

Abstract
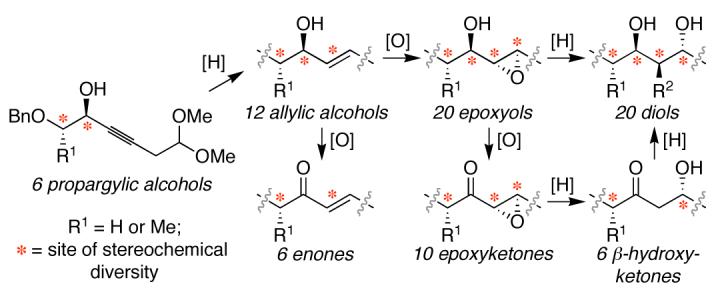
An efficient systematic approach to the diversity-oriented synthesis of polyketides has been developed to provide both skeletal and stereochemical diversity. Each synthetic intermediate is also a desired polyketide fragment and no protecting group manipulations are required. A first-generation synthesis provides a 74-membered polyketide library comprising six different skeletal classes, each in one to five steps from propargylic alcohol precursors. A study of epoxyol opening reactions revealed unusual reactivity trends based on epoxide configuration.
Polyketide natural products exhibit a tremendous variety of biological activities and chemical structures, making them attractive starting points for the synthesis of natural product-based libraries.1 Biologically, polyketides are known to bind to a wide range of targets and, thus, can be considered an empirically ’privileged’ family of structures.2 Chemically, polyketides present significant challenges in diversity-oriented synthesis, in that flexible, highly efficient synthetic approaches are required to allow systematic modification of biologically active compounds identified from the resulting libraries. To access this biological and chemical diversity, several approaches to polyketide libraries have been advanced.3 While efforts to date have focused primarily on accessing the polypropionate 1,3-diol motif,4 an ideal diversity-oriented synthesis would provide access to a wider range of skeletal motifs found in polyketide natural products. Indeed, generating libraries with both skeletal5 and stereochemical diversity6 is a key current challenge in diversity-oriented synthesis.7 Toward these ends, we report herein a unified approach to the diversity-oriented synthesis of polyketides and a first implementation that provides 74 stereochemically diverse polyketides falling into six different structural classes.
At the outset of our efforts, we noted that the polyketide skeleton arises biosynthetically from reiterative couplings of simple malonate building blocks, with tailoring reactions generating most of the structural diversity, primarily through variations in oxidation state and stereochemical configuration.8 We envisioned a conceptually related synthetic approach in which simple precursors are transformed stereoselectively to a variety of polyketide structures through formal alterations in oxidation state, with each synthetic intermediate also representing a desired polyketide fragment. Such an approach can be considered ’biomimetic’ under Breslow’s original broad definition.9 Importantly, in contrast to polyketide total synthesis, in which a variety of synthetic approaches can be interchanged as necessary, diversity-oriented synthesis requires that these various polyketide motifs be accessed in a unified, systematic fashion.
Thus, we formulated the general synthetic strategy outlined in Scheme 1, using propargylic alcohols 3 as versatile synthetic precursors10 that could be transformed to a variety of polyketide structures. These key intermediates could be generated from α-alkoxycarbonyl compounds 1and alkynes 2, potentially bearing R1 and R3 substituents. Terminal benzyl ether and dimethyl acetal groups were selected as functionalities that are directly compatible with biological assays,11 and are also potential handles for further functionalization. Subsequent reductive and oxidative transformations would then provide six different polyketide skeletal motifs: allylic alcohols (4), enones (5), epoxyols (6), epoxyketones (7), 1,3-diols (8), and β-hydroxyketones (9).R2 substituents could potentially be installed in conjunction with any of the formal reduction steps and the 1,3-diols 8 could be accessed from either epoxyols 6 or β-hydroxyketones9. Alcohol deoxygenation reactions can also be anticipated to provide corresponding deoxypolyketide fragments (not shown).
Scheme 1.
