our crystallization attempts, as well as those undertaken by others (51, 52), have been unsuccessful. The reasoning became clear after our NMR study (14) confirmed that one of these fragments, bovine Aα374-538, contains a high proportion of unordered conformation, which most likely prevented crystallization and influenced the quality of NMR data. In the present study, we overcame this problem by truncating the unordered regions of the Aα374-538 fragment and established the complete NMR structure of the resultant Aα406-483 fragment.
This study also revealed that the Aα406-483 fragment, which is preferentially monomeric at the concentration used for the NMR study, forms soluble oligomers at higher concentrations. Although this fragment includes only part of the αC-domain, our previous experiments indicate that a larger Aα374-568 fragment corresponding to the complete domain seems to also have a propensity to form oligomers. Indeed, Aα374-568 contained some oligomers at 2.4 mg/mL, as revealed by analytical ultracentrifugation (47), and precipitated upon its concentration above 5 mg/mL (results not shown), most probably due to formation of larger aggregates. The observed oligomerization of the αC-domain fragments provides the first direct evidence for the previously hypothesized homophylic interaction between the αC-domains in fibrin(ogen) (10). It also indicates that the presence of the αC-connector is not necessary for such interaction to occur. The concentration-dependence and reversibility of the oligomerization process suggest that the interaction between monomeric units in Aα406-483 oligomers is specific and therefore could be utilized for formation of αC polymers in fibrin.
Our NMR and unfolding experiments indicate that the Aα406-483 fragment is intrinsically unstable and at physiologic temperatures should be mostly unfolded. Although the stability of the larger fragments corresponding to the full-length αC-domain was found to be higher (13, 14), it is still not sufficient to preserve compact structure at physiologic temperatures. At the same time, our previous studies revealed that in the parent molecule the αC-domains are quite stable and their unfolding starts far above 37 °C (9, 10). To explain such a difference in stability, we hypothesized that in fibrinogen and fibrin the αC-domains are stabilized by the intra- and intermolecular interactions, respectively, and that these interactions are thermodynamically driven (14). The substantial increase in the thermal stability of the Aα406-483 fragment upon self-association (oligomerization) observed in this study provides direct evidence for this hypothesis. This is further reinforced by the analysis of sedimentation equilibrium data, which suggests that addition of a monomeric unit to an assembling Aα406-483 oligomer results in substantial increase of the stabilizing free energy.
Another important finding of this study is that the affinity of Aα406-483 molecules to each other upon self-association is rather low. The association constants determined by the analysis of sedimentation equilibrium data obtained at 20 °C was found to be 14.4 × 103 M−1, giving a value for the dissociation constant equal to 69 μM. Such an affinity suggests that at physiologic concentrations of fibrinogen (~9 μM) the αC-domains should preferentially be monomeric in the parent molecule. This is in agreement with the hypothesis that in fibrinogen the αC-domains are kept together by the interactions with each other and with the central region of the molecule, preferentially through fibrinopeptide B, and that removal of the latter results in their dissociation (29, 32). The low affinity also suggests that in polymeric fibrin, in which local concentration of the αC-domains dramatically increases, the αC-domains should self-associate to form αC polymers. However, these suggestions may be valid only if one assumes that the affinity of the full-length αC-domains to each other is similar to that of the Aα406-483 fragment. Therefore, identification of conditions in which the full-length αC-domain fragment forms soluble oligomers and determination of the association constant for its self-association is required to verify this assumption.
Having established the lowest energy conformation and stability of the monomeric Aα406-483 fragment one can speculate about a possible mechanism of its self-association. The structure consists of a mixed four-stranded parallel/anti-parallel 2↑1↓3↓4↑ β-sheet in which two β-hairpins interact through parallel β-strands. Such topology was proposed to be generally unfavorable because it would require that the two strands in the middle be involved in both parallel and antiparallel hydrogen bonding, and therefore to accommodate them in a short stretch of β-sheet may impose serious stress on the geometry of the β-sheet, rendering the whole structure less stable (53). This is in agreement with the detected low stability of Aα406-483 monomer and may also explain why the structure of its loose β-hairpin is so distorted. At the same time, the oligomeric form of this fragment, which according to the CD data (Figure 6, inset B) preserves the β-sheet structure, is more stable. Although formation of an extended β-sheet through a side-to-side interaction between four-stranded Aα406-483 β-sheet monomers is possible, such an oligomer would still preserve the stress on the geometry and thereby be less stable. A more stable structure can be obtained if upon self-association of the monomers the distorted β-hairpin would switch its orientation to become a part of an antiparallel β-sheet. This can be achieved by 3D domain swapping, which was found in a number of proteins, and was proposed as a mechanism for dimer/oligomer formation and polymerization of soluble proteins into amyloid fibrils (54, 55). This mechanism can easily explain how the monomeric αC-domains form both αC dimer in fibrinogen and αC oligomers in fibrin through swapping their β-hairpins, as presented schematically in Figure 8. Further experiments are required to test whether this mechanism is utilized by the αC-domains to form more stable dimeric and polymeric structures.
