

Abstract
Oxidative stress is strongly implicated in the progressive decline of cognition associated with aging and neurodegenerative disorders. In the brain, free radical-mediated oxidative stress plays a critical role in the age-related decline of cellular function as a result of the oxidation of proteins, lipids and nucleic acids. A number of studies indicate that an increase in protein oxidation and lipid peroxidation is associated with age-related neurodegenerative diseases and cellular dysfunction observed in aging brains. Oxidative stress is one of the important factors contributing to Alzheimer’s disease (AD), one of whose major hallmarks includes brain depositions of the amyloid beta-peptide (Aβ) derived from amyloid precursor protein (APP). Mutation in the APP and PS-1 genes, which increases production of the highly amyloidogenic amyloid β-peptide (Aβ42), is the major cause of familial AD. In the present study, protein oxidation and lipid peroxidation in the brain from knock-in mice expressing human mutant APP and PS-1 were compared with brain from wild type, as a function of age. The results suggest that there is an increased oxidative stress in the brain of wild type mice as a function of age. In APP/PS-1 mouse brain, there is a basal increase (at 1 month) in oxidative stress compared to the wild type (1 month), as measured by protein oxidation and lipid peroxidation. In addition, age-related elevation of oxidative damage was observed in APP/PS-1 mice brain compared to that of wild type mice brain. These results are discussed with reference to the importance of Aβ42-associated oxidative stress in the pathogenesis of AD.
Introduction
Amyloid β-peptide (Aβ) is the main component of senile plaques, which are a pathologic hallmark of Alzheimer’s disease (AD) brain [1]. Our laboratory, along with others, suggested that Aβ neurotoxicity is mediated through its ability to produce free radical oxidative stress, including protein oxidation and lipid peroxidation [2–5]. Presenilin is part of the γ-secretase complex, which together with β-secretase cleaves the Aβ peptide from amyloid precursor protein (APP). Mutations in APP, presenilin-1 (PS-1) and presenilin-2 (PS-2) genes lead to altered metabolism and increased production of amyloid-beta [Aβ (1–42)] [6–10] and have been shown to cause familial AD (FAD).
Several studies on the levels of oxidatively modified biomacromolecules such as proteins, lipids, DNA, and RNA in AD and MCI strongly suggest that oxidative stress plays a role in AD [2–5]. Oligomeric Aβ is likely the cause of memory dysfunction [11]. These findings are consistent with the notion that aggregation of Aβ plays an important role in the development of AD. However, the precise mechanism of pathogenesis in AD is still unclear. In order to determine if in vivo production of Aβ leads to oxidative damage, APP/PS-1 double mutant human knock-in mice were used in the current study. APP/PS-1 mice had elevated levels of Aβ (1–42), sufficient to cause Aβ (1–42) deposition (without over-expression of APP) beginning at 6 months of age [12] and had normal expression of PS-1 mRNA [12]. The regional distribution of Aβ deposition was similar in the APP/PS-1 mouse and in AD [12]. The early progression and regional distribution of Aβ deposition were also remarkably similar for the APP/PS-1 and Tg2576 mouse [12]. We reasoned that with increased age, Aβ production in APP/PS-1 mice would lead to increased oxidative damage relative to brain from wild type mice.
METHODS
Animals
The APP/PS-1 mice used in this study are the APP NLh/APP NLh X PS-1 P264L/ PS-1 P264L double mutant mice generated by using the Cre-lox knock-in technology to humanize the mouse Aβ sequence and to create a PS-1 mutation identified in human AD [13, 14]. All mice used in this study were males. These mice were maintained on a CD-1/129 background. All mice were maintained on a 12 h light: dark cycle in Bioclean units with sterile-filtered air and provided food and water ad libitum. All protocols were implemented in accordance with NIH guidelines and approved by the University of Kentucky Institutional Animal Care and Use Committee. The body weights of the old mice ranged from 32 to 35 g and of the young mice from 19 to 24 g. Following euthanasia with CO2, the brain was removed quickly, weighed and snap frozen in liquid nitrogen prior to analysis. Five animals/age groups (1, 2, 3, 6, 9, 12 and 15 month old) for both wild-type and APP/PS-1 double knock-in mice were used in this study. The selection of the above age groups in this study was chosen to accommodate a wide range of ages that bracket Aβ deposition and neuritic plaque formation [12,59], with implications to protein oxidation and lipid-peroxidation.
