

Abstract
Modafinil is approved for use in the treatment of excessive daytime sleepiness. The precise mechanism of modafinil action has not been elucidated, although both dopamine (DA) and norepinephrine (NE) systems have been implicated. To explore the roles of DA and NE in the mechanism of modafinil-induced arousal, dopamine β-hydroxylase knockout (Dbh −/−) mice were examined in behavioral paradigms of arousal (photobeam breaks and behavioral scoring of sleep latency). Dbh −/− mice completely lack NE but have hypersensitive DA signaling. It was hypothesized that Dbh −/− mice would be unresponsive to modafinil if the compound acts primarily via NE, but would be hypersensitive to modafinil if it acts primarily via DA. Dbh −/− mice had increased sensitivity to the locomotor-activating and wake-promoting effects of modafinil. Paradoxically, the α1-adrenergic receptor antagonist, prazosin, attenuated the effects of modafinil in control mice, but not in Dbh −/− mice. Blockade of DA receptors with flupenthixol decreased modafinil-induced locomotion and wake in both control and Dbh −/− mice. These results suggest that both NE and DA are involved in the behavioral effects of modafinil in control mice, but the requirement for NE can be bypassed by hypersensitive DA signaling.
Keywords: modafinil, sleep, locomotion, norepinephrine, dopamine, dopamine β-hydroxylase
1. Introduction
It has been estimated that 50–70 million Americans suffer from chronic disorders of sleep and wakefulness that adversely affect health and longevity, making sleep disorders second only to pain in the number of patients seeking medical attention (Colten and Altevogt, 2006). Accordingly, the development of medications to treat sleep disorders is a high priority. One such drug is modafinil, which is approved for use in the treatment of the excessive daytime sleepiness associated with narcolepsy and other sleep disorders. Modafinil has a behavioral phenotype distinct from that of more traditional stimulants such as amphetamine or cocaine in that it does not produce rebound hypersomnolence and has limited dependence liability (Ballon and Feifel, 2006; Edgar and Seidel, 1997; Minzenberg and Carter, 2008). The mechanism of action(s) underlying the wake-promoting effects of modafinil remain to be elucidated (Minzenberg and Carter, 2008).
Early research from the 1990s implicated the catecholamines, NE and DA, in the mechanism of action of modafinil but the animal data are confusing, with the role that each neurotransmitter plays yet to be clarified. Of more than 100 neurotransmitter and enzyme targets, the only consistent finding in vitro is a low affinity (Ki = 2–7 μM) inhibition of the DA transporter (DAT) (Mignot et al., 1994). While DAT knockout mice show decreased responsiveness to modafinil (Wisor et al., 2001) other data indicated that DA receptor antagonists were ineffective in blocking modafinil-induced arousal (Duteil et al., 1990; Lin et al., 1992; Simon et al., 1995). The compounds that consistently block the effects of modafinil are α1-adrenergic receptor (α1AR) antagonists (Duteil et al., 1990; Hermant et al., 1991; Lin et al., 1992) while α1bAR knockout mice show an attenuated response to the drug (Stone et al., 2002). However, modafinil does not bind α1ARs (Mignot et al., 1994). Modafinil is also a weak inhibitor of the norepinephrine transporter (NET) in vitro and in vivo (IC50 ~ 36 μM); (Madras et al., 2006). However the in vivo assessment of this interaction was limited as data from only one animal was available for the most effective modafinil dose (Madras et al., 2006). While many neurotransmitters are involved in producing and maintaining arousal states, these transporters remain the only known biochemical targets of modafinil. Thus, the focus of these experiments was on the catecholaminergic systems and their interaction.
A novel tool for studying the relative roles of NE and DA in behavioral phenotypes are dopamine β-hydroxylase knockout (Dbh −/−) mice. These mice completely lack NE and epinephrine (EPI), but show a compensatory increase in high-affinity state DA receptors and are hypersensitive to the behavioral effects of both direct (e.g. quinpirole) and indirect (e.g. amphetamine, cocaine) DA agonists (Schank et al., 2006; Weinshenker et al., 2002b). Previous studies examining arousal behaviorally or by electroencephalgram (EEG) have revealed that exploratory activity in a novel environment, latency to sleep following handling or exposure to environmental stimuli, and wake bout duration were attenuated in Dbh −/− mice, suggesting that NE is important for maintaining vigilance (Hunsley and Palmiter, 2003, 2004). Thus, Dbh −/− mice were used to explore the role of catecholamines in modafinil-induced arousal.
It was hypothesized that if modafinil acts primarily via a noradrenergic mechanism, Dbh −/− mice should be non-responsive since they completely lack NE. In contrast, if modafinil acts mainly through DA systems, these mice should be hypersensitive. Modafinil was tested in Dbh −/− mice using both locomotor and sleep latency paradigms as behavioral readouts, and NE-DA interactions were further explored by examining the effect of NE and DA receptor antagonist pretreatments.
2. Methods
2.1 Animals and housing
Dbh −/− mice, maintained on a mixed 129/SvEv and C57BL/6J background, were developed and generated as described (Thomas et al., 1995, 1998). Dbh −/− males were bred to Dbh +/− females. Pregnant Dbh +/− mice were given the AR agonists, isoproterenol and phenylephrine (20 μg/ml each) + vitamin C (2 mg/ml) from E9.5–E14.5, and L-3,4-dihydroxyphenylserine (DOPS; 2 mg/ml + vitamin C 2 mg/ml) from E14.5-birth in their drinking water to rescue the embryonic lethality associated with the homozygous Dbh −/− mutation. Because of this treatment, NE and EPI were present in Dbh −/− animals before but not after birth. Dbh −/− mice were identified by the delayed growth and ptosis phenotypes, which are 100% correlated with the Dbh −/− genotype. Genotypes were confirmed by PCR. Dbh +/− mice were used as controls as they had normal catecholamine levels and were indistinguishable from Dbh +/+ mice for all previously tested phenotypes, including locomotor activity (Bourdelat-Parks et al., 2005; Mitchell et al., 2006; Thomas et al., 1998; Thomas et al., 1995). Male and female mice aged 2–8 months were used in all experiments. No age or gender differences were found, and results were combined. All mice were reared in a specific pathogen-free facility with a 12 h light/dark cycle (lights on - 7 am; lights off - 7 pm). Food and water were available ad libitum except during behavioral testing. All experiments were carried out in a quiet, isolated behavior room between 8:00 and 15:00h. Mice were moved to this room at least 24 h before testing. Experimental protocols were approved by the Emory University IACUC and meet the guidelines of the American Association for Accreditation of Laboratory Animal Care.
