

Abstract
α4 integrins play a pivotal role in leukocyte migration and tissue-specific homing. The ability of integrins to bind ligand is dynamically regulated by activation-dependent conformational changes triggered in the cytoplasmic domain. An NMR solution structure defined a putative membrane-proximal salt bridge between the αIIbβ3 integrin cytoplasmic tails, which restrains integrins in their low-affinity state. However, the physiological importance of this salt bridge in α4 integrin regulation remains to be elucidated. To address this question, we disrupted the salt bridge in murine germ line by mutating the conserved cytoplasmic arginine RGFFKR in α4 integrins. In lymphocytes from knock-in mice (α4-R/AGFFKR), α4β1 and α4β7 integrins exhibited constitutively up-regulated ligand binding. However, transmigration of these cells across VCAM-1 and MAdCAM-1 substrates, or across endothelial monolayers, was reduced. Perturbed detachment of the tail appeared to cause the reduced cell migration of α4-R/AGFFKR lymphocytes. In vivo, α4-R/AGFFKR cells exhibited increased firm adhesion to Peyer patch venules but reduced homing to the gut. Our results demonstrate that the membrane-proximal salt bridge plays a critical role in supporting proper α4 integrin adhesive dynamics. Loss of this interaction destabilizes the nonadhesive conformation, and thereby perturbs the properly balanced cycles of adhesion and deadhesion required for efficient cell migration.
Introduction
Integrins, α/β heterodimeric glycoproteins, constitute the largest family of cell-surface adhesion molecules that mediate diverse cell-cell and cell-matrix interactions, supporting a wide range of physiologic processes including development, immune regulation, hemostasis, and wound healing.1,2 A hallmark of the integrin family of adhesion receptors is their unique ability to dynamically up- and down-regulate their adhesiveness and, thereby, to coordinate the orchestrated cellular movements observed in leukocyte migration.3 This ability is achieved largely through conformational changes that are coupled to ligand binding.3 Intracellular signals, elicited by other receptors that sense external stimuli, trigger conformational changes in the cytoplasmic domain, which are then propagated across the plasma membrane to the extracellular domain.1,2 Under resting conditions, integrins exist largely in a low-affinity, nonadhesive conformation, and upon stimulation are converted to high-affinity conformations. The integrin cytoplasmic domain plays a pivotal role in modulating the activity of the integrins to bind ligand.4,5 The cytoplasmic domains of the α- and β-subunits in integrins are thought to associate with each other at the membrane-proximal regions in latent low-affinity state (Figure 1A left). This αβ cytoplasmic association functions as a “clasp” that restrains integrins in a default low-affinity conformation. Upon activation, binding to the integrin cytoplasmic domains of signaling molecules (eg, talin) triggers “unclasping,” which leads to the conversion from low-affinity to high-affinity conformations (Figure 1A right).6
Figure 1.

A model for integrin activation regulated by the membrane-proximal cytoplasmic salt bridge and generation of α4-R/AGFFKR mice in which the cytoplasmic salt bridge is disrupted. (A) A positively charged RGFFKR (a circled plus) forms the membrane-proximal salt bridge with a negatively charged residue(s) (a circled minus) in the β-subunit. The salt bridge facilitates clasping of the α/β cytoplasmic domains, maintaining the extracellular domains in a default inactive conformation (left). Disruption of the salt bridge, which is induced via binding of cytoplasmic proteins such as talin (not shown), triggers unclasping of the cytoplasmic domains, converting the extracellular domains to the active conformation (right). The α- and β-subunits are labeled in the extracellular domains. The plasma membrane is shown with 2 parallel dashed lines. (B) Amino acid sequence alignment of the membrane-proximal cytoplasmic domains of selected integrin α-subunits. A conserved GFFKR motif is highlighted. The arginine residues in the GFFKR motif (RGFFKR) are shown in bold. (C) Targeted insertion of the floxed ACN cassette containing both the neor gene and the Cre-recombinase gene under the control of the sperm-specific ACE promoter,25 mutated exon 28 (28**), and UTR into the Itga4 locus, followed by deletion of the ACN cassette. The configurations of the targeting vector, α4 integrin genomic locus, the targeted allele following homologous recombination, and the ACN-deleted allele are shown. Exons are displayed as black boxes and the floxed ACN cassette, UTR, and diphtheria toxin cassette (DT) are displayed as white boxes. loxP sites are displayed as triangles. The external probe containing exon 24, which was used to verify the targeting event, is indicated. The map displays the following restriction sites: E indicates EcoRI; S, ScaI; Sa, SacI; X, XbaI; and X*, engineered XbaI. (D) Genotyping and confirmation of the deleted ACN cassette by PCR. PCR bands are shown for wild-type (WT/WT, 440 bp), heterozygous (KI/WT, 550 and 440 bp), and homozygous (KI/KI, 550 bp) samples.
Structural investigations using NMR revealed critical interactions at the association interface between α/β cytoplasmic domains.7 The arginine residue in the conserved GFFKR sequence (termed RGFFKR in this study, Figure 1B) at the membrane-proximal region of the α-subunit forms a putative salt bridge with the β-subunit. This putative membrane-proximal salt bridge is thought to constitute a critical interaction that contributes to clasping the α/β cytoplasmic domains and thereby restrains integrin activation (Figure 1A left). R/AGFFKR, the alanine substitution of RGFFKR that would disrupt the clasping salt bridge selectively, constitutively activated αLβ28 and αIIbβ35 in transfectants. In addition, β3-D723A, the alanine substitution of the conserved membrane-proximal aspartate in the β-subunit that would form a salt bridge with RGFFKR in the α-subunit, led to a constitutively active αIIbβ3 integrin in transfectants.5 Furthermore, these in vitro results using transfectants have been supported by findings in knock-in mice for αLβ2 and in patients for αIIbβ3. The deletion of the αL-GFFKR sequence in germ line persistently activated αLβ2 in a strain of knock-in mice (Lfa-1d/d).9 β3-D723H, the mutation found in a group of patients with inherited thrombocytopenia, increased activity of αIIbβ3 integrin in patients' platelets.10
In contrast to αIIbβ3 and αLβ2 integrins, the physiological role of the putative membrane-proximal salt bridge remains to be elucidated in α4 integrins (α4β1 and α4β7). α4 integrins play a pivotal role in lymphocyte adhesion to, and transmigration across, the endothelium during physiologic lymphocyte migration, including tissue-specific homing to gut-associated lymphoid tissues (GALTs) as well as pathologic leukocyte accumulation during inflammation.11,12 The activity of α4 integrins is dynamically regulated. The ability of α4 integrins to bind ligands is normally maintained in a low-affinity state, and rapidly up-regulated only upon activation with exogenous stimuli.13,14 Thus, it is of great importance to investigate the physiological significance of the putative membrane-proximal salt bridge in regulating α4 integrin activation.