Sample preparation
The brain samples were homogenized in lysis buffer (10mM HEPES, 137mM NaCl, 4.6mM KCl, 1.1mM KH2PO4, 0.6mM MgSO4) containing protease inhibitor leupeptin (0.5 mg/mL), pepstatin (0.7 µg/mL), trypsin inhibitor (0.5x µg/mL), and PMSF (40 µg/mL). Homogenates were centrifuged at 15,800 X g for 10 min to remove debris. The supernatant was extracted to determine the total protein concentration by the BCA method (Pierce, Rockford, IL).
Measurement of protein carbonyls
Protein carbonyls are an index of protein oxidation [15]. Samples (5 µl) were incubated for 20 min at room temperature with 5 µl of 12% sodium dodecyl sulfate (SDS) and 10 µl of 2,4-dinitrophenylhydrazine (DNPH) that was diluted 10 times with PBS (pH 7.5) from a 200 mM stock. The samples were neutralized with 7.5 µl of neutralization solution (2 M Tris in 30% glycerol). The resulting sample (250 ng) was loaded per well in the slot-blot apparatus. Samples were loaded onto a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.2% (v/v) Tween 20 (wash blot) for 1 h and incubated with a 1:100 dilution of anti-DNP polyclonal antibody in wash blot for 1 h. After completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8,000 in wash blot and added to the membrane for 1 h. The membrane was washed in wash blot three times for 5 min each and developed using Sigmafast Tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop (San Jose, CA), and quantitated with Scion Image.
Measurement of protein-bound 4-hydroxy-2-trans-nonenal (HNE)
Reaction of superoxide radical ion with nitric oxide results in the formation of peroxynitrite [15], which in the presence of CO2 leads to nitration of tyrosine residues [16, 17]. Lipid peroxidation results in formation of HNE [18]. Levels of protein-bound HNE were quantified by slot-blot analysis as described previously [18]. Anti-HNE antibody raised in rabbit was used as the primary antibody (5:1000 dilutions). The membrane was developed using Sigmafast tablets (BCIP/NBT substrate). The blot was dried, scanned with Adobe Photoshop, and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Measurement of 3-nitrotyrosine (3NT)
The sample (10 µl) was incubated with 10 µl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol. The resulting sample (250 ng) was loaded per well in the slot blot apparatus. Samples were loaded onto a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 3% (w/v) BSA in wash blot for 1 h and incubated with a 1:2000 dilution of 3-NT polyclonal antibody in wash blot for 90 min. Following completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8000 in wash blot and added to the membrane for 120 min. The membrane was washed in wash blot three times for 5 min and developed using sigmafast tablets (BCIP/NBT substrate). Blots were dried, scanned and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Statistics
Data are presented as the means ± S.E.M. One-way ANOVA was used to determine the effect of age on oxidative stress in the brain of wild type mice. Where necessary, Fishers PLSD was used for post hoc comparisons (by Statview software) to determine the effect of genotype (i.e., wild-type and APP/PS1) as a function of age to test whether there was a significant interaction between these variables. p values of <0.05 were considered significant.
Results
Increased oxidative stress in the wild type mice brain as a function of age
The whole brain from 1, 2, 3, 6, 9, 12 and 15 month old wild type mice were used for studying oxidative stress parameters by measuring the levels of protein carbonyls, protein bound HNE and 3-nitrotyrosine. The brain from the 1-month-old age group was taken as a control and the brains from mice of different age groups (as mentioned above) were compared with control. The results suggest that there is a significant gradual increase (approximately 20% at 2 and 3 months, 40% at 6 months, 70% at 9 months, 90% at 12 months and slightly more than 2-fold increase at 15 months) in protein carbonyls, protein-bound HNE, and 3-nitrotyrosine (Fig.1) in whole brain from wild type mice as a function of age.
Fig. 1.