2.2 Compounds
Modafinil (Cephalon, Inc., West Chester, PA) and the α1AR antagonist prazosin (0.5 mg/kg; Sigma-Aldrich, St. Louis, MO) were prepared by dissolving the compounds in warm 0.9% saline, 1.5% DMSO, and 1.5% cremophor EL. A 90 mg/kg dose of modafinil was reported to have identical effects in Dbh −/− and −/− control mice (Hunsley and Palmiter, 2004). Because Dbh −/− mice have altered responses to other stimulants that are most apparent at low to moderate doses (Weinshenker et al., 2002a; Schank et al., 2006), lower doses of modafinil (6.25 – 50 mg/kg) were selected for these experiments. The dose of prazosin was selected based on published reports that it can block the behavioral effects of stimulants in mice (Drouin et al., 2001; Weinshenker et al., 2002a). The non-selective DA receptor antagonist, cis-(Z)-flupenthixol dihydrochloride (0.025 or 0.25 mg/kg; Sigma-Aldrich) was dissolved in 0.9% saline. These doses of flupenthixol were selected based on motor behavior dose-response experiments (Rommelfanger et al., 2007). All compounds were administered i.p. in a volume of 10 ml/kg.
2.3 Sleep latency
Dbh +/− and Dbh −/− mice were individually housed in large plexiglass cages and allowed to acclimate for 4 h. Vehicle or modafinil (6.25, 12.5, or 25 mg/kg) was then administered, and mice were observed by a trained experimenter for behavioral signs of sleep. During sleep, mice exhibit a distinctive posture and breathing pattern that allows the observer to determine onset. Sleep was defined as 2 min of uninterrupted sleep behavior, and 75% of the next 10 min spent asleep (Hunsley and Palmiter, 2004). This behavioral scoring paradigm has been shown to reliably correlate with onset of sleep using EEG measurements (Hunsley and Palmiter, 2003, 2004). In a subset of animals, saline or the DA receptor antagonist, flupenthixol, (0.25 mg/kg) was administered 30 min prior to modafinil (25 mg/kg) administration. This dose of modafinil was used for the antagonist experiment because it was the only dose tested that significantly increased sleep latency in both control and Dbh −/− mice.
2.4 Locomotor activity
Locomotor activity was assessed using an automated system (San Diego Instruments, La Jolla, CA, USA) with photobeams that recorded ambulations (consecutive beam breaks). Mice were placed individually in the chambers and allowed to acclimate for 4 h, and were then administered vehicle or modafinil (6.25, 12.5, 25, or 50 mg/kg). Activity was recorded for an additional 2 h. This time frame was selected because by 2 h, locomotor activity had tapered off and was approaching baseline levels. In order to examine the effects of receptor antagonist pretreatment, vehicle, the α1AR antagonist prazosin (0.5 mg/kg), or the DA receptor antagonist, flupenthixol (0.025 or 0.25 mg/kg), were injected 30 min prior to modafinil (50 mg/kg) administration. This dose of modafinil was used because it produced comparable amounts of locomotor activity in control and Dbh −/− mice, thus making antagonist effects easier to compare between genotypes. All data were presented as total ambulations for the 2h following modafinil administration. To assess the effects of flupenthixol on exploratory activity in a novel environment, Dbh +/− and Dbh −/− mice were administered flupenthixol (0.25 mg/kg). Thirty minutes following injection, mice were placed in the locomotor chambers, and their activity was recorded for 2 h.
2.5 Statistical analysis
All data is presented as mean ± standard error of the mean. Student’s t-tests were used when comparing 2 groups with equal variance, Mann-Whitney tests were used when comparing 2 groups with unequal variance, and two-way ANOVA followed by Bonferroni post hoc tests were used when comparing more than 2 groups. ANOVA analysis assumes normally distributed data, and in two cases (Fig. 1, Fig. 2), the data was not normally distributed. Natural log transformation was performed, which resulted in normally distributed data, and statistics were conducted on the transformed data. Statistical analysis was conducted using Graphpad™ Prism 4.0c for Macintosh (San Diego, CA).
Fig. 1.
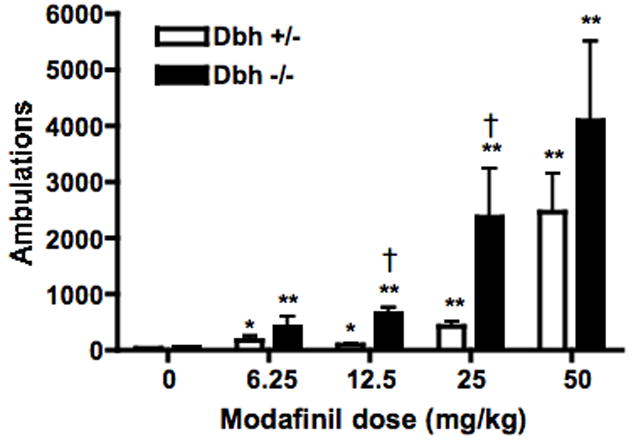
Effect of modafinil on locomotor behavior. Dbh +/− and Dbh −/− mice were placed in locomotor activity chambers. Four hours later, mice were injected with vehicle or modafinil (6.25, 12.5, 25, or 50 mg/kg, i.p.), and ambulations (consecutive beam breaks) were recorded for an additional 2 hours. Shown are the total ambulations for the two hours following modafinil administration. All data is presented as mean ± SEM. * p < 0.01, ** P < 0.001, compared to vehicle control for that genotype. † p < 0.001 compared to Dbh +/− mice for that dose.
Fig. 2.