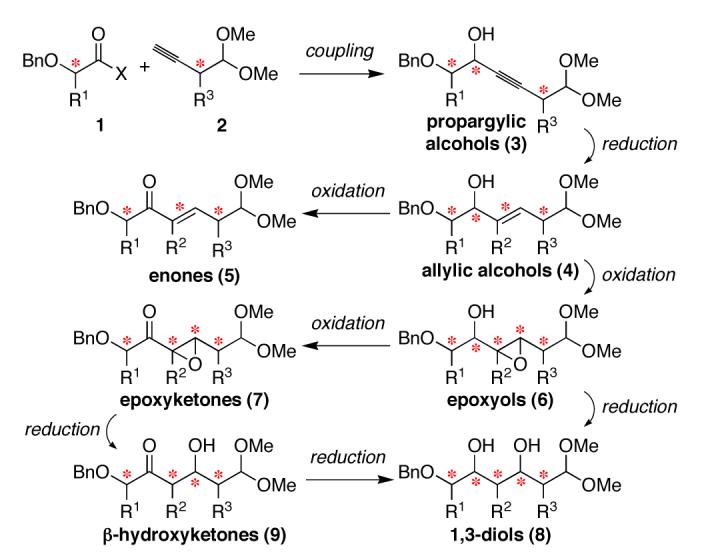
Unified Strategy for Synthesis of 6-Carbon Polyketide Fragments Having Skeletal and Stereochemical Diversity (4–9).a a R1, R2, R3 = H or Me; (*) = site of stereochemical diversity.
To begin exploring this concept, we synthesized propargylic alcohols 3 (R3 = H) by coupling of Weinreb amides 1 (R1 = H or Me, X = N[Me]OMe) and 3-butynal dimethyl acetal (2, R3 = H), followed by stereoselective alkynone reduction.12,13 We then investigated transformation of the key intermediates 3 to various polyketide motifs (Scheme 2). With a view toward future solid phase implementations, we sought homogeneous reaction conditions for stereospecific alkyne reduction to Z-allylic alcohols 10. Gratifyingly, Sato’s titanocene-catalyzed hydromagnesiation reaction provided an effective solution.14 Alkyne reduction with Red-Al also provided the complementary E-allylic alcohols 11.15
Scheme 2.

Synthesis of Six Different Classes of Polyketide Skeletal Motifs with Stereochemical Diversification.13 a Cp2TiCl2, i-BuMgBr, Et2O, 0 °C → rt, 8–48 h, 81–86%, ≥88:12 Z/E. b Red-Al, Et2O, 0 °C → rt, 3–8 h, 99%, ≥98:2 E/Z. c m-CPBA, NaHCO3, CH2Cl2, 0 °C → rt (R1 = H) or –4 °C (R1 = Me), 8–24 h, 85–93%, ≥90:10 dr. d D-or L-diethyl tartrate (matched), Ti(Oi-Pr)4, t-BuOOH, 4 Å MS, CH2Cl2, −20 °C (R1 = H) or −4 °C (R1 = Me), 2–3 d, 91–99%, 94:6 dr. e n-BuLi, Me3Al, 1,2-dichloroethane, 0 °C → rt, 2.5 h; then NaIO4, MeCN/H2O, 0 °C → rt, 2.5 h, 68– 73% (two steps), ≥98:2 dr. f MeMgCl, CuCl, THF/Et2O, −40 °C → rt, 4.5 h; then NaIO4, MeCN/H2O, 0 °C → rt, 1 h, 68–69% (two steps), ≥91:9 dr. g Dess— Martin periodinane, CH2Cl2, 0 °C → rt, 1.5–8 h, 85–100%. h SmI2, THF, −90 → −78 °C, 1.5 h, 65–93%. i NaBH(OAc)3, AcOH, CH3CN, −20 °C, 1 h, 87–95%, ≥93:7 dr.