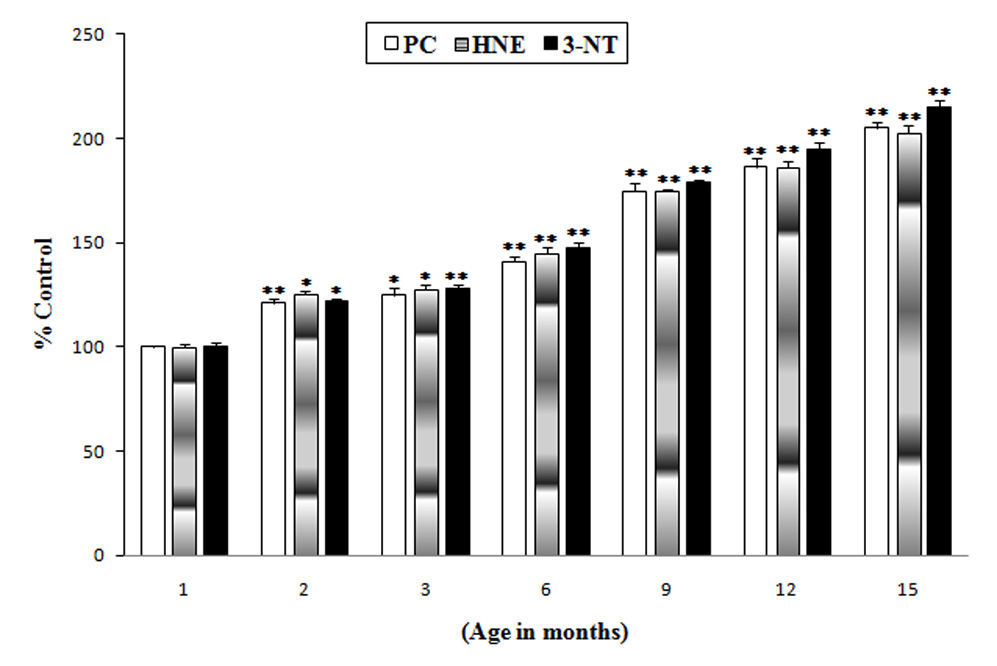
Increased oxidative stress in the brains of wild type mice as a function of age: Protein carbonyls (Plain bars), protein-bound HNE (gradient bars) and 3-NT (solid bars) levels in brains from mice of different age groups (1, 2, 3, 6, 9, 12 and 15 month old) were compared with the control (brains of 1 month-old wild type mice). Results shown are mean ±SEM obtained from five (5 animals/age group) independent preparations. Significance was assessed by one-way ANOVA. *p < 0.05, **p < 0.01.
Increased protein carbonyl levels in the brains of APP/PS-1 mice brain as a function of age
Protein carbonyls are elevated in vulnerable regions of AD brain [19–22] in brain of APP/PS1 double mutant mice [23], in APP/PS-1 mice neuronal culture [24] and in brain treated in vivo with Aβ (1–42) [25]. In the current study, a comparative analysis of protein oxidation indexed by protein carbonyls was carried out with brains from wild type and APP/PS-1 mice as a function of age. APP/PS1 double mutant mice brain demonstrated a basal significant increase (40%) in oxidative stress compared to wild type mice brain indexed by protein carbonyls (Fig. 2). That is, at one month of age, brain from APP/PS-1 mice has elevated protein oxidation compared to wild type. At each mouse age subsequently investigated, elevated protein carbonyls were found in brains from APP/PS-1 mice compared to controls (Fig. 2). The percentage increase in protein carbonyls in the brain of APP/PS-1 mice compared to controls (1 month old wild type mice) were 40%, 60%, 110%, 120%, 160% and 175% at 2, 3, 6, 9, 12, 15 months, respectively.
Fig. 2.