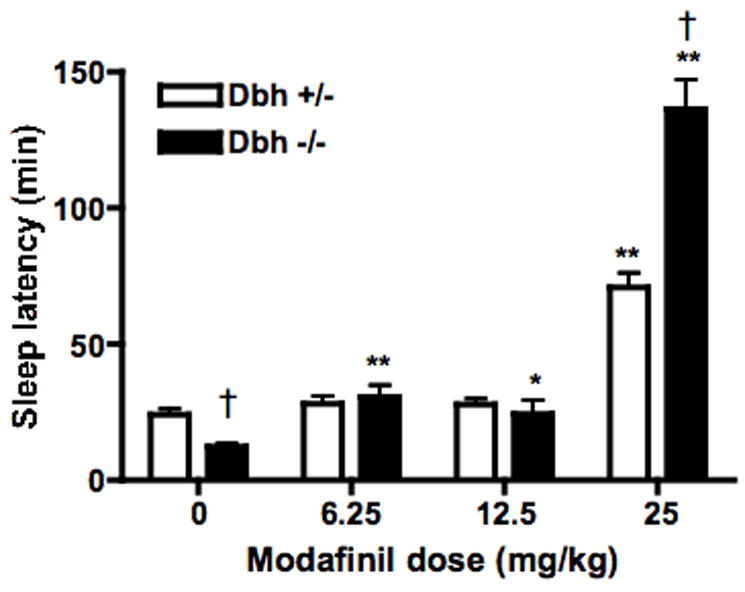
Effect of modafinil on sleep latency. Dbh +/− and Dbh −/− mice were placed in observation chambers, injected with vehicle or modafinil (6.25, 12.5, or 25 mg/kg, i.p.) 4 hours later, and observed until the onset of sleep. Shown is latency to sleep following the injection. * P < 0.05, ** P < 0.001 compared to vehicle control for that genotype. † P < 0.01 compared to Dbh +/− mice for that dose.
3. Results
3.1 Dbh −/− mice are hypersensitive to modafinil-induced locomotion and wake
Modafinil dose-dependently increased locomotor activity in both control (Dbh +/−) and Dbh −/− mice (Fig. 1). There was a main effect of modafinil dose (F(4,67) = 65.08, p < 0.0001), genotype (F(1,67) = 38.21, p < 0.0001), and a dose × genotype interaction (F(4,67) = 3.87, p < 0.007). Post hoc tests revealed a significant response to modafinil compared to vehicle in both Dbh −/− and control mice at all doses tested. Dbh −/− mice tended to have a greater response at all doses, and the genotype difference was significant at the 12.5 and 25 mg/kg doses (Fig. 1).
The results of the sleep latency experiments mirrored those of the locomotor activity experiments. Vehicle-treated Dbh −/− mice had a shorter sleep latency than vehicle-treated Dbh +/− mice, as previously reported (Hunsley and Palmiter, 2003) (Fig. 2). Modafinil dose-dependently increased sleep latency in both Dbh +/− and Dbh −/− mice, and Dbh −/− mice were hypersensitive to the wake-promoting effects of modafinil (Fig. 2). There was a significant effect of dose (F(3,66) = 65.53, p < 0.0001) and a dose × genotype interaction (F(3,66) = 8.800, p < 0.0001). All doses of modafinil tested significantly increased sleep latency in Dbh −/− mice, whereas only the highest dose tested (25 mg/kg) increased sleep latency in Dbh +/− mice. Furthermore, the highest dose of modafinil produced significantly longer sleep latency in Dbh −/− mice compared Dbh +/− mice.
3.2 Blockade of α1AR or DA receptors attenuates modafinil-induced locomotor activity and wake
Antagonists of α1ARs can attenuate the locomotor-activating and wake-promoting effects of modafinil in rodents and non-human primates (Duteil et al., 1990; Hermant et al., 1991; Lin et al., 1992). Consistent with these results, pretreatment of control (Dbh +/−) mice with the α1AR antagonist, prazosin (0.5 mg/kg), attenuated modafinil-induced (50 mg/kg) locomotor activity (Fig. 3). In contrast, prazosin pretreatment had no effect on modafinil-induced locomotor activity in Dbh −/− mice (Fig. 3).
Fig 3.
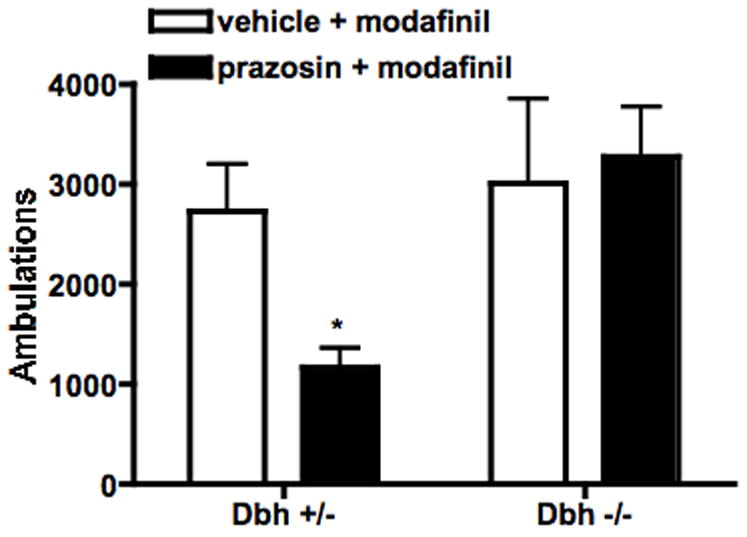
Effect of α1AR blockade on modafinil-induced locomotion. Dbh +/− and Dbh −/− mice were placed in locomotor activity chambers and injected with vehicle or prazosin (0.5 mg/kg, i.p.) 3.5 hours later. Thirty minutes following the pretreatment, mice were injected with modafinil (50 mg/kg, i.p.), and ambulations (consecutive beam breaks) were recorded for an additional 2 hours. Shown are the total ambulations for the 2 hours after modafinil administration. * P < 0.05 compared to vehicle control for that genotype.
To determine whether DA signaling was critical for the effects of modafinil, mice were pretreated with the non-selective DA receptor antagonist, flupenthixol (0.025 or 0.25 mg/kg), 30 min prior to modafinil (50 mg/kg for locomotor activity, 25 mg/kg for sleep latency). While the lower dose of flupenthixol had no effect (data not shown), the higher dose decreased modafinil-induced locomotor activity in both Dbh +/− and Dbh −/− mice (Fig. 4). This dose of flupenthixol decreased exploratory locomotor activity (main effect of treatment; F(1,28) = 7.74, p < 0.01), but to a lesser extent than modafinil-induced locomotor activity (49% and 46% decrease in exploratory activity in Dbh +/− and Dbh −/− mice versus 76% and 83% decrease in modafinil-induced activity) (Fig. 5). Flupenthixol also attenuated modafinil-induced wake in mice of both genotypes (Fig. 6).