Figure 8.

Schematic representation of a hypothetical dimerization and oligomerization of the Aα406-483 αC-domain fragment by β-hairpin swapping. A ribbon diagram of the monomeric Aα406-483 fragment containing a mixed parallel/anti-parallel 2↑1↓3↓4↑ β-sheet and its schematic representation are shown in panels A and B, respectively; individual β-strands are presented by arrows, a disulfide at the base of the first β-hairpin is in orange. This unstable structure (see text) can be re-arranged into a more stable dimer by swapping the second loose β-hairpin of the two monomeric units to form an anti-parallel β-sheet (panels C and D). Formation of a more stable oligomer consisting of an extended anti-parallel β-sheet can also be carried out by the same mechanism (panel E).
It should be noted that the structured region determined in this study (residues Aα420-479) represents about 80% of the studied Aα406-483 fragment, which, in turn, includes about three-quarters of the NH2-terminal half of the αC-domain. The remaining 20% include several extreme terminal residues, which are obviously unordered, and the 406-419 region of slower concerted motion (Figure 2). Although the structure of this region has not been determined here, there is a high probability that in the parent molecule or in αC oligomers it is ordered. In addition to the relaxation data, a more dense distribution of the conformations preceding this region in the larger Aα374-538 fragment (14) supports this hypothesis. Furthermore, the presence of a well defined turn (residues Aα420-422), which connects this region with the first β-hairpin (Figure 3C), and the size of this region both suggest that it may potentially form an additional β-strand antiparallel to the first β-hairpin. This hypothesis is also in agreement with the CD data (Figure 6, inset B) that suggest an increase in β-structure content upon oligomerization of the Aα406-483 fragment. It should also be mentioned that although the structure of the COOH-terminal half of the αC-domain is not identified yet (14), one cannot exclude that it may also form β-structure, at least upon polymerization of the αC-domains in fibrin. This prediction is based on the results of the previous studies indicating that the β-sheet structure increases significantly during the polymerization process, presumably due to lateral contacts between fibrin monomers through the αC-domains (56, 57). A putative propensity of the COOH-terminal half to form β-structures is also supported by the fact that mutations of its Val526 to Glu and Arg554 to Leu (human numbering) cause the fragments derived from this half to form amyloid fibrils in kidneys resulting in renal amyloidosis (25, 26). Further studies with fragments corresponding to the NH2- and COOH-terminal halves of the αC domain are required to test the above hypotheses.
In summary, this study established that the conformation of the Aα406-483 fragment, corresponding to NH2-terminal half of the bovine fibrinogen αC-domain, consists of an intrinsically unstable parallel/antiparallel β-sheet, that this fragment forms oligomers at increased concentrations, which may mimic αC polymers in fibrin, and that such oligomerization substantially increases its stability, suggesting that intra- and intermolecular interactions of the αC-domains in fibrinogen and fibrin are thermodynamically driven. Analysis of the structure and stability of both Aα406-483 monomers and oligomers suggests that in fibrin αC polymers may form extended β-sheets. Whether the COOH-terminal half of the αC-domain, which may also have a propensity to form β-sheets, adopts ordered conformation in fibrinogen and fibrin remains to be clarified.
Acknowledgments
We thank Dr. I. Pechik for helpful discussions and criticism.
Footnotes
This work was supported by National Institutes of Health Grant HL-56051 to L.M., American Heart Association, Mid-Atlantic Affiliate Grant-in-Aid 055527U to R.R.H., and by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute to N.T.
TBS, 20 mM Tris buffer, pH 7.4, with 150 mM NaCl; PBS, 20 mM potassium phosphate buffer, pH 6.5, 150 mM NaCl; CSI, Chemical Shift Indexing; RDC, Residual Dipolar Coupling; NOE, Nuclear Overhauser Effect; CD, Circular Dichroism.