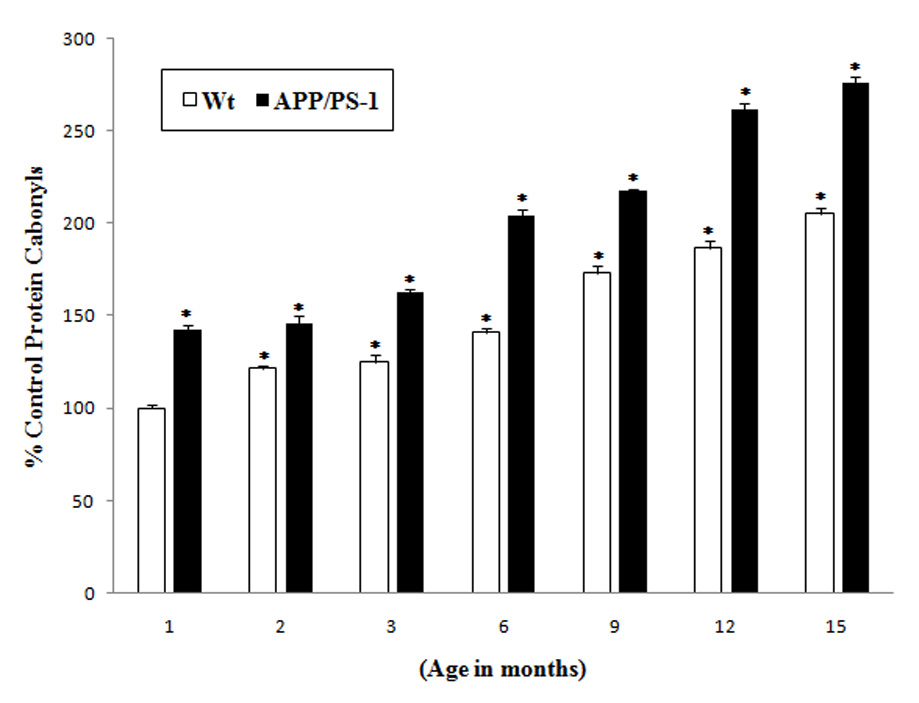
Increased protein carbonyl levels in the brains of APP/PS-1 mice: Protein carbonyl levels in brains from different age group (1, 2, 3, 6, 9, 12 and 15 month old) mice of APP/PS-1 (solid bars) and wild type mice (plain bars) were compared with the control (brain of 1 month old wild type mice). Results shown are mean ±SEM obtained from five independent preparations (5 animals/age group). Significance was assessed by two-way ANOVA followed by Scheff’s post-hoc test. *p < 0.05.
Increased protein-bound HNE levels in the brains of APP/PS-1 mice brain as a function of age
Lipid peroxidation indexed by HNE is elevated in AD brain [4, 18, 26, 27]. A comparative study on oxidative stress parameter assessed by protein-bound HNE was carried out in the wild type and APP/PS-1 mice brain as a function of age. As with protein carbonyls noted above, human APP/PS1 double mutant knock-in mice brain demonstrated a basal significant increase (40%) in protein-bound HNE levels compared to those in wild type mice brain (Fig. 3). At each subsequent age examined, elevated protein-bound HNE was observed in APP/PS-1 double mutant mice brain compared to wild type mouse brain. The percentage increase in HNE in the brain of APP/PS-1 mice compared to controls (1 month old wild type mice) were 40%, 50%, 100%, 120%, 160% and 175% at 2, 3, 6, 9, 12, 15 months, respectively.
Fig. 3.
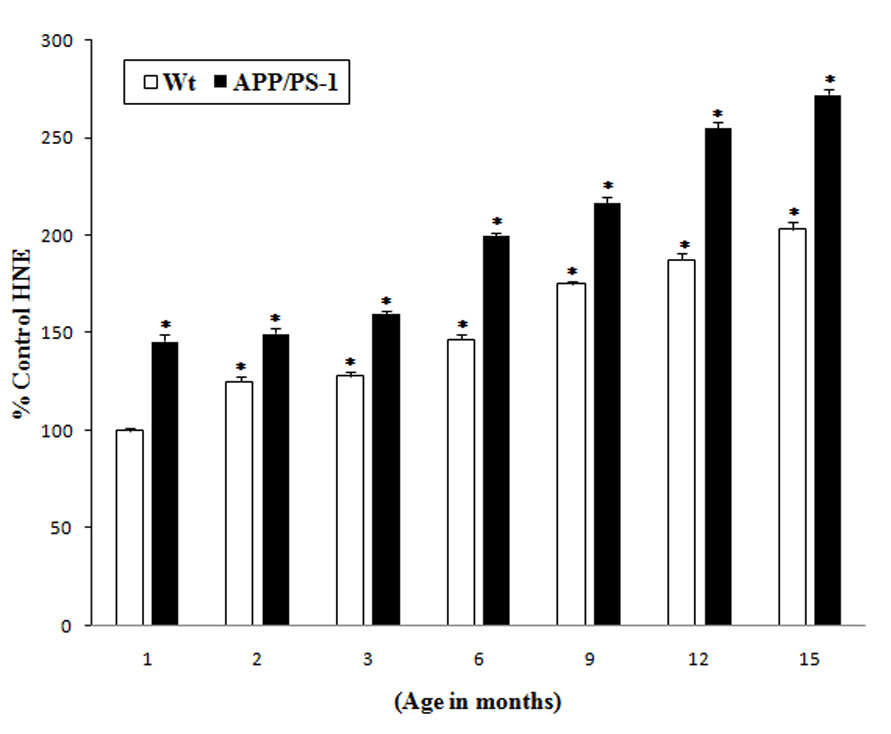
Increased protein-bound HNE levels in the brains of APP/PS-1 mice: Protein-bound HNE levels in brains from different age groups (1, 2, 3, 6, 9, 12 and 15 month old) of APP/PS-1 (solid bars) and wild type mice (plain bars) were compared with the control (brain of 1 month old wild type mice). Results shown are mean ±SEM obtained from five independent preparations (5 animals/age group). Significance was assessed by two-way ANOVA followed by Scheff’s post-hoc test. *p < 0.01.