Fig. 4.
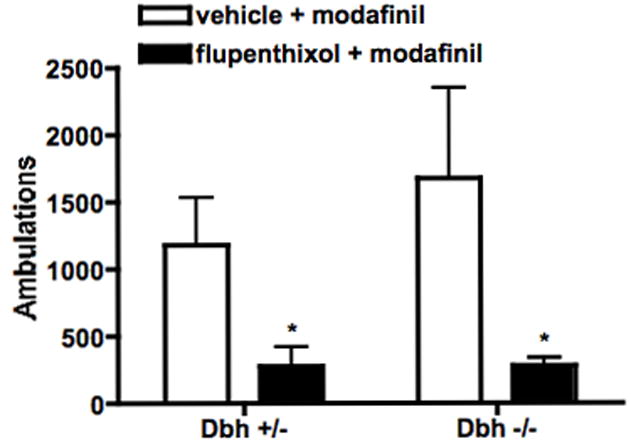
Effect of DA receptor blockade on modafinil-induced locomotion. Dbh +/− and Dbh −/− mice were placed in locomotor activity chambers and injected with vehicle or flupenthixol (0.25 mg/kg, i.p.) 3.5 hours later. Thirty minutes following the pretreatment, mice were injected with modafinil (50 mg/kg, i.p.), and ambulations (consecutive beam breaks) were recorded for an additional 2 hours. Shown are the total ambulations for the two hours after modafinil administration. * P < 0.05 compared to vehicle control for that genotype.
Fig. 5.
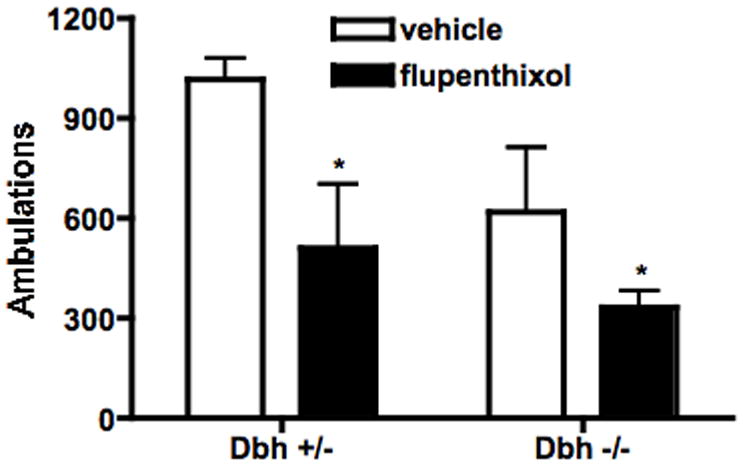
Effect of DA receptor blockade on locomotor behavior in a novel environment. Dbh +/− and Dbh −/− mice were injected with flupenthixol (0.25 mg/kg, i.p.), placed in locomotor activity chambers 30 minutes later, and ambulations (consecutive beam breaks) were recorded for an additional 2 hours. Shown are total ambulations for the two hours after being placed in the chambers. * P < 0.05 compared to vehicle control for that genotype.
Fig. 6.
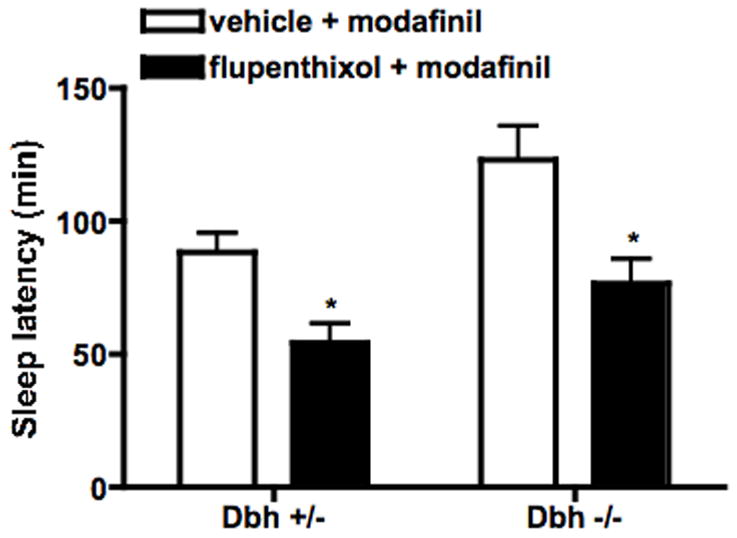
Effect of DA receptor blockade on sleep latency in modafinil-treated mice. Dbh +/− and Dbh −/− mice were placed in observation chambers, and injected with vehicle or flupenthixol (0.25 mg/kg, i.p.) 3.5 hours later. Thirty minutes following the pretreatment, mice were injected with modafinil (25 mg/kg, i.p.) and observed until the onset of sleep. Shown is latency to sleep following modafinil injection. * P < 0.05 compared to vehicle control for that genotype.
4. Discussion
Atlhough the effects of modafinil appear to involve both NE and DA, the exact contribution of these two monoamines to the mechanism of modafinil action remains unclear. Dbh −/− mice completely lack NE but have hypersensitive DA signaling. Thus, it was hypothesized that if modafinil acts primarily via NE, then the behavioral effects of modafinil would be attenuated in these mice, while if modafinil acts primarily via DA, Dbh −/− mice would be hypersensitive. As Dbh −/− mice were hypersensitive to both the wake-promoting and locomotor activating effects of modafinil, it is tempting to conclude that modafinil acts primarily via the dopaminergic system. However, this finding requires reconciliation with the reported effects of α1AR antagonists in attenuating the effects of modafinil. Lesioning of the locus coeruleus (LC), the major brainstem noradrenergic nucleus, using the selective noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4), had no effect on modafinil-induced wake behavior (Wisor and Eriksson, 2005). Because α1AR blockade attenuated the effects of modafinil in both intact and LC-lesioned animals, these authors proposed that modafinil acts by blocking DAT and increasing extracellular DA, which then directly stimulates α1ARs to promote wake (Wisor and Eriksson, 2005). However, there are a number of caveats to this model. Firstly, the potency of DA at cloned α1ARs is approximately 100-fold lower than that of NE (Zhang et al., 2004). Secondly, DSP-4 does not completely eliminate LC neurons, and in fact leaves ventral brainstem adrenergic and noradrenergic nuclei (e.g. A1, A2, C1, C2) intact (Fritschy and Grzanna, 1991). This is an important point, as projections from these nuclei provide the majority of the noradrenergic innervation to dopaminergic areas (i.e., ventral tegmental area, nucleus accumbens, periaquaductal grey (PAG)) and supplies NE and EPI to the hypothalamus (Jones, et al., 1977; Woulfe, et al., 1990; Delfs, et al., 1998) which is a likely site of the wake-promoting effects of modafinil (Delfs et al., 1998; Engber et al., 1998a; Jones et al., 1977; Lin et al., 1996; Scammell et al., 2000; Woulfe et al., 1990).