Here we attempt to disrupt the putative membrane-proximal salt bridge in the integrin α4 subunit in mouse germ line. The deletion of the entire GFFKR sequence, which was previously used in Lfa-1d/d knock-in mice,9 would not only disrupt the salt bridge via RGFFKR, but also substantially perturb the α/β cytoplasmic association interface. Thus, to selectively disrupt the putative membrane-proximal salt bridge, we knocked in a point mutation α4-R/AGFFKR. We have shown that compared with wild type (WT), lymphocytes from α4-R/AGFFKR mice exhibit constitutively enhanced cell adhesion to VCAM-1 and MAdCAM-1, thus demonstrating the importance of the membrane-proximal salt bridge in restraining integrin activation in α4β1 and α4β7. Furthermore, α4-R/AGFFKR lymphocytes exhibit perturbed lateral migration on, and transmigration across, VCAM-1 and MAdCAM-1 substrates as well as delayed transendothelial migration. In vivo, α4-R/AGFFKR lymphocytes display an increased capacity to firmly adhere to Peyer patch (PP) venules but a reduced capacity to home to the GALT. These data support the idea that the putative cytoplasmic membrane-proximal bridge plays a pivotal role in the balanced regulation of α4 integrin adhesiveness to facilitate cell migration.
Methods
Supplemental methods are available in Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
All experiments using mice were approved by the Institutional Review Board of the Immune Disease Institute.
Flow chamber assays
Flow chamber assays were performed as previously described15 using mouse VCAM-1-Fc (10 μg/mL), MAdCAM-1-Fc (10 μg/mL), or ICAM-1/Fc (50 μg/mL) immobilized on plastic. Cells were resuspended at 106/mL for interactions with VCAM-1 and ICAM-1, and at 4 × 106/mL for interactions with MAdCAM-1, in Hanks balanced salt solution, 10 mM HEPES, 0.5% BSA containing 1 mM Mg2+/Ca2+ or 1 mM Mn2+. Cells were infused into the chamber and allowed to accumulate on the substrate at a shear stress of 0.3 dyne/cm2 for 45 seconds. Wall shear stress was increased incrementally every 10 seconds. Microscopic images of cells under flow were recorded and analyzed off-line as previously described.16
Transwell assays
Transmigration was studied as described previously with minor modifications using a modified Boyden chamber assay with a 6.5-mm Transwell tissue culture system (Corning, Corning, NY) containing a permeable support insert with a 5-μm pore size.17 In brief, Transwell inserts were coated with either 40 μg/mL murine MAdCAM-1-Fc, VCAM-1-Fc, or ICAM-I-Fc. Splenocytes (106) were added to the upper chamber and allowed to transmigrate for 4 hours to the lower chamber that contained 2 μg/mL CXCL12. Transmigrated cells were counted using a FACScan flow cytometer (BD Biosciences, San Jose, CA).
Transendothelial migration under shear stress
Transendothelial migration (TEM) under shear stress was studied as previously described18 using bEnd.3 mouse brain endothelioma cells (passages 6 to 10; ATCC, Manassas, VA). bEnd.3 cells were plated at 0.5 × 106 cells per 40 mm on circular coverslips coated with 20 μg/mL fibronectin and cultured for 48 hours. The cells were stimulated for 24 hours with 50 ng/mL TNF-α, and 2 μg/mL CCL21 was overlaid for 15 minutes on the bEnd.3 cell monolayer. Coverslips with endothelium were then assembled into an FCS2 parallel wall laminar shear flow chamber (Bioptechs, Butler, PA). For transendothelial migration analysis, splenocytes were perfused for 2 minutes over the monolayer at 0.25 dyne/cm2 to allow accumulation. The flow rate was then increased to 2 dyne/cm2 and kept constant for an additional 15-minute period during which time the images were recorded. Cell migration was visualized using a 10× phase-contrast objective and images were recorded using a CCD camera at a rate of 1 frame per 5 seconds. Cell adhesion and transmigration were assessed in the resulting videos by counting the number of lymphocytes adherent and laterally migratory over the apical surface of the endothelium (bright diffraction in phase-contrast imaging), as well as those that underwent transmigration to the subendothelial space (transition to a dark appearance in phase-contrast imaging).
Live-cell imaging
Live-cell imaging of laterally migrating cells on VCAM-1 and MAdCAM-1 substrates was performed as previously described.19 Briefly, 20 μg/mL VCAM-1-Fc or MAdCAM-1-Fc with 16 μg/mL CXCL12 was coated on Delta T chambers (Bioptechs) at 4°C overnight. IL-15–treated memory/effector T cells were generated as previously described.19 After washing with L-15 medium (Cambrex Bioscience, East Rutherford, NJ), cells were added to the chambers and differential interference contrast (DIC) images were acquired using an Axiovert S200 epifluorescence microscope (Carl Zeiss, Heidelberg, Germany) equipped with a 63× oil objective coupled to an Orca CCD camera (Hamamatsu, Okayama City, Japan) at a frame rate of 15 seconds per frame. Cell migration was analyzed as previously described.20
Confocal image acquisition and processing
Fluorescent imaging was performed as previously described.19 Briefly, cells spread on 20 μg/mL VCAM-1 or MAdCAM-1 coimmobilized with 10 μg/mL CXCL12 were fixed and stained with mAb to α4 integrin (PS/2) followed by Cy3-conjugated goat anti–rat IgG and phalloidin–Alexa 488 for actin. Confocal imaging was conducted on a Radiance 2000 Laser-scanning confocal system (Bio-Rad Laboratories, Hercules, CA) using a microscope (model BX50BWI; Olympus, Melville, NY) with a 100× water-immersion objective. Imaging processing was performed with OpenLab software (Improvision, Waltham, MA). Multiple randomly selected fields were imaged for each condition by collecting serial sections through the entire sample Z-axis. These were then projected as complete stacks. For morphologic analysis, cell polarity was identified as the actin-rich crescent-shaped leading edge. The opposing trailing edge was assessed for the presence of extended (> 1 μm) tethers that were rich in both actin and α4-integrin staining. The number of such projections was counted for each of at least 20 cells in randomly selected fields and averaged.
Epifluorescence intravital microscopy
Intravital microscopy of PPs was performed as previously described.21 Briefly, C57BL/6J mice were anesthetized and a bowel segment of the small intestine was positioned for epifluorescence intravital microscopy, to record an individual PP. Preparations were transferred to an intravital microscope (IV-500; Mikron Instruments, San Diego, CA), equipped with a Rapp OptoElectronic SP-20 xenon flash lamp system (Hamburg, Germany) and QImaging Rolera-MGi EMCCD (Surrey, BC). WT or KI splenocytes were labeled with 1 ng/mL calcein-AM for 20 minutes at 37°C, and small boluses (10-50 μL; concentration of 10 × 106 cells/mL) of cells were injected via a left carotid artery catheter. Cell behaviors in PP venules were recorded in 10-minute recordings through 10× or 20× water-immersion objectives (Achroplan; Carl Zeiss). Rolling and sticking fractions in individual vessel segments were determined off-line by playback of digital video files. Rolling fraction was determined as the percentage of cells interacting with PP venules in the total number of cells passing through a vessel during the observation period. Sticking fraction was defined as the percentage of rolling cells that adhered in PP venules for 30 or more seconds.21
Competitive in vivo homing assay
A competitive homing assay was conducted as previously described22 using WT C57BL/6J-CD45.1+ congeneic recipient mice with or without gut inflammation. To induce gut inflammation, mice were fed for 6 days with 3% (wt/vol) DSS in drinking water.23 On day 5, the same numbers of donor splenocytes (2 × 107) from WT and KI mice labeled with different dyes (CFSE and CMTMR) were mixed and injected intravenously. Eighteen hours later, mice were killed and all lymphoid and nonlymphoid organs were harvested and analyzed as described.23 In some experiments, fluorescent dyes were switched.