Increased 3-NT levels in APP-PS-1 mice brain as a function of age
Another index of protein oxidation [15], 3-NT is reportedly elevated in AD brain [16, 17, 28]. 3-NT levels were compared in the wild type and APP/PS-1 mice brain as a function of age and, similar to the results from protein carbonyls, the results suggest that APP/PS1 double mutant mice brain demonstrated a basal significant increase (50%) in 3-NT levels compared to wild type mice at one-month age (Fig. 4). At each subsequent age studied, APP/PS-1 mouse brain had higher levels of 3-NT compared to controls. The percentage increase in 3-NT in the brain of APP/PS-1 mice compared to controls (1 month old wild type mice) were 50%, 60%, 100%, 125%, 150% and 175% at 2, 3, 6, 9, 12, 15 months, respectively.
Fig. 4.
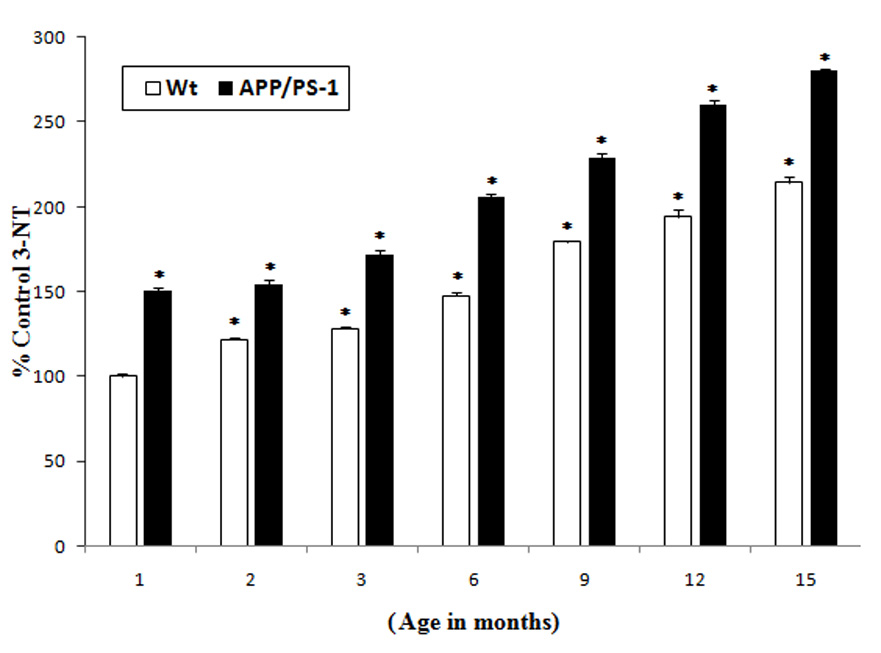
Increased 3-NT levels in the brains of APP/PS-1 mice: 3-NT levels in brains from different age groups (1, 2, 3, 6, 9, 12 and 15 month old) of APP/PS-1 (solid bars) and wild type mice (plain bars) were compared with the control (brain of 1 month old wild type mice). Results shown are mean ±SEM obtained from five independent preparations (5 animals/age group). Significance was assessed by two-way ANOVA followed by Scheff’s post-hoc test. *p < 0.05.
