

Abstract
The light-driven steps in the biogenesis and repair of the inorganic core comprising the O2-evolving center of oxygenic photosynthesis (photosystem II water-oxidation complex, PSII-WOC) are reviewed. These steps, known collectively as photoactivation, involve the photoassembly of the free inorganic cofactors to the cofactor-depleted PSII-(apo-WOC) driven by light and produce the active O2-evolving core comprised of Mn4CaOxCly. We focus on the functional role of the inorganic components as seen through the competition with non-native cofactors (“inorganic mutants”) on water oxidation activity, the rate of the photoassembly reaction, and on structural insights gained from EPR spectroscopy of trapped intermediates formed in the initial steps of the assembly reaction. A chemical mechanism for the initial steps in photoactivation is given that is based on these data. Photoactivation experiments offer the powerful insights gained from replacement of the native cofactors, which together with the recent X-ray structural data for the resting holoenzyme provide a deeper understanding of the chemistry of water oxidation. We also review some new directions in research that photoactivation studies have inspired that look at the evolutionary history of this remarkable catalyst.
Keywords: Bicarbonate, Calcium, Manganese, photosynthesis, water oxidation, oxygen-evolution, Photosystem II
Introduction
The process of oxygenic photosynthesis is responsible for producing nearly all atmospheric O2 on Earth by the light-driven oxidation of water[1]. However, oxidation of water to O2 is a mechanistically complex and highly energy demanding chemical reaction [2]driven by photooxidation of a chlorophyll with electrochemical reduction potential of ca. 1.2-1.3 V [3]. In nature, this reaction proceeds by using the visible light energy that is trapped and made chemically available by chlorophyll photo-pigments, and it is ultimately carried out at the inorganic active site (Mn4OxCa1Cly), the water-oxidizing complex (WOC), of a membrane spanning protein complex known as photosystem II (PSII) [4]. Although nature has devised a very efficient way for oxidizing water, the coupling of the water-splitting reaction to the PSII reaction center photochemistry (chlorophyll photooxidation) takes a heavy toll on the protein matrix due to the generation of highly reactive chemical intermediates under illumination. The damage done to the D1 subunit requires its resynthesis and replacement within the 19-31 subunits of PSII, as well as the reassembly of the inorganic catalyst. This process can occur as frequently as every 30 min at full solar flux [5, 6]. Reassembly of the inorganic cluster (Mn4Ca1OxCly) of the water-oxidizing complex (PSII-WOC) is known as photoactivation; named by George Cheniae and coworkers the discoverer of this phenomenon (reviewed in [7]).
In this article we review the chemistry behind this complex photoassembly process focusing on the inorganic principles and the functional role of the cofactors in catalyzing the water oxidation chemistry. This chemistry is responsible for the concerted four-electron oxidation of two water molecules to make O2 after injection of the fourth hole (electron vacancy) into the WOC. The thermodynamic efficiency of this chemistry has never been replicated in any abiotic catalyst of any kind. Unlike the reasonably well understood principles of single-electron and single-proton transfer chemistry, complex multi-electron/proton reactions control the overall energetics and thus the allowed pathway for catalysis [8, 9, 10]. Interested readers should also consult other recent reviews that cover in vitro and in vivo photoactivation of the WOC [11, 12, 13].
1.1 Photosystem II: A structural snapshot
The membrane-spanning PSII protein complex is assembled in vivo as a functional dimer with 19-31 subunits[14, 15] in each monomer, depending on the organism in question. Only within the last three years, has the structural arrangement of each subunit, with respect to each other, been resolved by X-ray crystallography to 3–3.5 Å resolution with considerable certainty (R-factor = 24 %) [16, 17]. PSII isolated from Thermosynechococcus sp. in its resting oxidation state has provided a more detailed structural model than was previously available from spectroscopic and mutagenesis studies alone. However, there has been noticeable differences in the two recent structures one at 3.0 Å [16] by Loll et al., and another at 3.5 Å by Ferriera et al. [17]. The structure of the active site of PSII along with the molecular modeling of the architecture near the water oxidation site from the 3.5 Å structure is shown in Fig 1. The modeling of the exact position of the metal ions inside the electron density representing the Mn4Ca-cluster has been especially debated. The Imperial College group has postulated an assignment for all the elements in the WOC at 3.5 Å, while the Berlin group has left it as an open question by showing only the heavy atom scatterers [16, 17]. A recent Mn EXAFS study of single crystals of these PSII core complexes has provided more evidence to distinguish between these models[18]. Also questions have arisen on the radiation damage to the cluster accrued by the X-rays used for the diffraction studies [19]. The Imperial College group has provided an alternative interpretation of the original electron density map that can be found in this special issue. However, the modeling of the protein subunits remains very similar from both groups. Importantly, it is now known how the extrinsic proteins are located with respect to the cluster. This information is valuable since it has been known that although an intact PSII core complex is needed for sustained O2 evolution in vivo, some PSII subcore complexes lacking the extrinsic subunits can also produce O2 in vitro. The smallest isolated reaction center (RC) complex that performs primary charge separation contains four subunits: D1 (psbA gene), D2 (psbD gene), and the two subunits of cytochrome b559, α (psbE gene) and β (psbF gene) along with another chloroplast encoded protein psbI [20, 21]. These four subunits along with two membrane spanning chlorophyll binding proteins CP47 and CP43 comprise what is called the PS II core. No complex without these six subunits had been isolated that evolved oxygen [22]. However, a complex completely devoid of CP43 was shown to be photoactivatable (i.e. showed O2 activity when the cluster was reconstituted), albeit with much lower photochemical quantum yield and to a lower level of activity[23]. The D1 polypeptide provides most of the amino acid residues comprising the active site surrounding the cluster, with a notable contribution by residues Arg357 and Glu354 from CP43 in the active site. The structures coupled with the numerous mutational experiments and spectroscopic measurements have provided a lot of constraints to the mechanism of water oxidation which are briefly touched below. However, detailed discussion of possible mechanisms is beyond the scope of this review (for earlier reviews [24, 25]).
Fig 1.
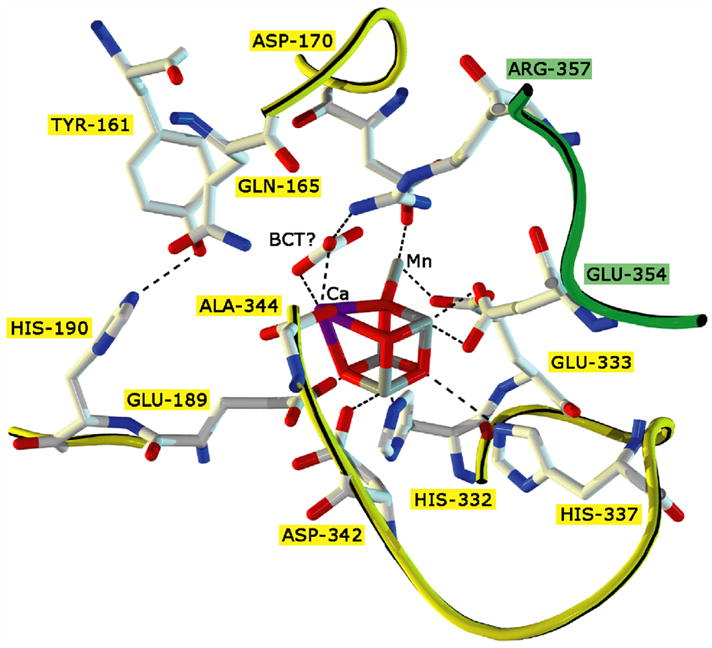
XRD structural model of the water oxidizing complex (WOC) in Photosystem II adapted from Ferriera et.al.(2004) at 3.5 Å resolution. In the center is the CaMn4O4-core surrounded by its direct ligands (purple = Ca, grey = Mn, red = O). All amino acids named in a yellow box belong to the D1 subunit, while those in a green box come from subunit CP43. Dashed lines are shown for visual guidance only.