Statistical analysis
Data are expressed as the mean plus or minus SEM for each group. The Student t test was used for statistical analyses unless otherwise indicated.
Results
Generation of knockin mice in which the membrane-proximal salt bridge is disrupted at the α4 cytoplasmic domain
To disrupt the critical salt bridge that stabilizes the α/β cytoplasmic association in α4 integrins in vivo, we used a replacement-type gene-targeting strategy24,25 to mutate RGFFKR to alanine in the integrin α4 gene (Itga4), thereby generating an Itga4-R/AGFFKR mutated allele in embryonic stem (ES) cells (Figure 1C). We obtained the correct integration of the Itga4-R/AGFFKR allele into the mouse germ line (Figure 1D and data not shown) and designated mice homozygous for the mutant α4 allele as α4-R/AGFFKR. These mice were born under normal Mendelian ratios, were fertile, and did not exhibit gross abnormalities.
Increased ligand binding by α4β1 and α4β7 in α4-R/AGFFKR lymphocytes
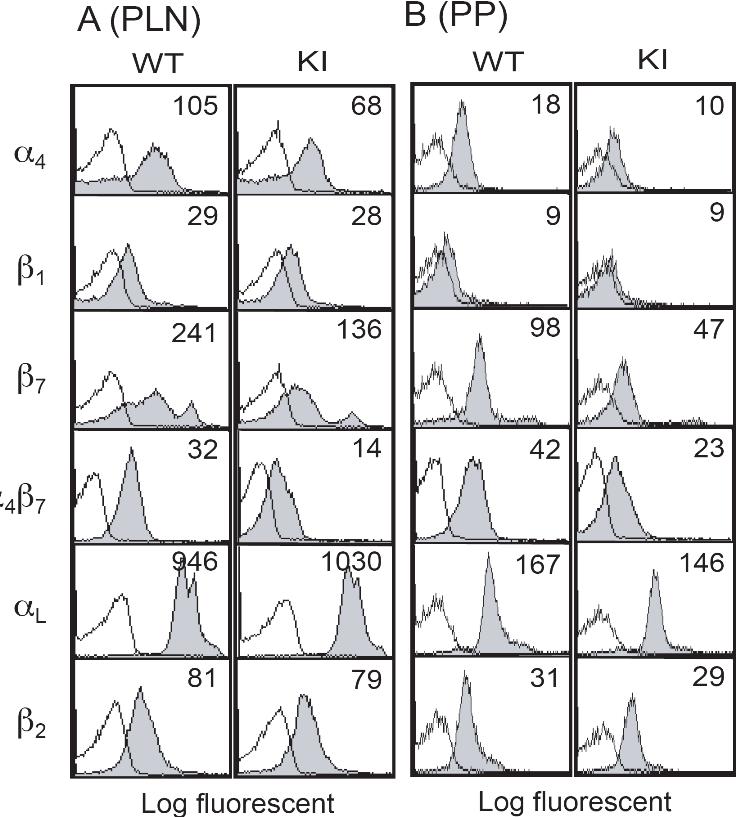
Lymphocytes from the spleen of α4-R/AGFFKR mice showed approximately 50% reduction of α4 and α4β7 expression on the cell surface, compared with WT cells (Figure 2A). Only a slight decrease in β1-subunit expression was observed. Expression of the αLβ2 integrin, L-selectin, and the activation markers CD69 and CD25 was not affected (Figure 2A and data not shown). Similar results were seen in lymphocytes from peripheral lymph nodes (PLNs) and Peyer patches (PPs) (Figure S1).
Figure 2.

Expression of integrins in α4-R/AGFFKR mice. (A) Cell-surface expression of integrins on splenic lymphocytes from wild-type (WT) and α4-R/AGFFKR (KI) mice. Numbers denote mean fluorescent intensity (MFI). Background binding of isotype control antibodies is shown with open histograms. (B) Cell-surface and total (cell surface plus intracellular) α4 protein expression in WT and KI splenocytes. Expression of α4 integrins was measured by immunofluorescent cytometry in the absence (for cell-surface expression) or presence (for total expression) of permeabilization treatment. Ratios of MFI values are shown. (C) α4 mRNA expression in spleen (SP) and peripheral lymph node (PLN) cells. (D) mRNA expression of GRP78 and the spliced form of XBP-1, markers to probe levels of UPR in WT and KI splenocytes. (C,D) mRNA expression was measured by a quantitative RT-PCR and normalized by GAPDH mRNA levels. (B-D) Data represent the mean plus or minus SEM of at least 3 independent experiments.
Despite the reduced cell-surface α4 integrin expression in α4-R/AGFFKR lymphocytes, WT and α4-R/AGFFKR cells showed comparable levels of total α4 integrin expression, which included cell-surface and intracellular expression, as determined by immunofluorescent cytometry analyses of permeabilized cells (Figure 2B). Quantitative reverse-transcription–polymerase chain reaction (RT-PCR) showed that mRNA expression levels of the α4 subunit in WT and α4-R/AGFFKR splenocytes were comparable (Figure 2C). Similar results with αLβ2 (ie, reduced cell-surface and comparable total expression) were also obtained in Lfa-1d/d knock-in mice, in which the αL GFFKR sequence had been deleted.9 As previously shown in the αL-subunit,8 heterodimer formation and subsequent translocation to the cell surface might be partially perturbed in the context of the mutant α4 subunit, possibly through effects on integrin conformation.
To investigate the possibility that a potential increase in an intracellular pool of mutant α4 integrin subunit might activate the unfolded protein response (UPR) that could modify cellular metabolism and functions,26 we studied UPR in WT and α4-R/AGFFKR splenocytes. Using quantitative RT-PCR, we measured the mRNA levels of GRP7827 and the spliced form of XBP-1,28 both of which are known to be up-regulated in response to UPR. We have shown that WT and α4-R/AGFFKR cells expressed comparable levels of GRP78 and the spliced XBP-1 (Figure 2D). Thus, at least in this experimental setting, we did not find evidence that α4-R/AGFFKR cells are subjected to an increased UPR, compared with WT cells.