References
- 1.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 2.Yee VC, Pratt KP, Cote HC, Trong IL, Chung DW, Davie EW, Stenkamp RE, Teller DC. Crystal structure of a 30 kDa C-terminal fragment from the γ chain of human fibrinogen. Structure. 1997;5:125–138. doi: 10.1016/s0969-2126(97)00171-8. [DOI] [PubMed] [Google Scholar]
- 3.Madrazo J, Brown JH, Litvinovich S, Dominguez R, Yakovlev S, Medved L, Cohen C. Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4- Å resolution. Proc Natl Acad Sci U S A. 2001;98:11967–11972. doi: 10.1073/pnas.211439798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pechik I, Madrazo J, Mosesson MW, Hernandez I, Gilliland GL, Medved L. Crystal structure of the complex between thrombin and the central “E” region of fibrin. Proc Natl Acad Sci U S A. 2004;101:2718–2723. doi: 10.1073/pnas.0303440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JH, Volkmann N, Jun G, Henschen-Edman AH, Cohen C. The crystal structure of modified bovine fibrinogen. Proc Natl Acad Sci U S A. 2000;97:85–90. doi: 10.1073/pnas.97.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 Å resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
- 7.Doolittle RF. Structural aspects of the fibrinogen to fibrin conversion. Adv Protein Chem. 1973;27:1–109. doi: 10.1016/s0065-3233(08)60446-5. [DOI] [PubMed] [Google Scholar]
- 8.Doolittle RF. Fibrinogen and fibrin. Annu Rev Biochem. 1984;53:195–229. doi: 10.1146/annurev.bi.53.070184.001211. [DOI] [PubMed] [Google Scholar]
- 9.Privalov PL, Medved LV. Domains in the fibrinogen molecule. J Mol Biol. 1982;159:665–683. doi: 10.1016/0022-2836(82)90107-3. [DOI] [PubMed] [Google Scholar]
- 10.Medved LV, Gorkun OV, Privalov PL. Structural organization of C-terminal parts of fibrinogen Aα-chains. FEBS Lett. 1983;160:291–295. doi: 10.1016/0014-5793(83)80985-5. [DOI] [PubMed] [Google Scholar]
- 11.Erickson HP, Fowler WE. Electron microscopy of fibrinogen, its plasmic fragments and small polymers. Ann N Y Acad Sci. 1983;408:146–163. doi: 10.1111/j.1749-6632.1983.tb23242.x. [DOI] [PubMed] [Google Scholar]
- 12.Weisel JW, Stauffacher CV, Bullitt E, Cohen C. A model for fibrinogen: domains and sequence. Science. 1985;230:1388–1391. doi: 10.1126/science.4071058. [DOI] [PubMed] [Google Scholar]
- 13.Tsurupa G, Tsonev L, Medved L. Structural organization of the fibrin(ogen) αC-domain. Biochemistry. 2002;41:6449–6459. doi: 10.1021/bi025584r. [DOI] [PubMed] [Google Scholar]
- 14.Burton RA, Tsurupa G, Medved L, Tjandra N. Identification of an ordered compact structure within the recombinant bovine fibrinogen αC-domain fragment by NMR. Biochemistry. 2006;45:2257–2266. doi: 10.1021/bi052380c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Credo RB, Curtis CG, Lorand L. Aα-chain domain of fibrinogen controls generation of fibrinoligase (coagulation factor XIIIa). Calcium ion regulatory aspects. Biochemistry. 1981;20:3770–3778. doi: 10.1021/bi00516a016. [DOI] [PubMed] [Google Scholar]
- 16.Procyk R, Bishop PD, Kudryk B. Fibrin-recombinant human factor XIII a-subunit association. Thromb Res. 1993;71:127–138. doi: 10.1016/0049-3848(93)90179-r. [DOI] [PubMed] [Google Scholar]
- 17.Sakata Y, Aoki N. Cross-linking of α2-plasmin inhibitor to fibrin by fibrin- stabilizing factor. J Clin Invest. 1980;65:290–297. doi: 10.1172/JCI109671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsurupa G, Medved L. Identification and characterization of novel tPA- and plasminogen-binding sites within fibrin(ogen) αC-domains. Biochemistry. 2001;40:801–808. doi: 10.1021/bi001789t. [DOI] [PubMed] [Google Scholar]
- 19.Tsurupa G, Ho-Tin-Noe B, Angles-Cano E, Medved L. Identification and characterization of novel lysine-independent apolipoprotein(a)-binding sites in fibrin(ogen) αC-domains. J Biol Chem. 2003;278:37154–37159. doi: 10.1074/jbc.M305154200. [DOI] [PubMed] [Google Scholar]