1.2 Mechanistic questions on water oxidation
For the water oxidation reaction to take place, the cluster cycles through five oxidation states (named, in order, as Si: i=0, 1,…, 4) starting from the resting state S1, before it releases a dioxygen molecule and total of four substrate protons (through the S-state cycle). Stabilization of the higher oxidation states (S2- and S3-states) in the Mn4Ca cluster is both due to the delocalization of positive charge among metal centers and also from a local electrostatic effect generated by the presence of strong sigma donor ligands such as aspartate, glutamate and histidine that surround the cluster in the active site. The role of the inorganic cofactors (Mn2+, Ca2+, Cl−) in actual water oxidation chemistry has been subject to immense scrutiny. Numerous synthetically inspired and structure based proposals have been formulated. Proposals ranging from homolytic O-O bond formation from two facial bridging electron-deficient oxo’s [25] to heterolytic O-O bond formation via nucleophilic-electrophilic oxo-coupling have been presented [26, 27]. And although various spectroscopic and structural markers (such as EPR and EXAFS)[28, 29, 30], mutagenesis [31] and water exchange rates of the individual S-states have been considered [32], no commonly agreed on mechanism of water oxidation has been convincingly proved. It is for these reasons that the photoassembly reaction of the Mn4Ca cluster provides special insights as it can dissect the roles of the individual cofactors and enables examination by substitution, a topic we refer to as “inorganic mutants”.
1.3 PSII damage and repair
1.3.1. Protein Assembly
All the mechanisms of water oxidation proposed to date involve highly reactive oxidized manganese ions. However, under normal environmental conditions such as high light flux and ambient atmospheric O2 pressure these intermediates can furnish radicals that are lethal and destroy the protein itself, leading to photoinhibition [33]. Loss of PSII activity by photoinhibition occurs as often as every 15-30 minutes under normal solar flux conditions. To cope with photoinhibition, oxygenic phototrophs have evolved repair and photo-protective mechanisms permitting a high rate of metabolic turnover of PSII polypeptides with the D1 protein exhibiting the highest rate of turnover. Since D1 appears to supply most of the protein-derived ligands to the Mn4Ca, the frequency of in vivo photoactivation must follow the D1 protein repair rate. The site of the light reactions of oxygenic photosynthesis in cyanobacteria, green algae and plants (in the chloroplast) is a specialized inner membrane called the thylakoid membrane system. In higher plants, the initial dissociation of a damaged PSII dimer into monomers in the stacked grana regions of the thylakoid membrane system and further migration to the stromal region (outside the thylakoid membrane), starts the process of repair. However, it is not clear whether photoactivation of the inorganic cofactors of the WOC occurs within PS II complexes bound to the stacked grana membranes or within the stromal region. In the PSII repair process in cyanobacteria, evidence for the initially assembled RC with D1, D2 and cyt b559 subunits in the (non-photosynthetic) plasma membrane has been established, but not with CP43 or CP47 subunits [6]. Also, these partially assembled reaction centers in the plasma membrane can carry out charge separation and single turnover electron transfer [34]. The absence of CP47 and CP43 indicates that assembly of a functioning WOC is highly unlikely in the plasma membrane. Recent claims have been put forth by Chow and Aro that in the repair process CP47 is probably only partially disassembled. They also state that CP43 is indisputably dissociated and re-attached along with PsbH and PsbL, both subunits are important in supporting CP43 re-assembly for repair in higher plants [33, 35]. This evidence then gives credence to the proposal that Mn4Ca could be photoassembled in the stromal region of the thylakoid membrane [33]. Observations on isolated PS II sub-complexes from which both CP43 and CP47 have been dissociated by detergents shows that these do not assemble a functional inorganic core during photoactivation, while the CP47RC sub-complex lacking only CP43 can be photoactivated, although the O2 evolution rate is 20% that of photoactivated PSII membranes and highly unstable [23].
In the vast majority of organisms containing PSII, the D1 protein is synthesized as a precursor protein (pD1 with a short peptide extension (8–16 amino acid residues) on the carboxy terminus (C-terminus) that is proteolytically removed during the assembly process [12, 36]. Experimental deletion of this segment still allows functional assembly of the WOC, provided the native mature C-terminal α-carboxylate at position 344 is available. The D1 C-terminal extension was shown not to be required for assembly of functional PS II complexes, nor for growth under optimal conditions in both Synechocystis 6803 [37] and Chlamydomonas [38]. However, the carboxyl extension must be cleaved beyond Ala 344 by a soluble protease, called ctpA, prior to assembly of an active inorganic core [37, 39]. This cleavage is essential for the assembly of the Mn4Ca-core and, hence, oxygen evolution. Alanine 344, which is at the terminus of the mature D1, is proposed to ligate the Mn4Ca through its backbone α-carboxyl moiety. While evidence is strong that the carboxyl group of D1–Alanine 344 is in close proximity to the Mn4Ca, it is not resolved whether it ligates Mn or Ca. In the X-ray structure models based on crystals diffracting to 3.0 Å and 3.5 Å, the 3.0 Å structure places the C-terminus as coordinating Mn, whereas the 3.5 Å structure model orients the C-terminal carboxy moiety towards a calcium atom. Although the 3.0 Å structure shows possible Ca2+ ligation, authors use results from FTIR light-dark difference spectroscopy to argue against it [16]. They conclude that Ala344 is ligated to one of the Mn-atoms. Controversially however, in Synechocystis 6803 cultures which lack the carboxyl extension or have a genetically engineered two-fold longer extension exhibit no difference in growth rates or O2 evolution rates versus wild type. For further discussion interested readers should consult the reviews by Burnap and by Chow and Aro, respectively [12, 33].
1.3.2. Intracellular Cluster re-assembly
The rate of the Mn4Ca cluster assembly depends critically on the local concentrations of free Mn2+, Ca2+, bicarbonate and the pH. Recently, estimates have been provided of intracellular concentrations of these ions in a few cyanobacteria. The Mn content of cyanobacterial cells is generally well in excess of that needed to supply all PS II complexes. However intracellular distribution and transport are not as well known. The capsular peptidoglycan comprising the cyanobacterial envelope (cell wall) plays a major role in concentrating free Mn2+ from extracellular solution [40]. Mature cells of Synechocystis sp. PCC 6803 take up or exchange free Mn2+ at a rate of about 2 × 106 Mn2+/cell in a period of 30 minutes [41]. There are two pools of intracellular Mn2+ that have been identified [42]. The largest pool, known as pool A, comprises ~108 atoms/cell, is found closely associated with the outer membrane possibly on either the outside or the inner leaflet (periplasm). This pool can be removed by EDTA, is electrogenically generated under light and can be prevented by electron transport inhibitors (DCMU) or abolished by proton uncouplers (like CCCP) that collapse the proton-motive force across the membranes. It thus can be inferred that uptake is energetically coupled to ATP hydrolysis. A tightly associated second pool B comprises ~1.5 × 106 atoms/cell, is not affected by EDTA, light or proton uncouplers and is found in the inner membrane (cytoplasm). Under Mn2+ starvation conditions, pool B contains about 105 Mn/cell which is essential for photosynthesis, of which the majority is targeted to PSII. By comparison, the marine cyanobacterium Synechococcus sp. PCC7002 contains 1.5 × 106 Mn/cell in pool B, while the non-oxygenic purple bacterium Rhodobacter capsulatus contains 1% of this amount. Also, two-component regulatory machinery for the sensing of extracellular Mn2+ concentrations and maintenance of intracellular Mn homeostasis in cyanobacteria and non-photosynthetic prokaryotes has been found recently [43].
Intracellular transport of transition metal ions used in enzymatic machinery is typically under tight control by proteins or small molecule chelators that ensure proper targeting of the metal to its final destination and also protect the nascent system from unwanted reactions. These ‘chaperone’ proteins are widely distributed in the case of iron and copper regulatory systems. The thermodynamic potentials for aqueous Fe2+/Fe3+ and Cu+/Cu2+ metals indicate that these metals are susceptible to redox changes within cells and thus will undergo major changes in speciation if not coordinated during transport. Thus, it may seem curious that no essential chaperone protein has yet been identified for delivery of Mn2+ to the apo-WOC-PS II complex during biogenesis. The explanation is likely due to the inaccessibility of the Mn3+ oxidation state in aqueous media at neutral pH owing to the large thermodynamic barrier which prevents Mn2+ oxidation (standard reduction potential for Mn3+ 1.2 V at pH 7 vs NHE). Moreover, divalent Mn2+ with its spherical electronic distribution and half-filled 3d5 electronic configuration forms relatively weak coordination complexes with all monodentate O, N, and S ligands [44]. Thus, free Mn2+ does not generally associate tightly with the internal machinery of the cell unless there are pre-organized chelation sites present with multiple ligand donors. Generally, the latter sites are specific for ions of a particular size, charge and dehydration energy so that biosynthetic discrimination is almost always ensured. A remarkable example of these rules is illustrated later in the example of the competition between Mn2+ and Cs+. Surface electrostatics also play a major role in guiding metal ions to their functional binding sites and this mechanism is important for steering Mn2+ to the apo-WOC-PS II site[45]. Binding of aquo-Mn2+ to the high affinity site in apo-WOC-PS II has been shown to be influenced by three factors: pH, bicarbonate ions and Ca2+, which are each discussed in the following sections. Although no known chaperones for Mn2+ insertion appear to exist as discussed above, repair of the PS II protein complex caused by light absorption does involve chaperone-like proteins [46, 47].