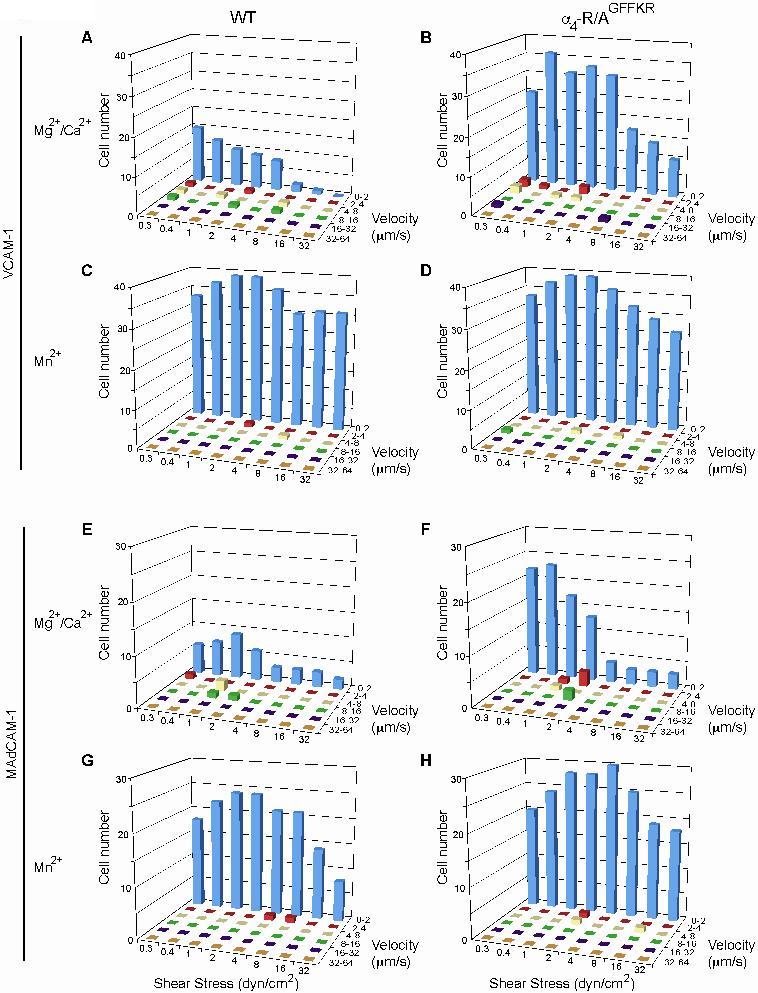
We investigated the adhesive interactions of α4 integrins with their ligands VCAM-1 and MAdCAM-1 under physiological shear conditions using a parallel wall flow chamber.16 A fraction of WT splenic lymphocytes firmly adhered to VCAM-1 and MAdCAM-1 substrates in approximately 1 mM Ca2+ + Mg2+, the cation condition in which integrins are predominantly maintained in a nonadhesive state (Figure 3A,B). Under the same setting, approximately 3 times as many α4-R/AGFFKR lymphocytes firmly adhered as did WT cells, demonstrating increased basal adhesiveness by α4 integrins in α4-R/AGFFKR lymphocytes (Figure 3A,B). In the current experimental setting, we observed only a small fraction of WT and α4-R/AGFFKR lymphocytes that rolled on VCAM-1 and MAdCAM-1 substrates (Figure S2). Increased adhesiveness was specific to the α4 integrins, as αLβ2 in α4-R/AGFFKR splenocytes remained as latent in binding to ICAM-1 as did WT cells (Figure S3). To study the relative contributions of α4β1 and α4β7 integrins to the enhanced adhesion by α4-R/AGFFKR lymphocytes, we used a panel of function-blocking mAbs. Adhesion of α4-R/AGFFKR and WT cells to VCAM-1 was blocked completely with anti-α4 mAb, and partially with either anti-β1 or anti-α4β7 blocking mAb, confirming that the interaction was mediated by both α4β1 and α4β7 integrins (Figure 3E,G,I). In the presence of either anti-β1 or anti-α4β7 blocking mAb, adhesion of α4-R/AGFFKR cells was greater than WT, demonstrating that the adhesiveness of both α4β1 and α4β7 integrins to VCAM-1 had increased (Figure 3E,G,I). By contrast, adhesion of α4-R/AGFFKR and WT cells to MAdCAM-1 was blocked completely with anti-α4 mAb, or by anti-α4β7 blocking mAb, but was rarely affected with anti-β1 blocking mAb, indicating that enhanced adhesion of α4-R/AGFFKR lymphocytes to MAdCAM-1 was solely mediated by the α4β7 integrin (Figure 3F,H,J). Upon stimulation with Mn2+ capable of mimicking inside-out signaling to induce high-affinity integrin conformations, WT cells exhibited increased firm adhesion to VCAM-1 and MAdCAM-1 (Figure 3C,D). Mn2+ stimulation enhanced firm adhesion of α4-R/AGFFKR cells to VCAM-1 and MAdCAM-1 (Figure 3C,D), suggesting that α4 integrins in α4-R/AGFFKR cells are not fully activated and are further activatable with agonists. These results convincingly demonstrated that the disruption of the α4 cytoplasmic membrane-proximal salt bridge increased the adhesiveness of both α4β1 and α4β7, despite the reduced cell-surface expression of mutant α4 integrins. Similar results were obtained with cells from PLNs and PPs (data not shown).
Figure 3.

Enhanced adhesive interactions of α4-R/AGFFKR splenocytes with VCAM-1 and MAdCAM-1 under shear stress. Splenocytes from wild-type (WT) and α4-R/AGFFKR (KI) mice were infused in 1 mM Mg2+/Ca2+ (A,B,E-J) or 1 mM Mn2+ (C,D) into a parallel wall flow chamber and allowed to accumulate on VCAM-1 (A,C,E,G,I) or MAdCAM-1 (B,D,F,H,J) substrates at 0.3 dyne/cm2 for 45 seconds. Shear stress was incrementally increased every 10 seconds from 0.5 to 32 dyne/cm2, and adhesive interactions of cells with the substrates were recorded and analyzed. In some experiments (E-J), cells were pretreated for 10 minutes at room temperature with blocking mAbs to α4 (PS/2, 30 μg/mL), α4β7 (DATK32, 30 μg/mL), or β1 (CBL 1333, 30 μg/mL) integrins or isotype control antibodies (IgG, 30 μg/mL). Data represent the mean plus or minus SEM of 3 independent experiments. P value less than .01 versus **KI in Mg2+/Ca2+ or *WT.