This hypothesis was tested in the present study by examining the effects of the α1AR antagonist, prazosin, in Dbh −/− mice. If modafinil acts by facilitating the ability of DA to directly stimulate α1ARs, then blocking α1ARs should attenuate the behavioral effects of modafinil whether or not NE is present. However, while prazosin attenuated modafinil-induced locomotor activity in control mice, it failed to do so in Dbh −/− mice. In contrast, the DA receptor antagonist, flupenthixol, attenuated the effects of modafinil in both control and Dbh −/− mice. Thus an alternate mechanism for modafinil-induced arousal may be proposed that partially depends on NE-DA interactions (Fig. 7, “right” pathway). The noradrenergic system provides excitatory drive onto DA neurons via α1ARs, which are critical for DA release and responses to dopaminergic drugs like psychostimulants (Weinshenker and Schroeder, 2007). This is consistent with the hypothesis that modafinil produces its behavioral effects via weak blockade of both DAT and NET (Gallopin et al., 2004; Madras et al., 2006). NET blockade increases extracellular NE, which in turn activates α1ARs and promotes the firing of DA neurons and DA release. DAT blockade prevents the reuptake of the released DA, which then promotes the behavioral effects of modafinil by activating DA receptors. NET blockade also increases NE in other brain regions involved in sleep-wake regulation, such as the hypothalamus (Fig. 7, “left” pathway). Although Dbh −/− mice lack NE, they can bypass the requirement for α1AR stimulation because of hypersensitive DA receptors.
Fig. 7.
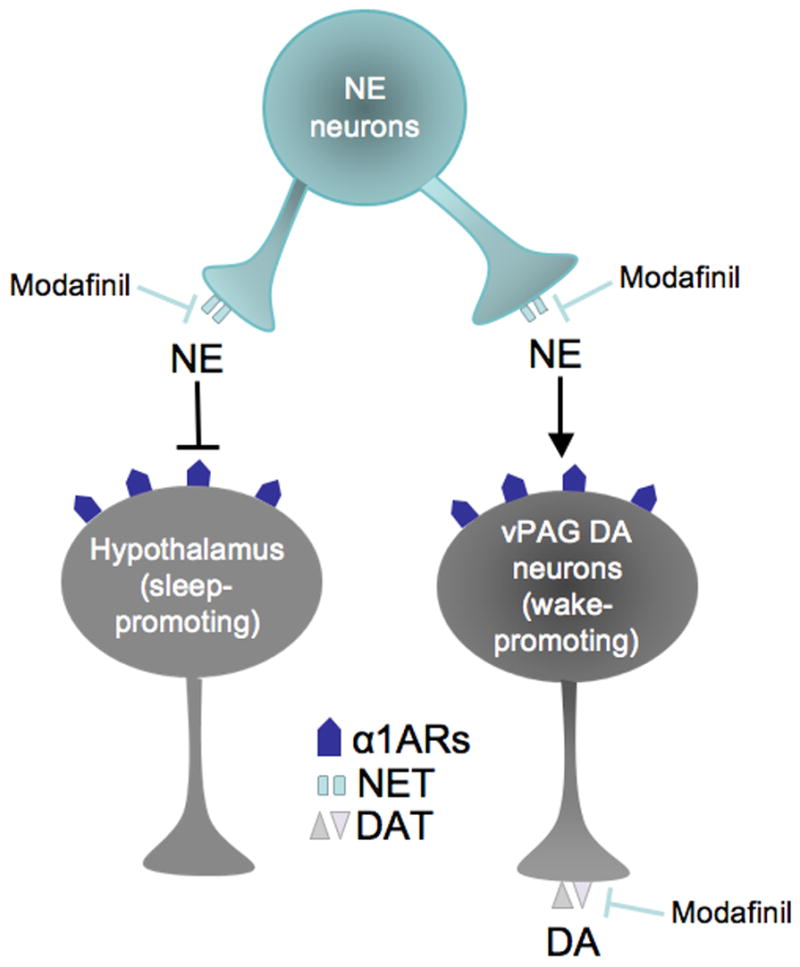
A hypothetical parallel pathway wiring diagram for modafinil-induced arousal. Modafinil blocks NET and DAT. In the wake-promoting pathway (right), the increased extracellular NE signals via α1ARs to activate wake-promoting DA neurons in the ventral periaqueductal gray (vPAG). The DAT blockade prevents the uptake of the released DA, thus facilitating DA transmission in vPAG projection areas. Simultaneously, NE inhibits sleep-promoting neurons in the hypothalamus (and perhaps other brain regions). Arrow to DA neurons signifies excitation, and bar to hypothalamic neurons signifies inhibition. NE, norepinephrine; DA, dopamine; α1ARs, α1-adrenergic receptors; NET, norepinephrine transporter; DAT, dopamine transporter.
Blockade of DAT and NET by modafinil
Early studies showed that modafinil interacts with DAT with low affinity (IC50 ~ 2–6 μM), but had no effect on NET (Mignot et al., 1994). However, subsequent studies showed that modafinil increased extracellular levels of both DA and NE in vivo, and in fact only NE was elevated in the hypothalamus (de Saint Hilaire et al., 2001). Modafinil was reported to inhibit catecholamine uptake via cloned human DAT and NET in human embryonic kidney cells, and displaced both DAT and NET PET ligands from primate brain in vivo (Madras et al., 2006) although there are concerns about the robustness of this effect of modafinil on NET as in vivo data were only reported for one animal. In an in vitro study, modafinil suppressed the activity of sleep-promoting neurons in the hypothalamus in a NE-dependent manner, and its effects were mimicked by the selective NET blocker, nisoxetine (Gallopin et al., 2004). These results support an integral part of the present model, that modafinil blocks DAT and NET and increases extracellular DA and NE. Moreover, modafinil has several similarities with bupropion, an existing NET/DAT blocker used both as an antidepressant as an anti-smoking pharmacotherapy (Wilkes, 2006). Bupropion increases locomotor activity in rodents (e.g. Mitchell et al., 2006) and wake in narcoleptic dogs (Nishino et al., 2006), and a common side effect of bupropion therapy is insomnia (e.g. Wilkes, 2006). Future studies directly comparing the wake-promoting effects of modafinil and bupropion might yield interesting information, as specific affinities for each drug at DAT and NET could underlie differences in clinical efficacy. Validation of our model will require a more complete understanding of modafinil-transporter affinities and interactions across species.