2.1 Apo-WOC-PSII preparation and instrumentation for sensitive O2 detection
In order to carry out in vitro studies of the photodamage and photorepair processes, numerous approaches have been reported to extract the Mn4Ca cluster from the native PSII. Here, we briefly summarize our most recently improved approach as compared to previous methods which have been found to either damage the protein as they cause low photoactivation yields, or cannot fully remove all Mn2+ bound to the protein complex. For a representative schematic of the process see Fig 2. An early reported method using TRIS buffer has been used extensively in this field, but has been found to be incapable of achieving even moderate photoactivation yield (supposedly, because of denaturation of the protein and also the generation of in situ TRIS radicals)[48] Also, hydroxylamine has been used to reduce the Mn ions and thus make the Mn4Ca cluster labile. In this method it is argued that only the 17 and 12 kDa proteins are removed similar to TRIS treatment. However, hydroxylamine has been found to be difficult to wash away and thus interferes with the re-assembly process. We and others have developed milder methods for removal of the inorganic cofactors and extrinsic proteins from PS II complexes (apo-WOC-PS II) which is essential for quantitative work leading to high photoactivation yields and reproducible kinetics [45, 49, 50]. The preferred method relies on short duration incubation of PSII particles at high pH and washing with excess Mg2+ to remove both manganese and calcium. When excess Mg2+ is used, it results in removal of all three extrinsic proteins. SDS gel chromatography has shown quantitative loss of the extrinsic polypeptides psbQ, psbO and psbP from spinach PSII [51].
Fig 2.

Scheme summarizing the methodology for removal of Mn4Ca cluster by high pH treatment, eg., isolation of apo-WOC-PSII particles from isolated spinach PSII membranes, and reconstitution by photoactivation.
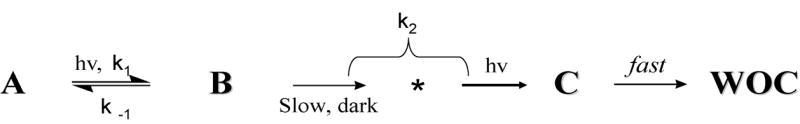
Our own studies of photoactivation were performed using a modified Clark-type electrochemical cell for the detection of dissolved O2 gas in photosynthetic samples[45, 52]. It enabled measurements of O2 concentration with exceptional sensitivity (50 femtomoles O2), micro-volume sample (5 μL), and 10-fold higher time-resolution than the commercially available Clark electrodes. The electronic layout and novel material used have been described and circuit diagrams are available upon request. Using this photoactivation instrument has enabled kinetic resolution of the first two steps prior to the rate-limiting step in the assembly of the inorganic cofactors [45, 52]. The steps that follow the rate-limiting step are not individually resolved, but their Mn stoichiometry needed to restore O2 evolution has been accurately determined. Photoactivation follows a two-step sequence, as originally postulated by Cheniae and Tamura on the basis of steady-state O2 yield measurements[53] and confirmed by Miller and Brudvig [54]. The O2 recovery kinetics is fitted to a two exponent fitting function that is a general solution to the model in Scheme 1 with rate constants k1, k-1 and k2. Kinetic resolution of these steps has revealed a reversible, light-dependent lag phase where no O2 is produced, followed by a single exponential recovery phase [55]. The kinetic resolution provides values of the three rate constants (k1, k-1 and k2). The sensitivity of the cell has permitted measurement of these rate parameters using sub-stoichiometric (up to hundreds of fold higher concentrations) of Mn2+/PSII.
Scheme 1.
2.2 Metal ion cofactor
2.2.1 Manganese
Mn appears to be unique in catalyzing O2 evolution in all oxygenic phototrophs. No other metal ions have been found to date that can replace it. We have examined several metal ions as potential surrogates for Mn2+ in the assembly process of spinach apo-WOC-PSII [11]. These studies were conducted by replacing Mn2+, while keeping all other components fixed at the optimal concentrations for Mn-dependent photoactivation (Cl−, Ca2+ and the electron acceptor ferricyanide). The following metal ions were found to inhibit Mn-dependent photoactivation with varying affinities and none were found capable of supporting O2 evolution in the absence of Mn2+ (Cs+, Ba2+, VO2+, V3+, Cr3+, Fe2+, Co3+, Ni2+, Cu2+, Zn2+, MoO42−, Ru3+, Rh3+, Re3+, UO22+). However, only a very limited range of conditions were studied: fixed pH 6 (optimal photoactivation yield using native cofactors) and concentrations of Ca2+, Cl− and electron acceptor. Other pH and concentration conditions ought to be examined before it is known for sure whether Mn can be replaced. The photoactivation process has been studied over a 250-fold range of Mn2+ concentrations and it was found that a Mn2+/Ca2+ ratio about 1:500 achieves optimal photoactivation yield, extending over the physiological range of concentrations[49]. At higher ratios an aberrant pathway is followed that photooxidizes too much Mn2+, while at lower ratio insufficient Mn2+ results in photodamage.
To date, manganese has been found invariably in all PSII-WOCs in vivo and is a required cofactor for photoactivation in vitro. Several factors may have conspired to make manganese the universal metal for catalyzing water splitting in all oxygenic phototrophs. Open shell transition metals are the clear winners versus main group atoms at storing/delivering large units of energy in the form of redox reactions, e.g., atom transfer reactions, in contrast to acid-base or electrostatic-induced chemistry (eg., Mg2+, Ca2+, Zn2+, Cl−, etc.) and ion concentration gradients (Na+, K+, H+). This preference arises because transition metals store and deliver energy in the form of both charge (the number of electrons) and bond strengths (potential and kinetic energy of electrons and nuclei). Their versatility originates from the greater number and smaller energy spacing of their valence d-orbitals which facilitates matching with substrate orbitals for maximal bond strength and accommodates a wider range of geometries[56]. A direct consequence is that the geometrical constraints can be relaxed in cases where the redox changes involve non-bonding d-orbitals. For the same reasons, the activation barriers between redox states in multi-step catalysis are usually lower.
Phototrophs take up Mn2+ but use it in the Mn3+ and Mn4+ oxidation states in the WOC. Mn2+ is highly soluble, has weak or modest affinity for simple ligands, speciates as the simple aqua cation in water over a wide pH range and is readily transported into cells by a variety of transport systems. Mn2+ is chemically stable in neutral solutions. It auto-oxidizes in O2-saturated solutions only above pH 9.5 where most phototrophs do not function, or at ambient pH only if exceptionally strong chelaters are accessible to stabilize the resulting Mn3+ (e.g., pyrophosphate). Hence, there is usually no need for chaperone proteins in the cell to deliver Mn2+ or to protect the cellular matrix from auto-oxidation. Chaperones for manganese have not been identified (yet) for assembly of the WOC during the PSII repair process. This simplification led to a great savings in energy and complexity for the primitive phototroph that first used Mn2+ over other more abundant redox metals (Fe2+) in the anaerobic world when oxygenic photosynthesis was invented. The redox potential for oxidizing Mn2+(aq) → Mn3+(aq) E0 = 1.2 V closely matches the redox potential of Chl-a containing reaction center(> 1.2 V), unlike the majority of other soluble transition ions. Thus, the emergence of Chl-a containing phototrophs enabled the photooxidation of Mn2+ to occur on the surface of Chl-a proteins. The resulting Mn3+ binds more tightly by ca. 1012–1015 and thus would be longer lived at the protein site. Moreover, loss of the redox energy by charge recombination is greatly slowed owing to the energy released upon dissociation of a proton to form [Mn3+(OH−)]2+ (pKa ~ 0). Energetically downhill direct electron-hole recombination without reprotonation is also greatly slowed owing to the unusually large vibronic distortion that Mn3+ produces with its ligand environment (e.g., 1 eV activation barrier for pseudo-rotation of the Jahn-Teller axes in MnF63-)[56]. Importantly, the photooxidation of the next Mn2+ is catalyzed by formation of the first [Mn3+(OH−)]2+ photoproduct, as the resulting OH− can serve as a better ligand than water to template the binding of the next Mn2+. The efficiency of this assembly process increases in the presence of bicarbonate which is a surrogate for hydroxide and can exist in neutral pH solutions at concentrations many orders of magnitude higher than hydroxide[13, 57].