Disturbed transmigration of α4-R/AGFFKR lymphocytes through VCAM-1 and MAdCAM-1
As an appropriate balance between adhesion and deadhesion is thought to be critical for efficiently coordinated cell migration, we studied how the persistently increased adhesion of α4-R/AGFFKR cells affected transmigration by using a Transwell assay.17 WT and α4-R/AGFFKR cells transmigrated equally well through an ICAM-1–coated Transwell insert into a chemokine CXCL12–containing lower chamber, demonstrating normal migration behavior in general and normal coordination of αLβ2 integrin adhesiveness, specifically in α4-R/AGFFKR (Figure 4A). In contrast, when Transwell inserts were coated with VCAM-1 or MAdCAM-1, α4-R/AGFFKR cells showed approximately 50% reduction of transmigrated cells compared with WT (Figure 4B,C).
Figure 4.

Perturbed transmigration of α4-R/AGFFKR splenocytes. (A-C) Transmigration of WT and KI splenocytes toward a CXCL12 gradient through ICAM-1–coated (A), VCAM-1–coated (B) or MAdCAM-1–coated (C) permeable inserts was examined using a modified Boyden chamber assay with a Transwell tissue culture system. (D) Flow-enhanced TEM through b.End.3 endothelial monolayers. TEM was studied in the presence of 2.0 dyne/cm2 shear stress. (A-D) Data are expressed as the mean plus or minus SEM of triplicates from 3 independent experiments. *P < .01 versus WT.
Disturbed transendothelial migration of α4-R/AGFFKR lymphocytes under shear stress
Transendothelial migration (TEM) is a critical step that regulates lymphocyte entry into lymph nodes as well as leukocyte accumulation into inflamed tissues. During lymphocyte TEM, α4 integrins cooperate with αLβ2 integrin and other adhesion molecules.29 The presence of shear stress enhances the capacity of lymphocytes to transmigrate through endothelial monolayers.30 To study transendothelial migration by WT and α4-R/AGFFKR lymphocytes under physiologic shear stress, we used monolayers of bEnd.3 mouse endothelial cells assembled into a flow chamber. The monolayers of bEnd.3 cells were treated with TNF-α to up-regulate surface expression of integrin ligands such as VCAM-1, MAdCAM-1, and ICAM-1. A CCL21 chemokine was immobilized on the endothelial surface to facilitate adhesion to, and transmigration through, endothelial cells. Identical numbers of WT and α4-R/AGFFKR cells were infused into the chamber and allowed to accumulate on endothelial monolayers for 2 minutes under 0.25 dyne/cm2 shear stress. The migratory behaviors of adherent lymphocytes were then recorded under 2.0 dyne/cm2 shear stress for 15 minutes. Although α4-R/AGFFKR cells were more adhesive to VCAM-1 or MAdCAM-1 in the absence of chemokine stimulation (Figure 3A,B), comparable numbers of WT and α4-R/AGFFKR cells accumulated on bEnd.3 endothelial monolayers (WT, 89.7 ± 4.0; KI, 94.3 ± 2.5 cells per field). Surface-displayed CCL21 is likely to activate αLβ2 and α4 integrins on lymphocytes to support firm adhesion on bEnd.3 cells that simultaneously express VCAM-1, MAdCAM-1, and ICAM-1. However, when allowed to transmigrate at 2.0 dyne/cm2 shear stress for 15 minutes, approximately 50% as few α4-R/AGFFKR cells underwent TEM as did WT cells (Figure 4D).
Perturbed lateral migration of α4-R/AGFFKR lymphocytes on VCAM-1 and MAdCAM-1 substrates
To investigate the mechanism(s) by which TEM of α4-R/AGFFKR lymphocytes was perturbed, we studied lateral migration with live-cell time-lapse video microscopy. Although we tried to use naive lymphocytes in which basal integrin adhesiveness is maintained at low levels, they failed to show an efficient lateral migration on integrin ligands in response to chemokine. Similar observations were made in previous reports.31 As an alternative, we used IL-15–treated memory/effector T cells, in which integrins are known to show little basal ligand binding, but readily up-regulate upon chemokine stimulation.32
WT T cells migrated smoothly on VCAM-1/CXCL12 substrates (Movie S1). By contrast, α4-R/AGFFKR T cells migrated poorly on VCAM-1/CXCL12 substrates (Video S2). Whereas WT cells migrating on VCAM-1 were polarized normally, displaying a typically hand mirror–like shape with a flattened leading edge followed by a short tail, α4-R/AGFFKR T cells migrating on VCAM-1 exhibited extremely stretched tails enriched in α4 integrin (Figure 5A). Close image analysis reveals (Figure 5B) that the most remarkable morphologic distinction between 2 types of cells is the strong presence of α4 integrin–rich trailing edge tethers (Figure 5B arrows) in α4-R/AGFFKR cells but not WT cells. Quantitative image analysis demonstrated that α4-R/AGFFKR cells displayed significantly more tethers than WT cells (Figure 5C). Similar results were obtained in T cells migrating on MAdCAM-1 substrates (Figure 5A,C). These results strongly suggest that the perturbed detachment of the tail accounts for decreased TEM of α4-R/AGFFKR cells across bEnd.3 endothelial monolayers.
Figure 5.

Suppressed migration of α4-R/AGFFKR T lymphocytes on VCAM-1 and MAdCAM-1 substrates. Lateral migration of T lymphocytes on VCAM-1 and MAdCAM-1 in response to chemokine CXCL12 was studied using live-cell imaging. (A) Representative confocal and DIC images of migrating cells stained for actin (Alexa 488) and α4 integrin (Cy3) are shown. (B) Morphologic analysis showing the strong presence of α4 integrin–rich trailing edge “tethers” (arrows) in α4-R/AGFFKR cells, but not WT cells, migrating on VCAM-1 and MAdCAM-1 (not shown). (C) The number of tethers per cell. Data are expressed as the mean plus or minus SEM of at least 20 cells from randomly selected fields. *P < .01 versus WT.
Increased firm adhesion of α4-R/AGFFKR lymphocytes to PP venules in vivo
We have demonstrated proof of principle that the aberrantly activated α4-R/AGFFKR perturbs TEM across bEnd.3 cell line under shear stress; however, TNF stimulation of, and apical addition of CCL21 to, bEnd.3 cells might not fully recapitulate in vivo phenotypes of HEVs in the gut during normal lymphocyte homing. To study the in vivo adhesive interactions of α4-R/AGFFKR lymphocytes with HEVs in the GALT, such as PPs, we performed an intravital microscopic investigation of PP venules.21
We injected calcein-labeled α4-R/AGFFKR or WT splenocytes into anesthetized WT syngeneic recipient mice that had been prepared for intravital microscopy of PPs. The injected cells were then observed and video recorded entering and interacting with PP venules. Adhesive interactions of α4-R/AGFFKR or WT donor splenocytes with PP venules were analyzed off-line. We found that α4-R/AGFFKR and WT cells rolled on PP venules at similar frequencies (Figure 6A) and comparable velocities (Figure 6C). However, α4-R/AGFFKR cells exhibited higher capacity to firmly adhere to PP venules than WT cells (Figure 6B). These data demonstrated that despite reduced cell-surface expression of α4 integrin, α4-R/AGFFKR cells showed increased firmed adhesion to PP venules in vivo.