We next sought to oxidize allylic alcohols 10 and 11 to epoxyols 12 and 13, respectively, using stereoselective epoxidation reactions. As hoped, syn epoxidation of Z-allylic alcohols 10 using m-CPBA provided the cis,syn-epoxyols 12.16 Conversely, anti epoxidation of E-allylic alcohols 11a and anti-11b under matched Sharpless conditions17 provided the trans,anti-epoxyols 13, although syn-11b proved unreactive in this case. Interestingly, however, syn-11b was amenable to syn epoxidation with m-CPBA in unusually high stereo-selectivity, providing one of the desired C2 epimeric epoxyols directly (not shown).13,16 Alcohol inversions using Mitsunobu or oxidation/re-reduction protocols then afforded the remaining epoxyol C2 epimers (not shown).13 In total, this approach provided serviceable access to all eight epoxyol diastereomers for R1 = H and 12 out of the 16 possible diastereomers for R1=Me.
We then explored nucleophilic epoxide-opening reactions to provide canonical polypropionate 2-methyl-1,3-diols 14a and 15a. Related epoxide-based approaches were pioneered by Kishi18 and have also been developed by others.19 We recognized that regioselectivity represented a key concern in these transformations.20 To address this issue, we evaluated a wide range of reaction conditions with bothcis- and trans-epoxyol substrates (Table 1). Notably, we discovered that epoxide configuration had a pronounced effect upon reaction outcome.
Table 1.
Nucleophilic Ring Opening of Epoxyols to 2-Methyl-1,3-diols.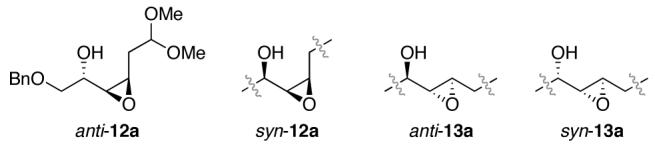
| product ratio (1,3-diol:1,2-diol)a | ||||
|---|---|---|---|---|
| conditionsb | anti-12a | syn-12a | anti-13a | syn-13a |
| n-BuLi/Me3Al | 79:9c | 88:12 | 0:0d | 4:20e |
| MeMgCl/CuCl | 32:9f | 33:60f | 77:23 | 78:22 |
| MeMgBr/CuBr | 26:7g | 33:62g | 68:32 | |
| MeMgBr/CuI | 12:19g | 28:51g | 48:26g | |
| MeMgBr/CuCN | 17:3g | 21:58g | 50:22g | |
| MeLi/CuI | 24:15h | 4:96 | 48:52 | |
| MeLi/CuCN | 17:12h | 2:6e | 58:42 | |
Determined by 1H-NMR.
5 equiv n-BuLi, 8 equiv Me3Al, 1,2-dichloroethane,0 °C → rt, 2.5 h; or 12 equiv Me[M], 6 equiv CuX,1:1 THF/Et2O, –40 °C → rt, 4.5 h.
Remainder 5-desmethoxy-2,5-dimethyl-1,3-diol.13
Debenzylated 13a (94%) and starting material (6%)recovered.
Remainder starting material.
Remainder 2-chloro-1,3-diol side product.
Remainder 2-bromo-1,3-diol side product and starting material.
Remainder unidentified decomposition products.
For cis-epoxyols anti-12a and syn-12a, epoxide opening with methyl cuprate nucleophiles generally favored formation of the undesired 1,2-diols and/or halogenated side products. However, Miyashita’s n-BuLi/Me3Al combination21 proved quite effective for these substrates, provided that at least 5 equiv of Me3Al were used to accommodate the five Lewis basic sites in the substrates. Interestingly, we discovered that the reaction required the use of chlorinated solvents, with 1,2-dichloroethane providing higher yields than methylene chloride. Further studies of the mechanistic implications of this intriguing finding are ongoing.
In contrast, for trans-epoxyols anti-13a and syn-13a, the n-BuLi/Me3Al combination was ineffective, leading only to debenzylation of the starting material in the case of anti-13a and to undesirable regioselectivity in the case of syn-13a. However, examination of a variety of methyl cuprates revealed that MeMgCl/CuCl provided effective access to the desired 1,3-diol products for these substrates. Interestingly, the choice of cuprate counterion had a significant effect upon regioselectivity, with chloride providing enhanced selectivity for 1,3-diol formation compared to bromides, iodides, and cyanides.