2.2.2. Calcium
Ca2+ is an essential ion in the photoassembly reaction in all native PSII-WOCs examined to date. If Ca2+ is left out of the solution, binding and photooxidation of many more Mn2+ occurs (up to 20) to the apo-WOC-PSII protein and no O2 evolution activity is observable[45, 58]. The relative binding affinities of various divalent metals (Mg2+, Ca2+, Mn2+, Sr2+), and the oxo-cation UO22+ have been characterized [11, 59, 60]. At low Ca/RC concentrations measurements of the kinetics of photoactivation reveal that the Ca2+ binding affinity increases following photooxidation of the first Mn2+ → Mn3+, during which 1 Ca2+ binds per PSII [55]. This photooxidation step and Ca2+ binding leads to a slow dark process that is uncharacterized but has been postulated to represent a protein conformational change. At high Ca/RC concentrations however, Ca2+ is already bound to its effector site and the protein conformation change is induced upon Mn2+ photooxidation. This process leads to more rapid uptake and photooxidation of the remaining 3 Mn2+ in kinetically unresolved steps [55]. Photoactivation kinetics and EPR spectroscopy reveal that when Ca2+ binds in the dark to its specific site it induces the formation of a new intermediate consistent with the bridged species [Mn2+-OH-Ca2+]. Illumination of this species gives rise to loss of a proton, consistent with a [Mn3+-O-Ca2+] intermediate, the existence of which was shown by a freeze-trap experiment and characterized by EPR (see below)[61]. Binding of Ca2+ to its effector site, eliminates this pH dependence and locks both geff and AZ of the photo-oxidized Mn3+ EPR signal at values observed in the absence of Ca2+ at alkaline pH. Thus, Ca2+ directly controls the coordination environment and binds close to the high-affinity Mn3+, probably sharing a bridging ligand. This Ca2+ effect and the pH-induced changes are consistent with ionization of a bridging water molecule, predicting that [Mn3+–(μ-O2−)–Ca2+] or [Mn3+–(μ-OH−)2–Ca2+] are the first light intermediates in the presence of Ca2+. Formation of this intermediate templates the apo-WOC-PSII for the subsequent rapid cooperative binding and photooxidation of 3 additional Mn2+ ions, forming the active water oxidase. The photoactivation and spectroscopic data noted above provide a self-consistent picture locating the Ca effector site as an integral part of the Mn4 core, in direct association with Mn probably via shared bridging ligands to Mn [28], [61].
Boussac discovered that Sr2+ can functionally replace Ca2+ in the intact holo-enzyme of PSII membranes and thus, restore O2 evolution activity [62]. Later it was found that there is a kinetic advantage for uptake of Sr2+ versus Ca2+ at the Ca-effector site during assembly of the apo-WOC enzyme and it was determined how each of the three rate constants measured by photoactivation are affected [11]. Sr2+ was shown to be five times more effective than Ca2+ in accelerating the rate of the first two assembly steps (k1 and k2) and two times better in retarding the deactivation step (k−1). Sr2+ competes with Ca2+ and thus occupies the Ca-effector site of the photoassembled PSII. A 65% lower O2 evolution yield per flash is obtained for the Sr-photoactivated enzyme, which is very close to the decreased steady-state O2 evolution rate in the Sr-exchanged holo-enzyme. This decrease in O2 flash yield is known to reflect a retardation of the rate of the final step, S3 → S0 + O2 [63]. Thus, the data taken together indicate that Ca2+ is clearly involved in accelerating the final step in the S-state catalytic cycle, involving the O-O bond formation reaction. No other metal ion other than Sr2+ has been found to functionally replace Ca2+ in water splitting. On this basis, it has been proposed that the non-directional bonding properties of these alkaline Earth ions and the ionic charge density of Ca2+ are critical for allowing formation of a peroxide intermediate, Ca2+(O22−), between oxo bridges in the step leading to O2 evolution from the S4 state of the Mn4O4Ca cluster[25, 28]. A role for Ca2+ as Lewis acid catalyst for proton ionization of substrate water and generation of a nucleophilic hydroxide has also been considered [24, 25], although this ignores the direct effect that Ca2+ has on the electronic energy of the Mn ions[28].
2.3 Inorganic Cofactors
2.3.1. Oxide bridges/Proton release
In order to build the Mn4Ca cluster from its free metal ions it is necessary to deprotonate four or more aqua ligands attached to the initially bound Mn2+. Photoactivation has provided important clues about how this process occurs. Alkaline pH accelerates the rate of Mn-dependent photoactivation by 10-fold between pH 5.5 and 7.5, while the final yield of O2 per flash follows closely the known pH dependence of the holo-enzyme (peaks at pH 6). The data indicate that protons are released during the first two steps of assembly [11, 45]. Direct measurements of proton stoichiometry released during photoactivation have been described using a commercial ISFET (ion-selective field-effect transistor) detector [11]. These measurements reveal that a single proton is released upon photooxidation of the high affinity Mn2+ to Mn3+ in the presence of Ca2+. The first light-induced assembly intermediate therefore contains one fewer proton than the dark precursor and, if this proton is assumed to originate from ionization of a water ligand, it can be formulated as, [Mn3+(O2−)Ca2+] or [Mn3+(OH−)2Ca2+] [61].
2.3.2 Bicarbonate ion
Bicarbonate ion is important not only as a substrate for carbon fixation in the Calvin cycle but also for its role in PSII electron transport efficiency and photoassembly of the WOC cluster in PSII. In recent years the requirement for bicarbonate to observe efficient in vitro photoassembly has been shown convincingly [49, 50]. In cyanobacteria, either bicarbonate or NaHCO3 is actively taken into the cell via ATP-dependent membrane pumps that control either intake or the expulsion of Na+. Consequently, bicarbonate uptake is tightly controlled and coupled to the light reactions via photophosphorylation. Three distinct transport systems known as carbon concentration mechanisms (CCM) are involved, including one for CO2 uptake [64]. The photosynthetic activity of the cyanobacterial CCM is modulated in response to inorganic carbon (Ci), with 50% stimulation at concentrations between a low-affinity, constitutive state (approximately 200 μM) and a high-affinity, induced state (10–15 μ is stored M). Ci intracellularly in the form of HCO3− and converted back to CO2 in structures called carboxysomes for fixation by the Calvin-Benson-Bassham reactions. This conversion uses enzymes known as carbonic anhydrase which exist in multiple subclasses, including a thylakoid membrane associated carbonic anhydrase[65]. Some alkalophilic cyanobacteria (Arthrospira genus) that originate from volcanic soda lakes prefer very high extracellular carbonate concentrations reaching 0.4 to 1.2 M NaCO3. These organisms lack carbonic anhydrases all together[66]} and possess a reversible bicarbonate site within the WOC that is required for O2 evolution ([67]). This site may be the same as the bicarbonate site associated with the Arg 357 residue of subunit CP43 which when mutated, gives rises to lowered O2 evolution activity, hypothesized as due to a more energetic two-electron oxidation pathway that forms H2O2 [68].
PS II appears to possess its own biochemical machinery for regulating the bicarbonate concentration within the lumenal space near the WOC. A low level of carbonic anhydrase (CA) activity, more than 103 slower than typical Zn2+-CA, has been found to be intrinsic to oxygen-evolving PS II membranes [69, 70]. A soluble CA activity was found associated with the 33 kDa extrinsic protein of PS II (PsbO) and depends on the concentration of Mn2+ and Cl−. However the latter results are at contradiction with earlier work showing that neither Mn2+ nor Ca2+ have specific binding sites on the spinach 33-MSP when studied as a free protein [71]. Given the low activity and requirement for Mn2+ there appears to be no clear physiological significance for this site of bicarbonate activity in PSII in vivo [65, 72].