Discussion
Age-related impairment of the central nervous system (CNS) is associated with increased susceptibility to the development of many neurodegenerative diseases such as AD and Parkinson's disease (PD) [29]. Oxidative stress is one of the important mediators in the progressive decline of cellular function during aging [30, 31]. In the brain, free radical-mediated oxidative stress plays a critical role in the age-related decline of cellular function as a result of the oxidation of nucleic acids, lipids, and proteins, which alters their structure and function [30–32]. The brain particularly is susceptible to oxidative stress because of its high content of peroxidizable unsaturated fatty acids, high oxygen consumption per unit weight, high levels of free radical-inducing iron/ascorbate, and relatively low levels of antioxidant defense systems [30, 31, 33].
A number of studies indicate a strong role for increase in protein oxidation as a primary cause of cellular dysfunction observed during aging as well as in age-related neurodegenerative diseases [15, 26, 31, 34]. Free radical-mediated damage to neuronal membrane components also are implicated in aging, as well as the etiology of AD [26,35] with increased protein oxidation in the brain in AD [16, 17, 21, 22, 26, 28]. Further, our laboratory earlier showed that the total level of protein oxidation increases in the brains of old mice when compared to young [36]. On these lines, the present study was performed in wild type as well as APP/PS-1 mice (AD-related rodent model) to investigate and compare the status of oxidative stress as a function of age (1, 2, 3, 6, 9, 12, 15 months old mice), by measuring protein oxidation (protein carbonyls and 3-NT formation) and lipid-peroxidation (HNE-adducts). Our results in the present study are in agreement with the above findings, suggesting that there is a significant increase (20%, 20%, 40% 70%, 90, 110% at 2, 3, 6, 9, 12, 15 months, respectively) in protein carbonyls in brains of wild type mice (Fig. 1) as a function of age. However, there is a significant basal increased protein carbonyl level (40%) in the brains of APP/PS-1 mice compared to wild type at 1 month-old of age. Increased markers of lipid peroxidation, including free and protein-bound HNE and acrolein, occur in AD brain [4, 18]. Our results on protein-bound HNE and 3-NT formation are consistent with this notion, as we found a significant increase in protein-bound HNE and 3-NT levels in the brains of wild type mice as a function of age. Additionally, there is a basal significantly increased HNE (40%) and 3-NT (50%) level in the brains of APP/PS-1 mice compared to wild type at 1 month-old mice. Also, for each age examined elevated protein oxidation (Fig. 2 and 4) and lipid-peroxidation (Fig. 3) is observed in the APP/PS-1 mice brain compared to wild type mice brain. The levels of protein oxidation increase with an increase in lipid peroxidation suggesting an inter-correlation for all outcome measures (protein carbonyls, protein-bound HNE and 3-NT). There is significant interaction (p< 0.001) between age and genotype suggesting that the APP/PS-1 mice show a more rapid accumulation of oxidative damage compared to wild type mice.
Insights into potential disease mechanisms of AD have been facilitated by the discovery of the genetic mutations that underlie inherited forms of early onset AD [7, 8]. Several mutations in the genes encoding the APP, PS-1, and PS-2 proteins have been shown to lead to familial AD [37, 38]. These mutations lead to the overproduction of Aβ (1–42) [24], which is followed by the extracellular deposition of Aβ in the brain [37, 38]. At present several transgenic mouse models of AD that carry APP and/or PS1 genes with mutations are used and most of these models develop progressive age-related Aβ neuropathology with amyloid plaques and elevated levels of Aβ [6, 39,59]. Previous studies involving in vivo and in vitro experiments showed that oxidative stress increases Aβ production and that Aβ increases oxidative damage [40–43]. Strain differences may also play a role in timing the onset of Aβ deposition. Earlier investigations showed that oxidative stress chronologically precedes Aβ deposition in human brain and increased levels of 8-hydroxyadenine (8OHG) in frontal cortex of familial Alzheimer's disease (FAD) with a mutation in PS-1 or AβPP gene, suggesting neuronal RNA-oxidation [44, 45]. Additionally, recent studies on MCI brain showed increased levels of 8-hydroxyadenine and fapyguanine comparable to the late AD (LAD) brain, suggesting that oxidative modification of nuclear-DNA and mitochondrial-DNA occurs early in the pathogenesis of AD [46]. Taken together, these alterations could contribute to alterations in protein production that further propagate neuron dysfunction[46]. However, in our study using APP/PS-1 mice, we demonstrated that these mice exhibit a significant basal increase in oxidative stress (as early as in 1 month old mice) compared to wild type mice and further increase as a function of age. The observed increase in oxidative measurements with age is likely caused by the combination of mutations with age factor, which has been shown to specifically increase the production of Aβ42 [24, 47].