Figure 6.

Increased firm adhesion of α4-R/AGFFKR lymphocytes in vivo to PP venules. Adhesive interactions of WT and α4-R/AGFFKR lymphocytes to PP venules in WT recipient mice were studied using epifluorescence intravital microscopy. Rolling (A) and sticking (B) fractions as well as rolling velocity (C) are shown. Data are mean plus or minus SEM of 9 different PP venules analyzed per group in 2 independent experiments. *P < .05 versus WT.
Suppression of α4-R/AGFFKR lymphocyte migration to the gut
α4 integrin plays a pivotal role in a physiologic lymphocyte recirculation through the GALT as well as a pathologic lymphocyte accumulation to inflamed gut.11,12 To study the impact of aberrantly activated α4 integrins on lymphocyte migration to the gut, we used a competitive homing assay. We began with investigating lymphocyte homing to noninflamed tissues. Splenocytes from WT and α4-R/AGFFKR mice were fluorescently labeled with CFSE and CMTMR, respectively. Equal numbers of WT and α4-R/AGFFKR cells were mixed and intravenously administered into C57BL/6J-CD45.1+ mice. Eighteen hours later, WT and α4-R/AGFFKR lymphocytes homed equally well to SP and PLNs (Figure 7A), and comparable fractions of WT and α4-R/AGFFKR lymphocytes were present in peripheral blood (WT, 0.59% ± 0.18%; and α4-R/AGFFKR, 0.57% ± 0.18%; of injected cells [mean ± SEM, n = 6]). By contrast, homing of α4-R/AGFFKR lymphocytes to the gut (including MLNs, PPs, small and large intestines) was reduced by approximately 50% compared with WT (Figure 7A). To study whether a subpopulation of α4-R/AGFFKR cells with regard to α4 surface expression might selectively be blocked to enter tissues, we compared α4 integrin expression of WT and α4-R/AGFFKR cells in donor cells before injection and those homed to tissues. The ratio of α4 expression in WT and KI cells is comparable in both donor cell populations (Table S1).
Figure 7.

In vivo homing of α4-R/AGFFKR lymphocytes to the gut is suppressed. Competitive homing assay to compare α4-R/AGFFKR (KI) and wild-type (WT) splenic lymphocytes. Equal numbers (2 × 107) of fluorescently labeled cells were mixed and injected into C57BL/6J-CD45.1+ congeneic mice in the absence (A) or presence (B) of DSS-induced colitis. Homing indices were determined 18 hours after injection. Data are expressed as the mean values plus or minus SEM of at least 5 independent experiments. *P < .05; **P < .01; and ***P < .001, versus SP. SP indicates spleen; PBL, peripheral blood lymphocyte; PLN, peripheral lymph node; MLN, mesenteric lymph node; PP, Peyer patch; SI, small intestine; LI, large intestine; BM, bone marrow; LIV, liver; and LUN, lung.
We then investigated lymphocyte homing to inflamed gut. We induced mild colitis in C57BL/6J-CD45.1+ recipient mice by feeding them for 6 days with 3% DSS. At day 5, mice exhibited approximately 4% body weight loss and intestinal edema formation (not shown). We performed a new set of in vivo competitive homing assays using the recipient mice with DSS-induced colitis. Eighteen hours after injection of equal numbers of fluorescently labeled WT and α4-R/AGFFKR cells, cells were harvested from inflamed gut and other organs, and the homing indices were then determined. WT and α4-R/AGFFKR lymphocytes homed equally well to SP and PLNs. However, α4-R/AGFFKR lymphocytes exhibited approximately 50% reduced homing to the gut, compared with WT (Figure 7B). These results demonstrated that the aberrant activation of α4 integrins perturbed lymphocyte homing to both uninflamed and inflamed gut.
α4-R/AGFFKR mice contain reduced gut lymphocytes
The capacity of lymphocytes to migrate/home represents an important factor in regulating the size and cellularity of lymphoid tissues,33 which could be affected by the perturbed migration of lymphocytes in α4-R/AGFFKR mice. Therefore, we studied the distribution of lymphocytes in lymphoid organs. Macroscopic examination showed that the PPs from α4-R/AGFFKR mice were smaller than those of WT (WT, 1.4 ± 0.1 mm; α4-R/AGFFKR, 0.9 ± 0.1 mm; diameter, P < .01), whereas SP, PLNs, and MLNs were indistinguishable from one mouse strain to another (data not shown). Consistent with the results of our in vivo homing assay, α4-R/AGFFKR mice contained significantly fewer lymphocytes in the gut (ie, PP and IEL compartments) compared with WT (Table 1).
Table 1.
Distribution of leukocytes
| Organ | Cell number (× 105) |
|||||
|---|---|---|---|---|---|---|
| Mononuclear cells |
CD3+ T-cells |
B220+ B-cells |
||||
| WT | α4-G/AGFFKR | WT | α4-G/AGFFKR | WT | α4-G/AGFFKR | |
| BM* | 260 ± 60 | 290 ± 60 | 2.7 ± 0.5 | 3.5 ± 0.1 | 59 ± 0.8 | 66 ± 2.3 |
| Spleen* | 650 ± 200 | 630 ± 120 | 160 ± 7 | 210 ± 50 | 370 ± 35 | 320 ± 5 |
| Thymus* | 850 ± 340 | 820 ± 270 | 180 ± 80 | 250 ± 49 | ND | ND |
| PLN† | 9.8 ± 1.8 | 12 ± 2.1 | 4.4 ± 0.8 | 5.4 ± 1.0 | 5.4 ± 1.0 | 6.6 ± 1.1 |
| MLN† | 6.0 ± 3.3 | 9.9 ± 5.2 | 4.1 ± 1.7 | 5.6 ± 3.4 | 2.6 ± 1.1 | 3.8 ± 1.7 |
| IEL* | 22 ± 5.2 | 7.0 ± 1.4‡ | 20 ± 4.2 | 6.0 ± 1.4‡ | ND | ND |
| PP† | 4.4 ± 0.8 | 1.3 ± 0.7‡ | 0.63 ± 0.2 | 0.11 ± 0.1‡ | 3.5 ± 0.5 | 1.1 ± 0.7‡ |
BM indicates bone marrow; PLN, peripheral lymph node; MLN, mesenteric lymph node; IEL, intraepithelial lymphocyte; PP, Peyer patch; and ND, not detected.
Numbers of cells per mouse.
Numbers of cells per node.
P < .01 versus WT.
Discussion
We have demonstrated that disruption of the putative cytoplasmic membrane-proximal salt bridge leads to constitutively activated α4β1 and α4β7 integrins. Our results with α4-R/AGFFKR knock-in mice have convincingly demonstrated that the membrane-proximal salt bridge plays a critical role in restraining default low-affinity state of α4 integrins in physiologically relevant settings.