Bicarbonate ions efficiently replace alkaline pH buffers by accelerating the rate of photoactivation, while increasing the yield of reactivated centers. This is contrary to the lower yield observed upon increasing the buffer pH above 6. This can be understood as follows: bicarbonate binds to two sites within apo-WOC complex as depicted in Fig 3 and described next. A high affinity bicarbonate site operates at concentrations as low as 10 μM in the presence of stoichiometric Mn2+/PSII concentrations, where it accelerates the first step of Mn2+ binding and/or photooxidation [50]. The high affinity bicarbonate site has been attributed to the ionization of protons from carboxylate residues or ion-pairing of cationic residues on the surface of the WOC protein complex. These accelerate the rate of photoactivation by electrostatically attracting Mn2+ from solution close to the WOC [49]. The rate stimulation is much like that seen with lipid soluble anions like tetraphenylboron which elevate the water soluble cation concentration at the membrane interface [11]. In more extensive studies covering 10 and 100 fold higher concentrations of Mn2+ and Ca2+, respectively, a second, lower affinity, bicarbonate binding site on apo-WOC-PS II has been identified that responds to changes in the concentrations of Mn2+, PSII and bicarbonate. This ternary site produces three-fold larger rate acceleration of photoactivation versus “bicarbonate free” samples containing only residual bicarbonate arising from atmospheric equilibration (6 μM). Recent EPR measurements on the trapped intermediates have shown that bicarbonate serves as an integral native cofactor for the initial steps (as described in detail later in this review). It was speculated that it could furnish an oxo/hydroxo bridge for the formation of the WOC [49]. Since its conjugate acid CO2 is highly soluble in the membrane phase, there will be higher concentration of bicarbonate near the thylakoid membrane than in bulk solution at equilibrium in any system which gets all of its inorganic carbon from CO2. At still higher (2-8 mM) concentrations of bicarbonate and Mn2+, the formation of free Mn2+-bicarbonate complexes occurs in solution [73]. The formation of these species coincides with a reduction in the rate of photoactivation (lower reconstitution of O2 yield). These free [Mn2+(CO3)n](2−n)+ complexes appear to serve as competitive electron donors to the reaction center. This competitive electron donation is eliminated if the extrinsic subunits are present in the native PSII-WOC, suggesting that a native role of these subunits is to protect the Mn4Ca core from external attack by cellular reductants.
Fig 3.
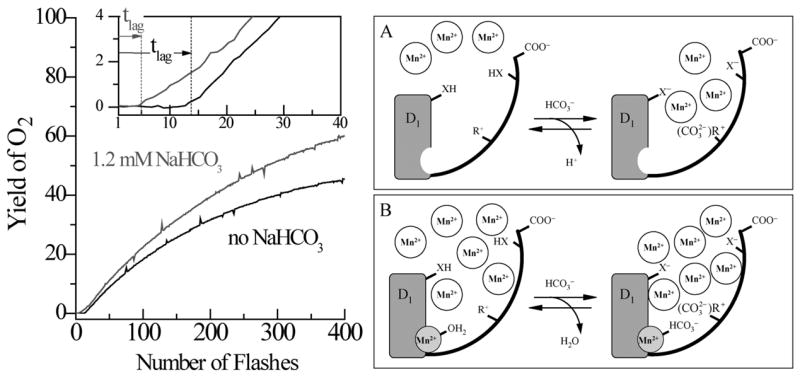
(Left side) Kinetics of reconstitution of O2 evolution capacity by photoactivation of the apo-WOC-PSII membranes in the presence of (1) 0 mM, and (2) 1.2 mM, added bicarbonate. Assay conditions are pH 6.0, 1 μM apo-WOC-PSII, 100 μM MnCl2, 100 mM CaCl2, and 1.8 mM K3Fe(CN)6. The insert expands the first 40 flashes (3 s repetition rate). The vertical dashed lines represent the lag time tlag for each experiment, as derived from fitting the entire kinetics (Baranov et.al. (2004)). (Right side) Two-site model for bicarbonate acceleration of the rate of the first step of photoactivation. (A) The high-affinity bicarbonate site: When the high affinity Mn2+ site is unoccupied at low Mn2+ concentrations ([Mn2+] < 30 μM = KD [61]), (B) The low affinity bicarbonate site: At high Mn2+ concentrations ([Mn2+] > 30 μM = KD), bicarbonate binds directly to the high-affinity Mn2+ binding site (Baranov et.al.)(Dasgupta et al., submitted).
The clear involvement of bicarbonate in the photoassembly process has been interpreted in terms of a possible evolutionary role for bicarbonate in promoting selectivity for Mn2+ binding and photooxidation over other divalent metals in the Archean eon when this chemistry was invented. This work is summarized elsewhere [13, 74, 75].
2.3.3. Chloride ion
The role of chloride ion in photoactivation is not clearly revealed, as its influence occurs only following the rate-limiting step and thus affects only the O2 evolution activity of the fully assembled cluster [45]. However, it should be stated that it is an essential cofactor, both for the O2 activity of the native enzyme, and for photoactivation yield, as substitution with nitrate and other surrogate ions inhibits the recovery of O2 activity of the native enzyme. A chloride binding site directly on the Mn4 cluster has been inferred by 14N ESEEM studies of competitive azide binding to the chloride site within intact PSII centers [76]. Evidence against a direct ligation to Mn4Ca cluster has also been put forth by FTIR on chloride depleted nitrate substituted PSII samples[77]. It has been speculated that the role of chloride in the water-splitting mechanism is to participate together with charged amino acid side chains in a proton-relay network, which facilitates proton transfer from the manganese cluster to the medium[78].
2.3.4. Non-native cofactors
Previous studies on photoactivation have also encompassed usage of additional non-native cofactors like tetraphenylboron, histidine and imidazole to accentuate or inhibit the in vitro assembly process[11]. Recently borate, B(OH)4−, a chemical analogue of bicarbonate, has been shown to affect the kinetics of photoassembly of the CaMn4Ox core at only one of the two aforementioned bicarbonate sites during photoactivation [75]. Borate addition in place of bicarbonate causes a 4 fold stimulation of the rate of photoactivation and a 50% increase in yield of photoassembled PSII centers when using the lowest concentrations of Mn2+ (≤ 10 μM Mn2+), at which the high affinity Mn2+ site is not occupied. However, unlike bicarbonate ion, when this latter site is occupied at 50–100 fold larger concentrations of Mn2+ (and sufficient Ca2+), borate addition shows no effect on the rate of the first Mn2+ photooxidation step or the yield of photoassembly. Borate, like bicarbonate, does not affect the second (rate-limiting) dark step of photoactivation which is attributed to a protein conformational change [79]. This indicates that borate acts only at the high affinity bicarbonate site (which is not involved in binding of Mn2+). The borate acceleration of photoactivation seen at the high affinity bicarbonate site is stronger than bicarbonate, which is consistent with its stronger basicity (pKa = 9.3) vs. bicarbonate (pKa = 6.3). At this site, borate serves as a base to promote proton ionization or ion-pairing with residues of PSII that are responsible for electrostatic steering of Mn2+ to the interface of the unfolded apo-WOC-PSII precursor. In this way, PSII utilizes borate and bicarbonate to elevate the local Mn2+ concentration well above the bulk solution concentration. This feature offers a clear advantage for building a multinuclear Mn cluster in which incompletely photoassembled intermediates deactivate by autoreduction unless sufficient Mn2+ is available for building the next stable intermediate of the cluster. The absence of a borate effect on the low affinity bicarbonate site indicates selectivity for bicarbonate at the high affinity Mn2+ binding site. This is fully consistent with the poor metal coordinating properties of the borate anion compared to bicarbonate anion in solution. The dissociation constant of the borate ion with Mn2+ at pH 6.0 is KD > 0.5 M [75], which is at least 10 fold weaker affinity than bicarbonate [73].
Cadmium ions (Cd2+) are known to inhibit reconstitution of O2 evolution in spinach PSII in competition with Ca2+ [80]. Recently it was shown that Cd2+ and Gd3+ also inhibit the photoactivation process by binding selectively to the Ca2+ effector site, but do not bind to the high affinity Mn2+ site [81]. This binding was shown to be competitive and reversible with Ca2+. In the presence of Cd2+ a new kinetic pathway of photoactivation occurs in competition with the normal two-step pathway. Paradoxically, this inhibition of O2 yield is accompanied by a large acceleration of the rate of recovery of O2 evolution, equal to a 5–10 x faster dark step (k2, Fig 5) and by a slightly slower deactivation rate of the first light-induced intermediate (reduction of photooxidized Mn3+). The data suggest that the binding of the first Mn2+ (e.g., the high affinity Mn site) increases by 5–10 fold in the presence of Cd2+ bound to the Ca2+ effector site, attributable to acceleration of the protein conformational change that limits the overall rate of photoassembly. These data reveal that metal ions that bind to the Ca2+ effector site interact with the high affinity Mn2+ through mutual positive cooperative binding, yielding greater thermodynamic stability and more efficient binding/photooxidation of the high affinity Mn2+.
Fig 5.

The sequence of kinetic intermediates (top) formed during assembly of the inorganic core of the WOC by photoactivation (A, B,C). (Bottom) Proposed chemical formulation for intermediates.