- 20.Cheresh DA, Berliner SA, Vicente V, Ruggeri ZM. Recognition of distinct adhesive sites on fibrinogen by related integrins on platelets and endothelial cells. Cell. 1989;58:945–953. doi: 10.1016/0092-8674(89)90946-x. [DOI] [PubMed] [Google Scholar]
- 21.Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Transglutaminase-mediated oligomerization of the fibrin(ogen) αC domains promotes integrin-dependent cell adhesion and signaling. Blood. 2005;105:3561–3568. doi: 10.1182/blood-2004-10-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lijnen HR, Soria J, Soria C, Collen D, Caen JP. Dysfibrinogenemia (fibrinogen Dusard) associated with impaired fibrin-enhanced plasminogen activation. Thromb Haemost. 1984;51:108–109. [PubMed] [Google Scholar]
- 23.Koopman J, Haverkate F, Grimbergen J, Egbring R, Lord ST. Fibrinogen Marburg: a homozygous case of dysfibrinogenemia, lacking amino acids Aα 461-610 (Lys 461 AAA→stop TAA) Blood. 1992;80:1972–1979. [PubMed] [Google Scholar]
- 24.Koopman J, Haverkate F, Grimbergen J, Lord ST, Mosesson MW, DiOrio JP, Siebenlist KS, Legrand C, Soria J, Soria C. Molecular basis for fibrinogen Dusart (Aα 554 Arg→Cys) and its association with abnormal fibrin polymerization and thrombophilia. J Clin Invest. 1993;91:1637–1643. doi: 10.1172/JCI116371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen Aα-chain. Nat Genet. 1993;3:252–255. doi: 10.1038/ng0393-252. [DOI] [PubMed] [Google Scholar]
- 26.Uemichi T, Liepnieks JJ, Benson MD. Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest. 1994;93:731–736. doi: 10.1172/JCI117027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uemichi T, Liepnieks JJ, Yamada T, Gertz MA, Bang N, Benson MD. A frame shift mutation in the fibrinogen Aα chain gene in a kindred with renal amyloidosis. Blood. 1996;87:4197–4203. [PubMed] [Google Scholar]
- 28.Francis CW, Marder VJ. Rapid formation of large molecular weight α-polymers in cross-linked fibrin induced by high factor XIII concentrations. Role of platelet factor XIII. J Clin Invest. 1987;80:1459–1465. doi: 10.1172/JCI113226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weisel JW, Medved L. The structure and function of the αC domains of fibrinogen. Ann N Y Acad Sci. 2001;936:312–327. doi: 10.1111/j.1749-6632.2001.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 30.Medved LV, Gorkun OV, Manyakov VF, Belitser VA. The role of fibrinogen αC-domains in the fibrin assembly process. FEBS Lett. 1985;181:109–112. doi: 10.1016/0014-5793(85)81123-6. [DOI] [PubMed] [Google Scholar]
- 31.Veklich YI, Gorkun OV, Medved LV, Nieuwenhuizen W, Weisel JW. Carboxyl-terminal portions of the α chains of fibrinogen and fibrin. Localization by electron microscopy and the effects of isolated αC fragments on polymerization. J Biol Chem. 1993;268:13577–13585. [PubMed] [Google Scholar]
- 32.Gorkun OV, Veklich YI, Medved LV, Henschen AH, Weisel JW. Role of the αC domains of fibrin in clot formation. Biochemistry. 1994;33:6986–6997. doi: 10.1021/bi00188a031. [DOI] [PubMed] [Google Scholar]
- 33.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 36.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 37.Hantgan RR, Paumi C, Rocco M, Weisel JW. Effects of ligand-mimetic peptides Arg-Gly-Asp-X (X = Phe, Trp, Ser) on alphaIIbbeta3 integrin conformation and oligomerization. Biochemistry. 1999;38:14461–14474. doi: 10.1021/bi9907680. [DOI] [PubMed] [Google Scholar]