Activation of α1ARs receptors is involved in modafinil-induced arousal
It has previously been reported that modafinil-induced arousal is reduced by α1AR antagonists and in α1b knockout mice (Duteil et al., 1990; Hermant et al., 1991; Lin et al., 1992; Wisor and Eriksson, 2005; Wisor et al., 2001), and this was confirmed in our experiment with prazosin in control mice (Fig. 3). However, prazosin failed to block modafinil-induced locomotion in Dbh −/− mice. This result is reminiscent of the pattern of response to typical dopaminergic agents; Dbh −/− mice have an increase in high affinity-state DA receptors and are hypersensitive to amphetamine, cocaine, and direct DA agonists, and these behavioral responses cannot be blocked by α1AR antagonists as they are in control mice (Schank et al., 2006; Weinshenker et al., 2002b). Thus, α1AR signaling appears critical for modafinil-induced arousal in normal animals via its effect on DA neuron activity and DA release, but the requirement for NE can be bypassed by hypersensitive DA receptors. We believe this is because NE, acting primarily via α1ARs, can provide excitatory drive onto midbrain DA neurons and facilitate DA release (Weinshenker and Schroeder, 2007). This supports the idea that the activation of α1ARs is important for DA transmission, as proposed in our model (Fig. 7, “right” pathway). These results suggest that the increase in extracellular NE following modafinil administration promotes arousal by activating α1ARs and facilitating DA transmission (Fig. 7, “right” pathway).
Activation of DA receptors is involved in modafinil-induced arousal
Early data indicated that DA antagonists could not block the behavioral effects of modafinil. For example, it was reported that D1 and D2 antagonists failed to prevent modafinil-induced locomotor activity in rodents (Duteil et al., 1990; Simon et al., 1995). However, additional examination of the actual data indicates that DA antagonists can attenuate modafinil responses under certain conditions. Both the D1 antagonist, SCH23390 (Simon et al., 1995) and the D2 antagonist, haloperidol (Duteil et al., 1990; Simon et al., 1995) partially inhibited modafinil-induced locomotor activity. It was argued that the antagonists did not suppress modafinil-induced locomotor activity to a greater extent than baseline locomotor activity. An additional caveat was that neither of these studies simultaneously examined D1 and D2 inhibition or examined the effects of DA antagonists on the wake-promoting effects of modafinil. More recently, it was shown that activation of D2 autoreceptors with quinpirole, which can inhibit DA release, attenuated the wake-promoting effects of modafinil (Wisor and Eriksson, 2005). In the present study, the D1/D2 antagonist flupenthixol had a larger suppressive effect on modafinil-induced locomotor activity than it did on exploratory activity. Furthermore, when the confound of DA antagonist effects on locomotor activity were controlled for by examining sleep latency, flupenthixol still attenuated the effects of modafinil. The fact that flupenthixol was effective in Dbh −/− mice indicates that the DA antagonist was acting downstream of NE signaling. These results are consistent with the final part of the proposed model, that the increased extracellular DA (brought about by dual DAT blockade and α1AR activation) promotes arousal by activating DA receptors. Although DA is not traditionally thought of as a regulator of the sleep-wake cycle, recent evidence indicates that DA can influence sleep states and promote wake (Berridge, 2006; Dzirasa et al., 2006; Isaac and Berridge, 2003; Lu et al., 2006; Wisor et al., 2001).
Anatomical correlates of the model
Where in the brain might these noradrenergic-dopaminergic interactions be occurring? Lu and colleagues recently identified a population of wake-active DA neurons in the ventral periaqueductal gray (vPAG) that receive noradrenergic and adrenergic innervation (Lu et al., 2006). α1ARs are expressed in the vPAG (Jones et al., 1985; Pieribone et al., 1994), and α1AR agonists depolarize nearly all vPAG neurons (Vaughan et al., 1996). Thus, modafinil may increase NE in the vPAG and activate the wake-promoting DA neurons, which innervate other brain regions implicated in arousal such as the hypothalamus and prefrontal cortex. DAT blockade by modafinil in these regions could further amplify the wake-promoting signal (Fig. 7, “right” pathway).
Limitations of the model
The primary caveat of our model is that we do not know the extent of direct NET blockade by modafinil in vivo. Modafinil can elevate extracellular NE in rats, and its electrophysiological effects can be mimicked by a selective NET blocker in brain slices (de Saint Hilaire et al., 2001; Gallopin et al., 2004). However, there are conflicting data sets on in vitro NET blockade (Madras et al., 2006; Mignot et al., 1994), and only one report of in vivo NET blockade (measured in a single monkey; (Madras et al., 2006)), and species differences may exist (our unpublished data). It is also possible that modafinil cannot block NET in vivo at physiological doses but can increase extracellular NE via indirect pathways.