2.4 Role of protein subunits in assembly
As noted above, the D1, D2, CP47, CP43 and cytochrome b559 subunits comprise the minimal oxygen-producing core of native PS II (a number of small subunits that lack cofactors are also generally present in cores)[22]. A stable CP47RC subcore complex assembles within the thylakoid membrane of cyanobacteria in the deletion mutant lacking the psbC gene for CP43 [82]. These complexes can be isolated and are active in electron transport but inactive in O2 evolution, presumably due to an incomplete Mn cluster. A stable CP47RC subcore complex has been prepared from spinach by removal of CP43 with detergents. Although the CP47RC subcore is devoid of Mn and inactive in O2 evolution, it is capable of substantial activity (20% vs native control) after reconstitution of the Mn cluster by photoactivation in the presence of the free inorganic cofactors [23]. The aforementioned high sensitivity O2 electrode/photoactivation cell was absolutely essential for observing this reconstitution. The successful reconstitution of the CP47RC subcore together with the subunit positioning data and XRD of the Mn binding site suggest that a RC subcore lacking CP47, might also be capable of O2 evolution. However, to date no stable subcore complex lacking CP47 has been isolated to test this hypothesis. The implication is that both CP43 and CP47 play an essential role in creating the conditions needed for efficient photoassembly of a stable Mn4Ca cluster.
The peripheral protein subunits including the manganese stabilizing protein (MSP) have been shown to play an important role in the rate and quantum yield of photoactivation in vivo. Burnap and coworkers have identified that in Synechocystis sp. PCC 6803 MSP deletion or mutant altering the binding affinity of MSP responds with higher photoactivation quantum efficiency in whole cells. They argue that absence of MSP [83, 84] or, in this case, the altered binding of MSP renders the active site (Mn2+ high affinity site) more accessible to incoming Mn2+. Increased accessibility of the active site of H2O oxidation is shown by a variety of studies showing that the extrinsic PSII polypeptides shield the Mn4Ca catalyst from attack by exogenous reductants, and the carboxyl ligands to the Mn4Ca from chemical modification. The increased accessibility of the active site was hypothesized to result in a higher probability usage of the oxidizing equivalents generated by the reaction center for the photooxidation and ligation of Mn atoms rather than becoming dissipated via charge recombination. Another reason for increased quantum yield that they provide was that MSP binding affects protonation/deprotonation of the critical protons necessary for photoassembly. Hence, the absence of MSP may permit more facile deprotonation of the critical ionizable groups and/or shift their pKa of the ionized/un-ionized equilibrium toward the deprotonated state [85].
Mechanism of Photoactivation
3.1 Two-quantum model and extended models
Through the pioneering work of Cheniae and his coworkers, it was shown that photoactivation is a multi-quantum process and the basic kinetic scheme derived from these original studies remains intact[12], although the molecular details have been significantly extended. Photoactivation involves the formation of two unstable intermediates separated by a dark rearrangement step. The basic features of this model were developed by studying the yield of photoactivation in Mn-depleted samples as a function of both light intensity and flash interval in a train of single-turnover flashes. Cheniae’s observation was that the quantum yield for producing O2-evolving centers was low at very low light intensities, reached a maximum at intermediate intensities, and was again low at high light intensities. In all cases, the quantum yield of photoactivation was observed to be much lower than the quantum yield of photosynthetic oxygen evolution. They reasoned that photoactivation required at least two quanta of light per reaction center with one of the steps occurring with very low quantum yield. When photon fluxes were low, labile photoproducts decayed before the next quantum of light could advance the photoactivation reaction to the next intermediate. On the other hand, intermediate light intensities suppressed the decay due to photooxidation to the next intermediate in the sequence. The fact that high light intensities also gave low quantum yields led them to postulate that a dark relaxation process occurs after the absorption of the first quantum and must take place before the next quantum can be productively utilized. This interpretation was secured by the outcome of paired flash experiments, which demonstrated that two quanta were sufficient to advance the assembling centers through the low quantum yield phase and required the prediction of both the forward dark rearrangement and the decay process. Experiments from Burnap’s lab have used paired flash experiments to reveal a third kinetic phase of photoactivation, presumably representing an alternate pathway to the second intermediate, IM2[86]. The new rate is 5-10 times faster than the rate-limiting dark rearrangement step that is normally observed. Assuming that the dark rearrangement step is a protein conformational change, Burnap asserts the possibility that a fraction of the centers may already exist in the rearranged state during or shortly after the first light-induced step. These centers are thus able to process the second quantum without the prior delay required by the conformational change.
Since the development of the fast oxygen electrode [52], O2 evolution measurements have enabled kinetic resolution of these intermediates, as described in a series of publications that have been reviewed [11, 13]. Next we review more recent work aimed at trapping these intermediates for spectroscopic characterization.
3.2 Trapping assembly intermediates
Initial attempts to trap the intermediates for spectroscopic characterization have been carried out mainly in three labs (Cheniae, Brudvig, and Dismukes). Apo-WOC-PSII samples prepared by TRIS washing, NH2OH treatment or chelator treatment all suggested the presence of a dimanganese intermediate that was claimed to be a true kinetic intermediate in overall photoactivation (IM2) and thus productive in formation of the final Mn4Ca cluster. However, all these trapping experiments disagreed on one major criterion: the preparation of the initial apo-WOC-PSII sample could not supporting full reconstitution upon photoactivation. Britt and coworkers later showed that photooxidation of Mn2+ by apo-WOC-PSII isolated from Synechocystis sp. PCC 6803 leads to a trappable Mn3+ intermediate characterized by EPR which was assigned to the first light intermediate (IM1)[87]. Again however, systematic measurements of the fate of this detectable Mn3+ intermediate in photoactivation were not reported.
In more recent studies, we have used pulsed-EPR of the Mn2+-apo-WOC-PSII dark intermediate and parallel-mode EPR of the photoconverted Mn3+ form to characterize the high affinity Mn intermediate at low temperature. We have used spinach apo-WOC-PSII samples that reconstitute 60% or more of O2 evolution activity in the photoactivation reaction [49]. The EPR method measured the reversible binding of the photooxidizable Mn2+ in the dark—e.g., without interference from light dependent reactions at this site and thus it gives a true measure of the equilibrium dissociation constant for this Mn2+ (KD = 40–50 μM at pH 7.5). This Mn2+ site possesses the highest affinity of apo-WOC-PSII and therefore, we associate this photooxidizable site with the previously described “high-affinity Mn2+ site” involved in electron donation [88, 89] and photoactivation [54, 90]. It is noteworthy that all methods that report a considerably higher affinity (KD ~ 0.1–3 μM) rely on photooxidation/illumination and thus are not reversible by definition.
3.2.1. Ca2+ bound intermediates
We used EPR spectroscopy to characterize the ligand coordination environment of the first photoactivation intermediate (IM1 and IM1* in Fig 5), corresponding to a photooxidized Mn3+ species bound to apo-WOC-PSII [61]. The interconversion of the intermediates detected by EPR are schematically shown in Fig 4. In the absence of any added Ca2+, this species was shown to exist in two pH-dependent forms differing in terms of strength and symmetry of their ligand fields and whose populations are in equilibrium via a pH-dependent process. Transition from an EPR-invisible low-pH form to an EPR-active high-pH form occurs by deprotonation of an ionizable ligand bound to Mn3+, postulated to be a water molecule: [Mn3+(OH2)]↔[Mn3+(OH−)]. The EPR-active Mn3+ exhibits a strong pH dependence (pH ~ 6.5–9) of its ligand field symmetry deduced from changes to both geff of the Mn3+ EPR signal and independently the 55Mn magnetic hyperfine splitting (ΔAZ = 22%) and attributed to a protein conformational change. The application of ligand field theory to the change in geff indicates the rhombicity changes by Δδ = 10%, where δ is the mixing coefficient of the 5B1g ground state (d4 hole in the d(x2-y2) orbital) with the excited 5A1g state (d4 hole in the d(z2) orbital) caused by orthorhombic distortions of a predominantly tetragonal ligand field). The binding of Ca2+ at its effector site nearly eliminates this pH-dependence of the Mn3+ spectral parameters, with δ becoming constant (4.05 ± 0.05) and AZ smaller and with greatly reduced variation (4%) in the pH range 6.5 to 9.0. Importantly, both δ and AZ approximate to those observed at high pH ~ 9 in the absence of Ca2+, and were assigned to ionization of a water ligand, [Mn3+(OH−)]. It was implicated thus that Ca2+ binding induces proton ionization from a water ligand in the coordination shell of Mn3+ and also organizes the coordination shell to make it more uniform and less susceptible to global protein conformational changes induced by pH. The data are consistent with the formation of an oxide (or di-μ2-hydroxide) bridge, [Mn3+(O2−)Ca2+]. Here, Ca2+ is proposed to induce ionization of a second water ligand bound to Mn3+, or more plausibly, a second ionization of the bridging hydroxo ligand resulting in a bridging oxide ion. Ionization of the water ligand is coupled to the availability of a local base, B−, with pKa~ 6.7 as can be estimated from the pH titration curve. Finally, formation of this intermediate [Mn3+(O2−)Ca2+], templates the apo-WOC-PSII for the subsequent rapid cooperative binding and photooxidation of 3 additional Mn2+ ions, forming the active water oxidase. However, no experimental evidence characterizing these subsequent intermediates is yet available.