The importance of the putative membrane-proximal salt bridge in β1 integrins including α4β1 remains to be questioned. Czuchra et al previously knocked in an alanine substitution, in the β1 subunit, of a membrane-proximal aspartate D759 that would form a salt bridge with RGFFKR in the α-subunit.34 Despite the hypothesis that the mutation would activate β1 integrins, β1-D759A keratinocytes failed to show an increase in cell adhesion to β1 integrin ligands; and β1-D759A bone marrow cells did not show activation-dependent conformational changes in the β1-subunit. These results led Czuchra et al to argue that the membrane-proximal salt bridge might not be important for physiologic regulation of β1 integrins. It has yet to be directly demonstrated whether α4β1 adhesiveness in β1-D759A mice is enhanced; however, the effectiveness of perturbing the membrane-proximal salt bridge in α4β1 might substantially differ in β1-D759A and α4-R/AGFFKR. The NMR solution structure for the αIIbβ3 cytoplasmic domain showed that RGFFKR (ie, αIIb-R995) interacts not only with the membrane-proximal aspartate (β3-D723), but also with 2 other membrane-proximal conserved residues, histidine (β3-H722) and glutamate (β3-E726).7 Therefore, in β1-D759A mice, perturbation by the D759A mutation might be at least partially compensated by intact β1-H758 and/or β1-E762, thereby creating a much milder effect on the adhesiveness of β1 integrins. By contrast, the R/AGFFKR mutation disrupts all 3 electrostatic interactions, thereby inducing more robust activating phenotypes. Alternatively, R/AGFFKR mutation might destabilize the cytoplasmic α/β association via perturbing the local structural integrity of the α4 cytoplasmic domain. Future studies are necessary to determine the structure of α4β1 and α4β7 cytoplasmic domains.
Persistently increased adhesion of α4-R/AGFFKR cells perturbed transmigration through VCAM-1 and MAdCAM-1. An appropriate balance between adhesion at the leading edge and deadhesion at the trailing edge is important for efficient cell migration.3 As shown by elongated tails of R/AGFFKR cells laterally migrating on VCAM-1 and MAdCAM-1, perturbed detachment at the trailing edge is likely to suppress directional forward movements of α4-R/AGFFKR cells. Our results are consistent with previous studies involving either other strains of Lfa-1d/d knock-in mice expressing constitutively active αLβ29 or β7 (D146A) knock-in mice expressing constitutively active α4β7 integrin via a mutation in the negative regulatory metal-binding site ADMIDAS (adjacent to metal ion-dependent adhesion site).19 Lymphocytes from both knock-in strains showed disturbed lateral migration on, and transmigration across, substrates bearing the corresponding integrin ligands. In addition, aberrantly activated αLβ2 or α4β7 integrin delayed TEM in Transwell assays performed in the absence of shear stress. Since the presence of shear stress, a more physiologically relevant condition, promotes TEM, it is of great interest to study the impact of aberrantly activated integrins on shear stress–enhanced TEM. We have shown the proof of principle that aberrantly activated α4 integrins interfered with shear stress–enhanced TEM. Although the ability of αLβ2 to support TEM appears to be intact in α4-R/AGFFKR, impaired detachment of α4 integrins could interfere with αLβ2-driven diapedesis and migration of α4-R/AGFFKR cells.
In cooperation with αLβ2, α4β7 plays a dominant role over α4β1 in supporting lymphocyte homing to the GALT.35,36 Thus, although α4-R/AGFFKR mice exhibited constitutive activation of both α4β1 and α4β7 integrins, aberrantly activated adhesion of α4β7 to MAdCAM-1 appears to play a major role in the reduced homing of α4-R/AGFFKR cells to the GALT in uninflamed gut. By contrast, adhesive interactions of aberrantly activated α4 integrins with not only MAdCAM-1 but also VCAM-1 might be involved in reduced lymphocytes homing to DSS-induced inflamed gut.37,38 How did aberrantly activated α4 integrin reduce lymphocyte homing to the gut? The most plausible explanation is that TEM through high endothelial venules (HEVs) in the GALT is perturbed by aberrantly activated α4β7 integrins that suppress detachment at the trailing edge. This is supported by in vitro TEM experiments under shear stress, which showed delayed TEM by α4-R/AGFFKR cells. In homing assays, α4-R/AGFFKR cells that had become firmly adhered to HEVs, but had failed to enter GALT interstitial spaces, might eventually become detached. Although the reduced expression of α4β7 in α4-R/AGFFKR might reduce lymphocyte adhesion to gut HEVs and, thereby, homing to the gut, our intravital microscopic observation that α4-R/AGFFKR cells exhibited in vivo an increased cell adhesion to PP venules favors the alternative mechanism (ie, aberrant activation). We believe that the aberrant activation of α4β7 represents the primary mechanism interfering with gut homing. As α4 integrins in α4-R/AGFFKR cells are persistently adhesive, independent of activation, it is possible that α4-R/AGFFKR lymphocytes are entrapped in α4 integrin ligand-expressing organs outside the GALT, thereby reducing cell homing to the GALT. However, similar numbers of α4-R/AGFFKR and WT donor cells remained in the peripheral blood 18 hours after injection. This result suggests that both WT and α4-R/AGFFKR cells in circulation were equally available to the GALT and other lymphoid tissue. Thus, entrapment is unlikely to be the major cause of reduced GALT homing by α4-R/AGFFKR cells. Future investigations would be required to better understand how the aberrantly activated α4 integrin perturbs lymphocyte migration to the gut.
In summary, using knock-in mice we have demonstrated that the putative membrane-proximal cytoplasmic salt bridge is critical for maintaining default nonadhesive states of α4β1 and α4β7 integrins and consequently in supporting properly regulated integrin adhesive dynamics. The membrane-proximal salt bridge constitutes an important regulatory component within the integrin, one that facilitates the appropriate adhesion and deadhesion balance required for efficient and coordinated migration of lymphocytes to the gut.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Timothy A. Springer for his valuable advice, Dr Mario R. Capecchi for the self-excision cassette used in pACN-TV, and Dr Klaus Rajewsky for Bruce-4 ES cells. We thank Dr Xiaohui Zhang, Dr Koichi Yuki, Ms Mary Mohrin, Ms Ronnie Yoo, and Ms Whitney Silkworth for their technical assistances.
This work was supported by grants from the Arthritis Foundation (Atlanta, GA; C.V.C.) and the National Institutes of Health (Bethesda, MD), AI063421 and HL048675 (M.S.). Y.I. was partially supported by fellowships from the Nakajima Foundation (Tokyo, Japan), the Japan Intractable Diseases Research Foundation (Tokyo, Japan), the Yamada Science Foundation (Osaka, Japan), and the Uehara Memorial Foundation (Tokyo, Japan).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.I., E.J.P., and C.V.C. designed and performed research, analyzed data, and wrote the paper; D.P., A.P., and G.C. designed and performed research and analyzed data; U.H.A. designed research and analyzed data; and M.S. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Motomu Shimaoka, 200 Longwood Avenue, Boston, MA 02115; e-mail: shimaoka@idi.harvard.edu.