In both the cis- and trans-epoxyol series, residual undesired 1,2-diols were then conveniently separated after NaIO4 cleavage.22 NMR analyses of 1,3-diol acetonide derivatives using Rychnovsky’s 13C-NMR method23 and NOESY experiments were then used to confirm relative stereochemical assignments for both the epoxyol precursors and 2-methyl-1,3-diol products.13 Importantly, this approach provided comprehensive access to all eight 2-methyl-1,3-diol diastereomers in the R1 = H series (14a,15a).
Analogous efforts at nucleophilic opening of the more hindered epoxyols in the R1 = Me series (12b, 13b) were thwarted by complete regioselectivity for 1,2-diol formation under all conditions tested. However, expansion of the synthetic route to include additional desired polyketide motifs provided a useful alternative solution. Dess—Martin oxidation of allylic alcohols 10 and 11provided enones 17 and 18, respectively, while oxidation of epoxyols 12 and 13 provided epoxyketones 19 and 20, respectively. The epoxyketones could then undergo reductive epoxide opening with SmI2 to provide β-hydroxyketones 21.24 All three of these structural classes represent desired polyketide motifs. NOESY analysis of 1,2-diol cyclic carbonates derived from β-hydroxyketones 21b provided direct confirmation of relative stereochemical assignments for the epoxyol precursors in the R1 = Me series.13 At this point, directed reductions of β-hydroxyketones 21 provided anti-1,3-diols 16 and their syn-1,3-diol congeners (not shown), representing all 12 possible 1,3-diol diastereomers for R2 = H.13,25,26
In conclusion, we have developed an efficient, unified diversity-oriented synthesis of polyketides that provides both skeletal and stereochemical diversity. Our first-generation synthesis has provided a 74-membered polyketide library comprising six different skeletal classes. Overall, this work addresses three important current challeges in the field of diversity-oriented synthesis. First, our synthesis provides substantial skeletal diversity, and in an unusual context involving acyclic molecules. Second, our route provides a unified approach to these various polyketide motifs. Third, the synthetic route is highly efficient, providing each polyketide motif in only one to five steps from propargylic alcohols 3, with each synthetic intermediate also representing a desired polyketide motif and with no protecting group manipulations required. While this has been accomplished by strategic application of established chemistry, the extensive synthetic investigations undertaken to achieve these high yields and stereoselectivities required in diversity-oriented synthesis have revealed interesting new avenues for further chemical investigations. Further expansion of this synthetic approach to provide more comprehensive access to these and other polyketide motifs and screening of the resulting polyketide libraries against a variety of targets is ongoing. The availability of this unified synthetic approach will allow systematic evaluation of skeletal and stereochemical modifications to biologically active compounds identified therein. Moreover, the results of these screens will provide important insights into the effectiveness of natural product-based libraries at addressing diverse biological targets.
Supplementary Material
Acknowledgment
We thank Prof. Jae-Sang Ryu (Ewha University) for helpful discussions and Dr. George Sukenick, Hui Fang, Hui Liu, Sylvi Rusli, and Anna Dudkina for expert mass spectral analyses. D.S.T. is a NYSTAR Watson Investigator, H.I. is a Visiting Scientist from Daiichi Sankyo, L.M.A. was a Summer Undergraduate Research Program student. We gratefully acknowledge financial support from the NIH (P41 GM076267), DOD (CM030085), William Randolph Hearst Fund in Experimental Therapeutics, William H. Goodwin and Alice Goodwin and the Commonwealth Foundation for Cancer Research, and MSKCC Experimental Therapeutics Center.