Numerous lines of genetic and biochemical evidence suggest that Aβ is central to the pathogenesis of AD [2, 26, 35, 48], and Aβ-associated oxidative stress induces damage to neurons in vitro [49–51] and in vivo [25, 26, 51–53]. Increased levels of oxidative damage have also been detected in a number of aging-related neurodegenerative diseases including AD [2–5, 26, 28, 44,45, 54–57], which may aggravate accumulation and deposition of Aβ [58]. Recent studies with this mouse show oligomeric Aβ contributes to memory deficits [11]. The APP/PS-1 mice used in our study had elevated levels of Aβ (1–42), sufficient to cause Aβ (1–42) deposition beginning at 6 months of age [12,59]. Our results (Fig. 2, 3 and 4) are in agreement with the above findings, suggesting that there is a sudden elevation of protein oxidation (protein carbonyls and 3-NT) and lipid peroxidation (HNE) in brains of APP/PS-1 mice about 6 months of age and further increases exponentially up to 15 months of age. This effect may be due to the increased production of Aβ at physiological levels at 6 months onwards that primes the brain of APP/PS1 mice to enhanced oxidative stress [59]. The increased production of oligomeric Aβ in 6-month old APP/PS-1 mice relative to brain in wild type mice [59] is consonant with the notion that oligomeric Aβ induces oxidative damage [53] and may be related to memory loss [60]. Our finding of increased protein oxidation (Fig. 2, 3 and 4) and lipid peroxidation (Fig. 2, 3 and 4) is consistent with this notion in 6 months old Tg2576 mice [42], suggesting that oxidative damage contributes to AD pathogenesis before Aβ accumulation in the AD brain [24, 44, 61].
In conclusion, this study provides further evidence that age-related alteration in Aβ leads to elevated oxidative damage in brain. We hypothesize that a similar process occurs in AD, with oligomeric Aβ leading to oxidative damage that in turn affects memory and cognition. The APP/PS-1 mice may allow both an improved understanding of crucial relationships between these phenotypic traits and oxidative stress as a function of age leading to neurodegeneration in AD brain. Reducing oxidative damage in brain potentially can be considered a promising strategy for therapeutic intervention in AD.
Table. 1.
Increase in the brain levels of protein carbonyls, protein-bound HNE and 3-NT in APP/PS-1 compared to the wild type (1 month old mice brain) as a function of age
| Age (in months) | Protein carbonyls* | HNE * | 3-NT * |
|---|---|---|---|
| 1 | 40% | 40% | 50% |
| 2 | 40 % | 40% | 50% |
| 3 | 60% | 50% | 60% |
| 6 | 110% | 100% | 100% |
| 9 | 120% | 120% | 125% |
| 12 | 160% | 160% | 150% |
| 15 | 175% | 175% | 175% |
Refers to the percentage increase of the values relative to the respective levels of protein carbonyls, protein-bound HNE and 3-NT of 1 month-old wild type mice brain.
Acknowledgments
This research was supported in part by NIH grants AG-10836 and AG-029839 to D.A.B. and AG-05119 to D.A. B., D. S. C., and W. R. M. We thank Cephalon, Inc., Frazer, PA, for the donation of the animals used in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This manuscript is dedicated to the life of Earl R. Stadtman (1919–2008), a dear friend who gave such elegant understanding of protein oxidation to the scientific community.