Fig 4.

Schematic representation of the trapped intermediates in the first steps of photoactivation in presence of Ca2+. Protein-derived and non-changing water ligands to Mn2+/Mn3+ are not shown. Brackets [ ] represent the high-affinity Mn2+ site of apo-WOC-PSII. The CW EPR signals from [Mn3+-O2−- Ca2+] in red and [Mn2+-O2− - Ca2+] in blue is shown. See text for details.
3.2.2. Bicarbonate effect on intermediates IM1 and IM1*
Bicarbonate coordination of free Mn2+ in water lowers the oxidation potential for photoconversion to Mn3+ by a large amount (0.5–0.6 V vs. NHE)[91]. This thermodynamic stabilization is thought to be responsible for the increased stability of the Mn3+ photoactivation intermediate that forms in the presence of bicarbonate, as seen by slowing of the recombination reaction that causes its decay[49]. However, evidence for direct interaction between the high affinity Mn2+/Mn3+ forms and bicarbonate has been lacking until recently [75]. Two lines of evidence have shown that bicarbonate forms a ternary complex by coordinating directly to Mn2+ at the high affinity site in apo-WOC-PSII and increasing the yield of photooxidation to form Mn3+. Parallel-mode CW-EPR spectroscopy has shown that bicarbonate controls the ligand field strength of the high affinity Mn3+ ion [92]. In the absence of bicarbonate, geff decreases monotonously by 0.8% with increasing pH between 6 and 9. This correlation means that the rhombicity of the ligand field of Mn3+ decreases with increasing pH. The shallow slope and linearity over 3 pH units suggests a continuous range of protein conformational changes within PSII that gradually transform the local ligand field symmetry at the high-affinity Mn3+ site to a more axially symmetric environment as the pH increases. Upon addition of 2–10 mM bicarbonate this pH dependence of geff disappears and is locked into a more rhombic ligand field environment that is no longer in equilibrium with the bulk pH. A similar conclusion can be drawn from the pH dependence of the 55Mn hyperfine splitting, AZZ. In the absence of added bicarbonate, AZZ decreases monotonically by 16–22 % as pH increases from 6 to 9. Addition of 2–10 mM bicarbonate eliminates this pronounced pH dependence, and locks AZZ within a narrow range, which is equal to the maximum AZZ observed at acidic pH in the absence of bicarbonate. Comparison to ligand field calculations and experimental data available for other transition metal complexes indicates that the decrease of AZZ can be attributed to either expansion of the Mn3+ d-orbitals bearing unpaired spins, arising from weakening of electron repulsion with its ligands (lower dipolar hyperfine), or alternatively, to an increase of the ligand-metal covalency at higher pH (lower isotropic hyperfine). To summarize, addition of bicarbonate causes both geff and AZZ of Mn3+ to confine to a narrow range that is independent of solution pH between 6 and 8. This binding produces a fixed inner coordination environment at the high affinity Mn3+ site that is considerably more resistant to environmental pH variations. This effect is specific for bicarbonate as other anions including borate do not affect the Mn site. These data provide compelling evidence that bicarbonate is a native cofactor for the assembly of the Mn4Ca-core.
The complementary pulsed EPR approach electron spin echo envelop modulation (ESEEM) spectroscopy has been applied to answer whether bicarbonate binds directly to the high affinity Mn2+ located in apo-WOC-PSII [75, 93]. By comparison of ESEEM data for isotopically labeled 13C- and 12C- bicarbonate, a weak (~1.45 MHz) electron-nuclear hyperfine interaction was clearly resolved at the 13C Larmor frequency. Good simulation of the line-shape and intensity profile was achieved and together with comparison to model transition metal-bicarbonate complexes[73] resulted in the following description of this site: (bi)carbonate is bound as a direct ligand in the first coordination sphere of Mn2+ in a bi-dentate mode and thus likely speciating as the carbonate form.
3.2.3 Mechanism of Photoactivation
Fig 5 shows the current molecular mechanism of photoactivation, culminating in formation of the active Mn4CaOx core, based on the evidence accumulated from both O2 recovery kinetics and EPR spectroscopy on the trapped intermediates. Formation of the first Mn3+ intermediate involves: 1) diffusion of Mn2+ to PSII enhanced by electrostatic attraction from anionic amino acid residues and bound bicarbonate ions; 2) Mn2+ binding to the “high affinity” site in the D1 protein likely as [Mn2+(OH2)]. This intermediate is denoted IM0 and may consist of three dark equilibrated species, including bicarbonate bound, water bound and Ca2+ bound forms; 3) A single turnover flash (that starts the assembly process) or photoaccumulation at −20 °C leads to the formation by photooxidation of this bound Mn2+ precursor to Mn3+ with concomitant loss of a proton (IM1). This intermediate is unstable until a protein conformational change that occurs in the dark takes place and forms the kinetically more stable intermediate IM1* [Mn3+-O-Ca2+]; 4) Binding of Ca2+ in the dark to its own specific site occurs either before or after this step, albeit with higher affinity after photooxidation forms Mn3+. This increased affinity reflects cooperative binding energy between Mn3+ and Ca2+. Since bicarbonate addition does not affect the k2 step and also there is no evidence yet for a bicarbonate bound form in this state, the bound bicarbonate from the first photooxidation process may dissociate or be released as CO2. Ultimately the active site is primed for uptake of another Mn2+ which forms a spin-coupled cluster (composition Mn2+Mn3+Ca2+) that has not yet been detected by EPR. More detailed spectroscopy has to be carried out in order to trap and characterize the second photooxidation intermediate, IM2 and those that follow. However, to date no kinetic resolution has been observed in the final photoassembly steps forming IM2 or later intermediates nor the protein conformational change. Titrations of the four Mn2+ ions required for photoassembly has previously shown that they bind irreversibly once the active Mn4Ca core is assembled[45], implying that the “fast” assembly of the remaining 3 Mn2+ reflects favorable interaction energy between Mn ions (cooperative binding). Some evidence has been provided by Dau and coworkers using EXAFS of another trapped intermediate. They have investigated a thermal disassembly pathway of the native PSII complex which retains a Mn-Mn interaction (~ 2.7 Å separation) in the active site [94]. Activity in photoactivation was not examined in this case.
4. Future Directions
The complexity of the assembly process has inspired two new approaches in order to sort out questions regarding the inorganic chemistry involved. Use of synthetic Mn-complexes as sources of Mn2+ for the photoactivation of apo-WOC-PSII is becoming popular. All of the organic ligands that constitute these complexes are more lipophilic and tend to give complexes that have poor solubility in aqueous buffer.
Recently, Klimov and co-workers reported multiple classes of N-donor (bipy-, picolinate- and pyrazine-class) and O-donor (acetates) ligands which can be used to deliver Mn and found that dimeric complexes lead to higher yields of photoactivation [95, 96]. Also Liu and coworkers reported 79 % photoactivation yield of apo-WOC-PSII with a Mn2+ complex prepared with imidazolyl and phenolic ligands [97]. Although these numbers are impressive, the mechanisms of photoactivation by such complexes have not been studied, so there is no information yet on what sort of intermediate forms during assembly, how many Mn are ultimately bound and the chemical speciation of Mn (Mn2+ is a labile ion) in the system. Systematic studies have to carried out on such systems in order to learn ultimately how to assemble complexes on protein scaffolds directly from inorganic complexes.
Another emerging topic is designing artificial photoactivatible systems with specific Mn-binding sites within photosystems that possess an intrinsic chromophore. These permit light induced redox chemistry that drives the binding and photooxidation of Mn2+. This method has not yet been explored fully with artificial peptides. However, Allen and co-workers have demonstrated that a modified bacterial reaction center complex from Rhodobacter sphaeroides can be produced that photooxidizes Mn2+, but only in the presence of bicarbonate [74, 98]. Their results provide the first evidence that a stable Mn3+ binding site can be formed in a bacterial reaction center. This suggests a feasible route for evolution of a primitive precursor to the Mn4Ca core found universally in contemporary oxygenic reaction centers [13, 57]. Construction of biological-inspired completely abiotic synthetic representations of the PSII protein scaffolding and catalytic WOC site are underway in some laboratories[99].