References
- 1.Hynes RO. Integrins: bi-directional, allosteric, signalling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez-Madrid F, del Pozo MA. Leukocyte polarization in cell migration and immune interactions. EMBO J. 1999;18:501–511. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu C, Takagi J, Springer TA. Association of the membrane-proximal regions of the α and β subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276:14642–14648. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- 5.Hughes PE, Diaz-Gonzalez F, Leong L, et al. Breaking the integrin hinge. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 6.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin β tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 7.Vinogradova O, Velyvis A, Velyviene A, et al. A structural mechanism of integrin αIIbβ3“inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 8.Lu C, Springer TA. The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1 (LFA-1). J Immunol. 1997;159:268–278. [PubMed] [Google Scholar]
- 9.Semmrich M, Smith A, Feterowski C, et al. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. 2005;201:1987–1998. doi: 10.1084/jem.20041850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghevaert C, Salsmann A, Watkins NA, et al. A non-synonymous SNP in the ITGB3 gene disrupts the conserved membrane-proximal cytoplasmic salt bridge in the {alpha}IIb{beta}3 integrin and co-segregates dominantly with abnormal proplatelet formation and macrothrombocytopenia. Blood. 2008;111:3407–3414. doi: 10.1182/blood-2007-09-112615. [DOI] [PubMed] [Google Scholar]
- 11.Wagner N, Muller W. Functions of α4- and β7-integrins in hematopoiesis, lymphocyte trafficking and organ development. Curr Top Microbiol Immunol. 1998;231:23–32. doi: 10.1007/978-3-642-71987-5_2. [DOI] [PubMed] [Google Scholar]
- 12.Rose DM, Han J, Ginsberg MH. Alpha4 integrins and the immune response. Immunol Rev. 2002;186:118–124. doi: 10.1034/j.1600-065x.2002.18611.x. [DOI] [PubMed] [Google Scholar]
- 13.Chigaev A, Zwartz G, Graves SW, et al. Alpha4beta1 integrin affinity changes govern cell adhesion. J Biol Chem. 2003;278:38174–38182. doi: 10.1074/jbc.M210472200. [DOI] [PubMed] [Google Scholar]
- 14.Chan JR, Hyduk SJ, Cybulsky MI. Detecting rapid and transient upregulation of leukocyte integrin affinity induced by chemokines and chemoattractants. J Immunol Meth. 2003;273:43–52. doi: 10.1016/s0022-1759(02)00417-9. [DOI] [PubMed] [Google Scholar]
- 15.Salas A, Shimaoka M, Chen S, Carman CV, Springer TA. Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin LFA-1. J Biol Chem. 2002;277:50255–50262. doi: 10.1074/jbc.M209822200. [DOI] [PubMed] [Google Scholar]
- 16.Chen JF, Salas A, Springer TA. Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat Struct Biol. 2003;10:995–1001. doi: 10.1038/nsb1011. [DOI] [PubMed] [Google Scholar]
- 17.Röhnelt RK, Hoch G, Reiss Y, Engelhardt B. Immunosurveillance modelled in vitro: naive and memory T cells spontaneously migrate across unstimulated microvascular endothelium. Int Immunol. 1997;9:435–450. doi: 10.1093/intimm/9.3.435. [DOI] [PubMed] [Google Scholar]
- 18.Shulman Z, Pasvolsky R, Woolf E, et al. DOCK2 regulates chemokine-triggered lateral lymphocyte motility but not transendothelial migration. Blood. 2006;108:2150–2158. doi: 10.1182/blood-2006-04-017608. [DOI] [PubMed] [Google Scholar]
- 19.Park EJ, Mora JR, Carman CV, et al. Aberrant activation of integrin alpha4beta7 suppresses lymphocyte migration to the gut. J Clin Invest. 2007;117:2526–2538. doi: 10.1172/JCI31570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W, Carman CV, Kim M, Salas A, Shimaoka M, Springer TA. A small molecule agonist of an integrin, alphaLbeta2. J Biol Chem. 2006;281:37904–37912. doi: 10.1074/jbc.M606888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bargatze RF, Jutila MA, Butcher EC. Distinct roles of L-selectin and integrins α4β7 and LFA-1 in lymphocyte homing to Peyer's patch-HEV in situ: the multistep model confirmed and refined. Immunity. 1995;3:99–108. doi: 10.1016/1074-7613(95)90162-0. [DOI] [PubMed] [Google Scholar]
- 22.Mora JR, Bono MR, Manjunath N, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 23.Park EJ, Takahashi I, Ikeda J, et al. Clonal expansion of double-positive intraepithelial lymphocytes by MHC class I-related chain A expressed in mouse small intestinal epithelium. J Immunol. 2003;171:4131–4139. doi: 10.4049/jimmunol.171.8.4131. [DOI] [PubMed] [Google Scholar]
- 24.Kraus M, Pao LI, Reichlin A, et al. Interference with immunoglobulin (Ig)a immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation modulates or blocks B cell development, depending on the availability of an Igb cytoplasmic tail. J Exp Med. 2001;194:455–469. doi: 10.1084/jem.194.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR. Targeting genes for self-excision in the germ line. Genes Dev. 1999;13:1524–1528. doi: 10.1101/gad.13.12.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 27.Cho K, Thomas RL, Greenhalgh DG. CD14-dependent regulation of Grp78 in the liver and lungs of mice after burn injury. Exp Mol Pathol. 2003;75:148–153. doi: 10.1016/s0014-4800(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 28.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 29.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 30.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 31.Woolf E, Grigorova I, Sagiv A, et al. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8:1076–1085. doi: 10.1038/ni1499. [DOI] [PubMed] [Google Scholar]
- 32.Manjunath N, Shankar P, Wan J, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic t lymphocytes. J Clin Invest. 2001;108:871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanov II, Diehl GE, Littman DR. Lymphoid tissue inducer cells in intestinal immunity. Curr Top Microbiol Immunol. 2006;308:59–82. doi: 10.1007/3-540-30657-9_3. [DOI] [PubMed] [Google Scholar]
- 34.Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R. Genetic analysis of beta1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamann A, Andrew DP, Jablonski-Westrich D, Holzmann B, Butcher EC. Role of α4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol. 1994;152:3282–3293. [PubMed] [Google Scholar]
- 36.Berlin-Rufenach C, Otto F, Mathies M, et al. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J Exp Med. 1999;189:1467–1478. doi: 10.1084/jem.189.9.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teramoto K, Miura S, Tsuzuki Y, et al. Increased lymphocyte trafficking to colonic microvessels is dependent on MAdCAM-1 and C-C chemokine mLARC/CCL20 in DSS-induced mice colitis. Clin Exp Immunol. 2005;139:421–428. doi: 10.1111/j.1365-2249.2004.02716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soriano A, Salas A, Sans M, et al. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab Invest. 2000;80:1541–1551. doi: 10.1038/labinvest.3780164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}