Although the proposed model can explain many of the previous findings on the role of catecholamines in modafinil-induced arousal, it cannot explain all of them. Most prominently, it fails to account for some of the observed behavioral, neurochemical and molecular differences between modafinil and typical psychostimulants like amphetamine. For example, while Dbh −/− mice are hypersensitive to all doses of modafinil and high doses of amphetamine, they are actually resistant to the wake-promoting effects of low amphetamine doses (Hunsley and Palmiter, 2003). Canonical psychostimulants induce robust c-Fos expression in the striatum, while modafinil may not (Engber et al., 1998b; Lin et al., 1996). Furthermore, modafinil increased extracellular NE, but not DA, in the hypothalamus, which appears to be an important site of action for modafinil (de Saint Hilaire et al., 2001). Finally, modafinil suppressed sleep-promoting neurons in the hypothalamus in a NE-dependent manner, and its effects were mimicked by the selective NET blocker, nisoxetine (Gallopin et al., 2004). These data are consistent with previous studies indicating that NE potently increased wake via α1ARs in the hypothalamus and other brain regions (Berridge et al., 2003; Berridge and O’Neill, 2001). Taken together, these results indicate that NE plays a dual role in modafinil-induced arousal. Firstly, acting via α1ARs it facilitates DA transmission and promotes wake. Secondly, there appears to be a noradrenergic component of modafinil action that is independent of its effects on DA transmission and may involve suppression of sleep-promoting neurons in the hypothalamus. Further experiments will be needed to confirm various aspects of this model. There is also growing evidence that histamine release is an important mediator of modafinil-induced wakefulness. Because NE and histamine can reciprocally facilitate each other’s release, it is possible that a positive feedback loop exists between these two neurotransmitters (Bealer, 1993; Prast et al., 1991). Thus, histamine may be acting in conjunction with the catecholamines to produce modafinil-induced arousal, but whether it occurs in parallel to or downstream of DAT/NET blockade remains to be elucidated (Ishizuka et al., 2003; Lin et al., 2008; Minzenberg and Carter, 2008).
Acknowledgments
We thank Sumitomo Pharmaceuticals (Osaka, Japan) for generous donation of the DOPS needed to maintain our Dbh mouse breeding program. This study was supported in part by Cephalon, Inc.
Footnotes
Disclosure/conflict of interest
D. Bozyczko-Coyne and M. Williams are employees of Cephalon, Inc., and provided research funds for this study and partial support of D. Weinshenker and H. Mitchell.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ballon JS, Feifel D. A systematic review of modafinil: Potential clinical uses and mechanisms of action. J Clin Psychiatry. 2006;67:554–66. doi: 10.4088/jcp.v67n0406. [DOI] [PubMed] [Google Scholar]
- Bealer SL. Histamine releases norepinephrine in the paraventricular nucleus/anterior hypothalamus of the conscious rat. J Pharmacol Exp Ther. 1993;264:734–8. [PubMed] [Google Scholar]
- Berridge CW. Neural substrates of psychostimulant-induced arousal. Neuropsychopharmacology. 2006;31:2332–40. doi: 10.1038/sj.npp.1301159. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Isaac SO, Espana RA. Additive wake-promoting actions of medial basal forebrain noradrenergic alpha1- and beta-receptor stimulation. Behav Neurosci. 2003;117:350–9. doi: 10.1037/0735-7044.117.2.350. [DOI] [PubMed] [Google Scholar]
- Berridge CW, O’Neill J. Differential sensitivity to the wake-promoting actions of norepinephrine within the medial preoptic area and the substantia innominata. Behav Neurosci. 2001;115:165–74. doi: 10.1037/0735-7044.115.1.165. [DOI] [PubMed] [Google Scholar]
- Bourdelat-Parks BN, Anderson GM, Donaldson ZR, Weiss JM, Bonsall RW, Emery MS, Liles LC, Weinshenker D. Effects of dopamine beta-hydroxylase genotype and disulfiram inhibition on catecholamine homeostasis in mice. Psychopharmacology (Berl) 2005;183:72–80. doi: 10.1007/s00213-005-0139-8. [DOI] [PubMed] [Google Scholar]
- Colten HR, Altevogt B IoMotNA . Committee on Sleep Medicine and Research. Washington, D.C.: The National Academies Press; 2006. Sleep disorders and sleep deprivation: an unmet public health problem. [PubMed] [Google Scholar]
- de Saint Hilaire Z, Orosco M, Rouch C, Blanc G, Nicolaidis S. Variations in extracellular monoamines in the prefrontal cortex and medial hypothalamus after modafinil administration: a microdialysis study in rats. Neuroreport. 2001;12:3533–7. doi: 10.1097/00001756-200111160-00032. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS. Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res. 1998;806:127–40. doi: 10.1016/s0006-8993(98)00672-6. [DOI] [PubMed] [Google Scholar]
- Drouin C, Blanc G, Trovero F, Glowinski J, Tassin JP. Cortical alpha 1-adrenergic regulation of acute and sensitized morphine locomotor effects. Neuroreport. 2001;12:3483–6. doi: 10.1097/00001756-200111160-00022. [DOI] [PubMed] [Google Scholar]
- Duteil J, Rambert FA, Pessonnier J, Hermant JF, Gombert R, Assous E. Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur J Pharmacol. 1990;180:49–58. doi: 10.1016/0014-2999(90)90591-s. [DOI] [PubMed] [Google Scholar]
- Dzirasa K, Ribeiro S, Costa R, Santos LM, Lin SC, Grosmark A, Sotnikova TD, Gainetdinov RR, Caron MG, Nicolelis MA. Dopaminergic control of sleep-wake states. J Neurosci. 2006;26:10577–89. doi: 10.1523/JNEUROSCI.1767-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar DM, Seidel WF. Modafinil induces wakefulness without intensifying motor activity or subsequent rebound hypersomnolence in the rat. J Pharmacol Exp Ther. 1997;283:757–69. [PubMed] [Google Scholar]
- Engber TM, Dennis SA, Jones BE, Miller MS, Contreras PC. Brain regional substrates for the actions of the novel wake-promoting agent modafinil in the rat: comparison with amphetamine. Neuroscience. 1998a;87:905–11. doi: 10.1016/s0306-4522(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Engber TM, Koury EJ, Dennis SA, Miller MS, Contreras PC, Bhat RV. Differential patterns of regional c-Fos induction in the rat brain by amphetamine and the novel wakefulness-promoting agent modafinil. Neurosci Lett. 1998b;241:95–8. doi: 10.1016/s0304-3940(97)00962-2. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Grzanna R. Experimentally-induced neuron loss in the locus coeruleus of adult rats. Exp Neurol. 1991;111:123–7. doi: 10.1016/0014-4886(91)90058-k. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P. Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep. 2004;27:19–25. [PubMed] [Google Scholar]