Footnotes
Supporting Information Available:Detailed experimental procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Shang S, Tan DS. Curr. Opin. Chem. Biol. 2005;9:248–258. doi: 10.1016/j.cbpa.2005.03.006. [DOI] [PubMed] [Google Scholar]
- (2).Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J. Med. Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- (3)(a).Paterson I, Donghi M, Gerlach K. Angew. Chem., Int. Ed. 2000;39:3315–3319. doi: 10.1002/1521-3773(20000915)39:18<3315::aid-anie3315>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (b) Bode JW, Fraefel N, Muri D, Carreira EM. Angew. Chem., Int. Ed. 2001;40:2082–2085. [PubMed] [Google Scholar]; (c) Smith AB, III, Walsh SP, Frohn M, Duffey MO. Org. Lett. 2005;7:139–142. doi: 10.1021/ol047792c. [DOI] [PubMed] [Google Scholar]; (d) Misske AM, Hoffmann HMR. Chem. Eur. J. 2000;6:3313–3320. doi: 10.1002/(sici)1521-3765(20000915)6:18<3313::aid-chem3313>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; (e) Arjona O, Menchaca R, Plumet J. J. Org. Chem. 2001;66:2400–2413. doi: 10.1021/jo001660p. [DOI] [PubMed] [Google Scholar]; (f) Reggelin M, Brenig V. Tetrahedron Lett. 1996;37:6851–6852. [Google Scholar]
- (4).Bode SE, Wolberg M, Mueller M. Synthesis. 2006:557–588. [Google Scholar]
- (5)(a).Lee D, Sello JK, Schreiber SL. J. Am. Chem. Soc. 1999;121:10648–10649. [Google Scholar]; (b) Lee D, Sello JK, Schreiber SL. Org. Lett. 2000;2:709–712. doi: 10.1021/ol005574n. [DOI] [PubMed] [Google Scholar]; (c) Kwon O, Park SB, Schreiber SL. J. Am. Chem. Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e. [DOI] [PubMed] [Google Scholar]; (d) Ding S, Gray NS, Wu X, Ding Q, Schultz PG. J. Am. Chem. Soc. 2002;124:1594–1596. doi: 10.1021/ja0170302. [DOI] [PubMed] [Google Scholar]; (e) Couladouros EA, Strongilos AT. Angew. Chem., Int. Ed. 2002;41:3677–3680. doi: 10.1002/1521-3773(20021004)41:19<3677::AID-ANIE3677>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (f) Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]; (g) Burke MD, Berger EM, Schreiber SL. J. Am. Chem. Soc. 2004;126:14095–14104. doi: 10.1021/ja0457415. [DOI] [PubMed] [Google Scholar]; (h) Taylor SJ, Taylor AM, Schreiber SL. Angew. Chem., Int. Ed. 2004;43:1681–1685. doi: 10.1002/anie.200353466. [DOI] [PubMed] [Google Scholar]; (i) Oguri H, Schreiber SL. Org. Lett. 2005;7:47–50. doi: 10.1021/ol047945w. [DOI] [PubMed] [Google Scholar]; (j) Kumagai N, Muncipinto G, Schreiber SL. Angew. Chem., Int. Ed. 2006;45:3635–3638. doi: 10.1002/anie.200600497. [DOI] [PubMed] [Google Scholar]; (k) Yeager AR, Min GK, Porco JA, Jr., Schaus SE. Org. Lett. 2006;8:5065–5068. doi: 10.1021/ol0618252. [DOI] [PubMed] [Google Scholar]
- (6)(a).Sutherlin DP, Armstrong RW. J. Am. Chem. Soc. 1996;118:9802–9803. [Google Scholar]; (b) Annis DA, Helluin O, Jacobsen EN. Angew. Chem., Int. Ed. 1998;37:1907–1909. [Google Scholar]; (c) Harrison BA, Gierasch TM, Neilan C, Pasternak GW, Verdine GL. J. Am. Chem. Soc. 2002;124:13352–13353. doi: 10.1021/ja027150p. [DOI] [PubMed] [Google Scholar]; (d) Kim Y.-k., Arai MA, Arai T, Lamenzo JO, Dean EF, III, Patterson N, Clemons PA, Schreiber SL. J. Am. Chem. Soc. 2004;126:14740–14745. doi: 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]; (e) Potuzak JS, Moilanen SB, Tan DS. J. Am. Chem. Soc. 2005;127:13796–13797. doi: 10.1021/ja055033z. [DOI] [PubMed] [Google Scholar]; (f) Moilanen SB, Potuzak JS, Tan DS. J. Am. Chem. Soc. 2006;128:1792–1793. doi: 10.1021/ja057908f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)(a).Tan DS. Nat. Chem. Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]; (b) Burke M,D, Schreiber S,L. Angew. Chem., Int. Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (c) Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- (8).Fischbach MA, Walsh CT. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- (9).Breslow R. Chem. Soc. Rev. 1972;1:553–580. [Google Scholar]
- (10).The potential utility of propargylic alcohols in diversity-oriented synthesis has been noted previously: Weber L. Curr. Opin. Chem. Biol. 2000;4:295–302. doi: 10.1016/s1367-5931(00)00092-2.