References
- 1.Selkoe DJ. Aging, amyloid, and Alzheimer's disease: a perspective in honor of Carl Cotman. Neurochem Res. 2003;28:1705–1713. doi: 10.1023/a:1026065122854. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 4.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 5.Mattson MP. Oxidative stress, perturbed calcium homeostasis, and immune dysfunction in Alzheimer's disease. J Neurovirol. 2002;8:539–550. doi: 10.1080/13550280290100978. [DOI] [PubMed] [Google Scholar]
- 6.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 7.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 8.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 9.Kurt MA, Davies DC, Kidd M, Duff K, Rolph SC, Jennings KH, Howlett DR. Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp Neurol. 2001;171:59–71. doi: 10.1006/exnr.2001.7717. [DOI] [PubMed] [Google Scholar]
- 10.Qi Y, Morishima-Kawashima M, Sato T, Mitsumori R, Ihara Y. Distinct mechanisms by mutant presenilin 1 and 2 leading to increased intracellular levels of amyloid beta-protein 42 in Chinese hamster ovary cells. Biochemistry. 2003;42:1042–1052. doi: 10.1021/bi0267590. [DOI] [PubMed] [Google Scholar]
- 11.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 12.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- 13.Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer's disease mutations and a "humanized" Abeta sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- 14.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butterfield DA, Stadtman ER. Protein oxidation processess in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- 16.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 17.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1–42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 19.Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 20.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 21.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 22.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 23.Mohmmad Abdul H, Wenk GL, Gramling M, Hauss-Wegrzyniak B, Butterfield DA. APP and PS-1 mutations induce brain oxidative stress independent of dietary cholesterol: implications for Alzheimer's disease. Neurosci Lett. 2004;368:148–150. doi: 10.1016/j.neulet.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 24.Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1–42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 25.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: implications for Alzheimer's disease. Neuroscience. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 26.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer's disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res. 2004;29:2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- 28.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Youdim MB, Buccafusco JJ. CNS Targets for multi-functional drugs in the treatment of Alzheimer's and Parkinson's diseases. J Neural Transm. 2005;112:519–537. doi: 10.1007/s00702-004-0214-z. [DOI] [PubMed] [Google Scholar]
- 30.Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals and brain aging. Clin Geriatr Med. 2004;20:329–359. doi: 10.1016/j.cger.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004;59:478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- 32.Beckman KB, Ames BN. Mitochondrial aging: open questions. Ann N Y Acad Sci. 1998;854:118–127. doi: 10.1111/j.1749-6632.1998.tb09897.x. [DOI] [PubMed] [Google Scholar]
- 33.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 34.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 35.Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 36.Poon HF, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Proteomic analysis of specific brain proteins in aged SAMP8 mice treated with alpha-lipoic acid: implications for aging and age-related neurodegenerative disorders. Neurochem Int. 2005;46:159–168. doi: 10.1016/j.neuint.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Hardy J. The Alzheimer family of diseases: many etiologies, one pathogenesis? Proc Natl Acad Sci U S A. 1997;94:2095–2097. doi: 10.1073/pnas.94.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price DL, Sisodia SS, Gandy SE. Amyloid beta amyloidosis in Alzheimer's disease. Curr Opin Neurol. 1995;8:268–274. doi: 10.1097/00019052-199508000-00004. [DOI] [PubMed] [Google Scholar]
- 39.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 40.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, et al. Non-enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, Zhao B, Yew DT, Kusiak JW, Roth GS. Processing of Alzheimer's amyloid precursor protein during H2O2-induced apoptosis in human neuronal cells. Biochem Biophys Res Commun. 1997;235:845–848. doi: 10.1006/bbrc.1997.6698. [DOI] [PubMed] [Google Scholar]
- 42.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M, Perry G. Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 43.Behl C, Davis J, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 44.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 45.Nunomura A, Chiba S, Lippa CF, Cras P, Kalaria RN, Takeda A, Honda K, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of familial Alzheimer's disease. Neurobiol Dis. 2004;17:108–113. doi: 10.1016/j.nbd.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer's disease. J Neurosci Res. 2007;85:3036–3040. doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]
- 47.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 48.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 49.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's A beta(1--42) and A beta(25--35) J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 50.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 51.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1–42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339–342; [DOI] [PubMed] [Google Scholar]
- 52.Boyd-Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM, Jr, Klein JB, Ferguson J, Link CD, Butterfield DA. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1–42): Implications for Alzheimer's disease. Neurobiol Aging. 2005 doi: 10.1016/j.neurobiolaging.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 54.Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 55.Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 56.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 57.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 58.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 59.Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer's disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]