- Hermant JF, Rambert FA, Duteil J. Awakening properties of modafinil: effect on nocturnal activity in monkeys (Macaca mulatta) after acute and repeated administration. Psychopharmacology (Berl) 1991;103:28–32. doi: 10.1007/BF02244069. [DOI] [PubMed] [Google Scholar]
- Hunsley MS, Palmiter RD. Norepinephrine-deficient mice exhibit normal sleep-wake states but have shorter sleep latency after mild stress and low doses of amphetamine. Sleep. 2003;26:521–6. [PubMed] [Google Scholar]
- Hunsley MS, Palmiter RD. Altered sleep latency and arousal regulation in mice lacking norepinephrine. Pharmacol Biochem Behav. 2004;78:765–73. doi: 10.1016/j.pbb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Isaac SO, Berridge CW. Wake-promoting actions of dopamine D1 and D2 receptor stimulation. J Pharmacol Exp Ther. 2003;307:386–94. doi: 10.1124/jpet.103.053918. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Sakamoto Y, Sakurai T, Yamatodani A. Modafinil increases histamine release in the anterior hypothalamus of rats. Neurosci Lett. 2003;339:143–6. doi: 10.1016/s0304-3940(03)00006-5. [DOI] [PubMed] [Google Scholar]
- Jones BE, Halaris AE, McIlhany M, Moore RY. Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurons. Brain Res. 1977;127:1–21. doi: 10.1016/0006-8993(77)90377-8. [DOI] [PubMed] [Google Scholar]
- Jones LS, Gauger LL, Davis JN. Anatomy of brain alpha 1-adrenergic receptors: in vitro autoradiography with [125I]-heat. J Comp Neurol. 1985;231:190–208. doi: 10.1002/cne.902310207. [DOI] [PubMed] [Google Scholar]
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, Perrin D, Robert P, Roux M, Lecomte JM, Schwartz JC. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin−/− mice and patients. Neurobiol Dis. 2008;30:74–83. doi: 10.1016/j.nbd.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Lin JS, Hou Y, Jouvet M. Potential brain neuronal targets for amphetamine-, methylphenidate-, and modafinil-induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci U S A. 1996;93:14128–33. doi: 10.1073/pnas.93.24.14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Roussel B, Akaoka H, Fort P, Debilly G, Jouvet M. Role of catecholamines in the modafinil and amphetamine induced wakefulness, a comparative pharmacological study in the cat. Brain Res. 1992;591:319–26. doi: 10.1016/0006-8993(92)91713-o. [DOI] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madras BK, Xie Z, Lin Z, Jassen A, Panas H, Lynch L, Johnson R, Livni E, Spencer TJ, Bonab AA, Miller GM, Fischman AJ. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther. 2006;319:561–9. doi: 10.1124/jpet.106.106583. [DOI] [PubMed] [Google Scholar]
- Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep. 1994;17:436–7. doi: 10.1093/sleep/17.5.436. [DOI] [PubMed] [Google Scholar]
- Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology. 2008;33:1477–502. doi: 10.1038/sj.npp.1301534. [DOI] [PubMed] [Google Scholar]
- Mitchell HA, Ahern TH, Liles LC, Javors MA, Weinshenker D. The effects of norepinephrine transporter inactivation on locomotor activity in mice. Biol Psychiatry. 2006;60:1046–52. doi: 10.1016/j.biopsych.2006.03.057. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Nicholas AP, Dagerlind A, Hokfelt T. Distribution of alpha 1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci. 1994;14:4252–68. doi: 10.1523/JNEUROSCI.14-07-04252.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prast H, Heistracher M, Philippu A. In vivo modulation of the histamine release in the hypothalamus by adrenoreceptor agonists and antagonists. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:183–6. doi: 10.1007/BF00167216. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, Miller GW, Weinshenker D. Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc Natl Acad Sci U S A. 2007;104:13804–9. doi: 10.1073/pnas.0702753104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Estabrooke IV, McCarthy MT, Chemelli RM, Yanagisawa M, Miller MS, Saper CB. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci. 2000;20:8620–8. doi: 10.1523/JNEUROSCI.20-22-08620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD, Liles LC, Seeman P, Weinshenker D. Dopamine beta-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacology. 2006;31:2221–30. doi: 10.1038/sj.npp.1301000. [DOI] [PubMed] [Google Scholar]
- Simon P, Hemet C, Ramassamy C, Costentin J. Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol. 1995;5:509–14. doi: 10.1016/0924-977x(95)00041-m. [DOI] [PubMed] [Google Scholar]
- Stone EA, Cotecchia S, Lin Y, Quartermain D. Role of brain alpha 1B-adrenoceptors in modafinil-induced behavioral activity. Synapse. 2002;46:269–70. doi: 10.1002/syn.10127. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem. 1998;70:2468–76. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–6. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Bandler R, Christie MJ. Differential responses of lateral and ventrolateral rat periaqueductal grey neurones to noradrenaline in vitro. J Physiol. 1996;490(Pt 2):373–81. doi: 10.1113/jphysiol.1996.sp021151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Miller NS, Blizinsky K, Laughlin ML, Palmiter RD. Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. Proc Natl Acad Sci U S A. 2002a;99:13873–7. doi: 10.1073/pnas.212519999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Schroeder JP. There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology. 2007;32:1433–51. doi: 10.1038/sj.npp.1301263. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, White SS, Javors MA, Palmiter RD, Szot P. Regulation of norepinephrine transporter abundance by catecholamines and desipramine in vivo. Brain Res. 2002b;946:239–46. doi: 10.1016/s0006-8993(02)02889-5. [DOI] [PubMed] [Google Scholar]
- Wilkes S. Bupropion. Drugs Today (Barc) 2006;42:671–81. doi: 10.1358/dot.2006.42.10.1025701. [DOI] [PubMed] [Google Scholar]
- Wisor JP, Eriksson KS. Dopaminergic-adrenergic interactions in the wake promoting mechanism of modafinil. Neuroscience. 2005;132:1027–34. doi: 10.1016/j.neuroscience.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci. 2001;21:1787–94. doi: 10.1523/JNEUROSCI.21-05-01787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woulfe JM, Flumerfelt BA, Hrycyshyn AW. Efferent connections of the A1 noradrenergic cell group: a DBH immunohistochemical and PHA-L anterograde tracing study. Exp Neurol. 1990;109:308–22. doi: 10.1016/s0014-4886(05)80022-6. [DOI] [PubMed] [Google Scholar]
- Zhang WP, Ouyang M, Thomas SA. Potency of catecholamines and other L-tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology. 2004;47:438–49. doi: 10.1016/j.neuropharm.2004.04.017. [DOI] [PubMed] [Google Scholar]