- 38.Hantgan RR, Rocco M, Nagaswami C, Weisel JW. Binding of a fibrinogen mimetic stabilizes integrin αIIbβ3’s open conformation. Protein Sci. 2001;10:1614–1624. doi: 10.1110/ps.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Philo JS. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 40.Philo JS. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal Biochem. 2006;354:238–246. doi: 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 41.Stafford WF., III Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 42.Laue TM, Shah BD, Ridgeway TM, Pelletier S. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. The Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- 43.Philo JS. An improved function for fitting sedimentation velocity data for low-molecular-weight solutes. Biophys J. 1997;72:435–444. doi: 10.1016/S0006-3495(97)78684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang F, Nau WM. A conformational flexibility scale for amino acids in peptides. Angew Chem Int Ed Engl. 2003;42:2269–2272. doi: 10.1002/anie.200250684. [DOI] [PubMed] [Google Scholar]
- 45.Koehl P, Levitt M. Structure-based conformational preferences of amino acids. Proc Natl Acad Sci U S A. 1999;96:12524–12529. doi: 10.1073/pnas.96.22.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radivojac P, Obradovic Z, Smith DK, Zhu G, Vucetic S, Brown CJ, Lawson JD, Dunker AK. Protein flexibility and intrinsic disorder. Protein Sci. 2004;13:71–80. doi: 10.1110/ps.03128904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsurupa G, Veklich Y, Hantgan R, Belkin AM, Weisel JW, Medved L. Do the isolated fibrinogen αC-domains form ordered oligomers? Biophys Chem. 2004;112:257–266. doi: 10.1016/j.bpc.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 48.Chun PW, Kim SJ, Stanley CA, Ackers GK. Determination of the equilibrium constants of associating protein systems. 3 Evaluation of the weight fraction of monomer from the weight-average partition coefficient (application to bovine liver glutamate dehydrogenase) Biochemistry. 1969;8:1625–1632. doi: 10.1021/bi00832a044. [DOI] [PubMed] [Google Scholar]
- 49.Na GC, Timasheff SN. Stoichiometry of the vinblastine-induced self-association of calf brain tubulin. Biochemistry. 1980;19:1347–1354. doi: 10.1021/bi00548a013. [DOI] [PubMed] [Google Scholar]
- 50.Hantgan RR, Lyles DS, Mallett TC, Rocco M, Nagaswami C, Weisel JW. Ligand binding promotes the enthropy-driven oligomerization of integrin αIIbβ3. J Biol Chem. 2003;278:3417–3426. doi: 10.1074/jbc.M208869200. [DOI] [PubMed] [Google Scholar]
- 51.Doolittle RF. Structural basis of the fibrinogen-fibrin transformation: contributions from X-ray crystallography. Blood Rev. 2003;17:33–41. doi: 10.1016/s0268-960x(02)00060-7. [DOI] [PubMed] [Google Scholar]
- 52.Doolittle RF, Kollman JM. Natively unfolded regions of the vertebrate fibrinogen molecule. Proteins. 2006;63:391–397. doi: 10.1002/prot.20758. [DOI] [PubMed] [Google Scholar]
- 53.Zhang C, Kim SH. The anatomy of protein β-sheet topology. J Mol Biol. 2000;299:1075–1089. doi: 10.1006/jmbi.2000.3678. [DOI] [PubMed] [Google Scholar]
- 54.Lui Y, Eisenberg D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaskolski M. 3D domain swapping, protein oligomerization, and amyloid formation. Acta Biochim Polonica. 2001;48:807–827. [PubMed] [Google Scholar]
- 56.Marx J, Hudry-Clergeon G, Capet-Antonini F, Bernard L. Laser Raman spectroscopy study of bovine fibrinogen and fibrin. Biochim Biophys Acta. 1979;578:107–115. doi: 10.1016/0005-2795(79)90118-1. [DOI] [PubMed] [Google Scholar]
- 57.Hudry-Clergeon G, Freyssinet JM, Torbet J, Marx J. Orientation of fibrin in strong magnetic fields. Ann N Y Acad Sci. 1983;408:380–387. doi: 10.1111/j.1749-6632.1983.tb23258.x. [DOI] [PubMed] [Google Scholar]
- 58.Ottiger M, Delaglio F, Marquardt JL, Tjandra N, Bax A. Measurement of dipolar couplings for methylene and methyl sites in weakly oriented macromolecules and their use in structure determination. J Magn Reson. 1998;134:365–369. doi: 10.1006/jmre.1998.1546. [DOI] [PubMed] [Google Scholar]