- (11)(a).For examples of stable, biologically active dimethyl acetals, see: Cho W-J, Kim E-K, Park IY, Jeong EY, Kim TS, Le TN, Kim D-D, Lee E-S. Bioorg. Med. Chem. 2002;10:2953–2961. doi: 10.1016/s0968-0896(02)00137-2.Springer RH, Scholten MB, O’Brien DE, Novinson T, Miller JP, Robins RK. J. Med. Chem. 1982;25:235–242. doi: 10.1021/jm00345a009. Kim CD, Kim HH, Kim YK, Kwak YK, Kim S-O, Yoo S-E, Hong KW. J. Pharmacol. Exp. Ther. 2001;296:1085–1090.
- (12).Initial attempts to access 3a directly by various asymmetric alkyne additions to the corresponding aldehydes (reviewed in: Pu L. Tetrahedron. 2003;59:9873–9886.) suffered from poor enantioselectivity.
- (13).See Supporting Information for full details.
- (14).Sato F, Ishikawa H, Watanabe H, Miyake T, Sato M. J. Chem. Soc., Chem. Commun. 1981:718–720. [Google Scholar]
- (15).Chan K-K, Cohen N, De Noble JP, Specian AC, Jr., Saucy G. J. Org. Chem. 1976;41:3497–3505. doi: 10.1021/jo00884a001. [DOI] [PubMed] [Google Scholar]
- (16).Adam W, Wirth T. Acc. Chem. Res. 1999;32:703–710. [Google Scholar]
- (17).Katsuki T, Martin VS. Org. React. 1996;48:1–299. [Google Scholar]
- (18)(a).Johnson MR, Nakata T, Kishi Y. Tetrahedron Lett. 1979:4343–4346. [Google Scholar]; (b) Finan JM, Kishi Y. Tetrahedron Lett. 1982;23:2719–2722. [Google Scholar]
- (19)(a).Murphy PJ, Procter G. Tetrahedron Lett. 1990;31:1059–1062. [Google Scholar]; (b) Miyashita M, Hoshino M, Yoshikoshi A. J. Org. Chem. 1991;56:6483–6485. [Google Scholar]; (c) Miyashita M, Yoshihara K, Kawamine K, Hoshino M, Irie H. Tetrahedron Lett. 1993;34:6285–6288. [Google Scholar]; (d) Esumi T, Fukuyama H, Oribe R, Kawazoe K, Iwabuchi Y, Irie H, Hatakeyama S. Tetrahedron Lett. 1997;38:4823–4826. [Google Scholar]
- (20).Tius MA, Fauq AH. J. Org. Chem. 1983;48:4131–4132. [Google Scholar]
- (21).Sasaki M, Tanino K, Miyashita M. Org. Lett. 2001;3:1765–1767. doi: 10.1021/ol010062+. [DOI] [PubMed] [Google Scholar]
- (22).Chakraborty TK, Joshi SP. Tetrahedron Lett. 1990;31:2043–2046. [Google Scholar]
- (23)(a).Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]; (b) Rychnovsky SD, Rogers BN, Richardson TI. Acc. Chem. Res. 1998;31:9–17. [Google Scholar]
- (24).Molander GA, Hahn G. J. Org. Chem. 1986;51:2596–2599. [Google Scholar]
- (25).Evans DA, Clark JS, Metternich R, Novack VJ, Sheppard GS. J. Am. Chem. Soc. 1990;112:866–868. [Google Scholar]
- (26).1,3-Diol 16a and its C2 epimer could also be accessed directly by reductive epoxide opening of 13a and its C2 epimer with Red-Al.13,18b
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.