5. Conclusions
This review highlights some of the considerable progress that has been made in understanding the molecular events leading to the biogenesis and repair of the Mn4Ca-cluster and clarification of the role of the inorganic cofactors including bicarbonate. It also raises several important unanswered questions regarding the kinetic and molecular aspects of the steps in this process. In particular unresolved questions remain that are well suited for future investigation by photoactivation: Why is the quantum yield for photoactivation so low (in plant PSII) and do more efficient photoassemblers exist among other oxygenic phototrophs? Can we engineer organisms for higher quantum efficiency of photoassembly? What is the nature of the protein rearrangement that takes place during the slow dark step? What is the minimal number of single turnover photooxidation steps needed to make an O2 molecule during photoactivation starting from Mn2+? What types of aborted chemical reactions occur instead of O2 production in partially assembled clusters or inorganic mutants—eg., does H2O2 form? In light of the recent EPR data on early trapped intermediates, it is clear that significant structural insight can be mined that could be applied to characterizing the nature of the final steps of the photoassembly process. This should encourage a more systematic attempt to explore the landscape of alternative metal ions as substitutes for Mn and Ca, consideration of a wider pH range to overcome chemical speciation limitations of the non-native cofactors, and extension to higher temperatures using thermophiles to overcome activation barriers (< 73 °C is conceivably possible).
Acknowledgments
We thank Drs. A.M. Tyryshkin, D. R. J. Kolling and S. V. Baranov for helpful discussions and careful reading of the manuscript.
ABBREVIATIONS
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- CW
continuous wave
- EPR
electron paramagnetic resonance
- EDTA
ethylenediaminetetraacetic acid
- ESEEM
electron spin echo envelop modulation spectroscopy
- EXAFS
extended X-ray absorption fine structure
- MSP
manganese stabilizing protein
- WOC
water oxidizing complex
Footnotes
PLEASE NOTE THE ORDER OF THE AUTHORS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holland HD. Chemical Evolution of the Atmosphere and Oceans. Princeton University Press; Princeton, USA: 1984. [Google Scholar]
- 2.Krishtalik LI. Biophysics. 1989;34:958–962. [Google Scholar]
- 3.Rappaport F, Guergova-Kuras M, Nixon PJ, Diner BA, Lavergne J. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 4.Renger G. Biochim Biophys Acta. 2001;1503:210–228. doi: 10.1016/s0005-2728(00)00227-9. [DOI] [PubMed] [Google Scholar]
- 5.Keren N, Berg A, VanKan PJM, Levanon H, Ohad I. Proc Natl Acad Sci USA. 1997;94:1579–1584. doi: 10.1073/pnas.94.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zak E, Norling B, Maitra R, Huang F, Andersson B, Pakrasi HB. Proc Natl Acad Sci USA. 2001;98:13443–13448. doi: 10.1073/pnas.241503898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frasch W, Sayre RT. Photosynth Res. 2001;70:245–247. doi: 10.1023/A:1014773304784. [DOI] [PubMed] [Google Scholar]
- 8.Gray HB, Winkler JR. Quart Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 9.Alstrum-Acevedo JH, Brennaman MK, Meyer TJ. Inorg Chem. 2005;44:6802–6827. doi: 10.1021/ic050904r. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y. Inorg Chem. 2006;45:9930–9940. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ananyev GM, Zaltsman L, Vasko C, Dismukes GC. Biochim Biophys Acta. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. [DOI] [PubMed] [Google Scholar]
- 12.Burnap RL. Phys Chem Chem Phys. 2004;6:4803–4809. [Google Scholar]
- 13.Dismukes GC, Blankenship RE. In: Photosystem II: The Water/Plastoquinone Oxido-Reductase in Photosynthesis. Wydrzynski T, Satoh K, editors. Vol. 22. Springer; The Netherlands: 2005. pp. 609–626. [Google Scholar]
- 14.Hankamer B, Morris E, Nield J, Gerle C, Barber J. J Struct Biol. 2001;135:262–269. doi: 10.1006/jsbi.2001.4405. [DOI] [PubMed] [Google Scholar]
- 15.Kashino Y, Lauber WM, Carroll JA, Wang QJ, Whitmarsh J, Satoh K, Pakrasi HB. Biochemistry. 2002;41:8004–8012. doi: 10.1021/bi026012+. [DOI] [PubMed] [Google Scholar]
- 16.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 18.Yano J, Kern J, Sauer K, Latimer MJ, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. Proc Natl Acad Sci USA. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nanba O, Satoh K. Proc Natl Acad Sci USA. 1987;84:109–112. doi: 10.1073/pnas.84.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yruela I, Miota F, Torrado E, Seibert M, Picorel R. Eur J Biochem. 2003;270:2268–2273. doi: 10.1046/j.1432-1033.2003.03597.x. [DOI] [PubMed] [Google Scholar]
- 22.Hankamer B, Barber J, Boekema EJ. Annu Rev Plant Physiol Mol Biol. 1997;48:641–671. doi: 10.1146/annurev.arplant.48.1.641. [DOI] [PubMed] [Google Scholar]
- 23.Buchel C, Barber J, Ananyev G, Eshaghi S, Watt R, Dismukes C. Proc Natl Acad Sci USA. 1999;96:14288–14293. doi: 10.1073/pnas.96.25.14288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta J, van Willigen RT, Dismukes GC. Phys Chem Chem Phys. 2004;6:4793–4802. [Google Scholar]
- 26.Siegbahn PEM. Curr Opin Chem Biol. 2002;6:227–235. doi: 10.1016/s1367-5931(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 27.McEvoy JP, Brudvig GW. Phys Chem Chem Phys. 2004;6:4754–4763. [Google Scholar]
- 28.Carrell TG, Tyryshkin A, Dismukes GC. J Biol Inorg Chem. 2002;7:2–22. doi: 10.1007/s00775-001-0305-3. [DOI] [PubMed] [Google Scholar]
- 29.Britt RD, Campbell KA, Peloquin JM, Gilchrist ML, Aznar CP, Dicus MM, Robblee J, Messinger J. Biochim Biophys Acta. 2004;1655:158–171. doi: 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Yano J, Pushkar Y, Glatzel P, Lewis A, Sauer K, Messinger J, Bergmann U, Yachandra V. J Am Chem Soc. 2005;127:14974–14975. doi: 10.1021/ja054873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debus RJ. In: Photosystem II: The Water/Plastoquinone Oxido-Reductase in Photosynthesis. Wydrzynski T, Satoh K, editors. Vol. 22. Springer; The Netherlands: 2005. pp. 261–284. [Google Scholar]
- 32.Hillier W, Wydrzynski T. Biochim Biophys Acta. 2001;1503:197–209. doi: 10.1016/s0005-2728(00)00225-5. [DOI] [PubMed] [Google Scholar]
- 33.Chow WS, Aro E-M. In: Photosystem II: The Light-driven Water:Plastoquinone Oxidoreductase. Wydrzynski TJ, Satoh K, editors. Vol. 22. Springer; Dordrecht: 2005. pp. 627–648. [Google Scholar]
- 34.Keren N, Liberton M, Pakrasi HB. J Biol Chem. 2005;280:6548–6553. doi: 10.1074/jbc.M410218200. [DOI] [PubMed] [Google Scholar]
- 35.Suorsa M, Regel RE, Paakkarinen V, Battchikova N, Herrmann RG, Aro EM. Eur J Biochem. 2004;271:96–107. doi: 10.1046/j.1432-1033.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- 36.Roose JL, Pakrasi HB. J Biol Chem. 2004;279:45417–45422. doi: 10.1074/jbc.M408458200. [DOI] [PubMed] [Google Scholar]
- 37.Nixon PJ, Trost JT, Diner BA. Biochemistry. 1992;31:10859–10871. doi: 10.1021/bi00159a029. [DOI] [PubMed] [Google Scholar]
- 38.Lers A, Heifetz PB, Boynton JE, Gillham NW, Osmond CB. J Biol Chem. 1992;267:17494–17497. [PubMed] [Google Scholar]
- 39.Trost JT, Chisholm DA, Jordan DB, Diner BA. J Biol Chem. 1997;272:20348–20356. doi: 10.1074/jbc.272.33.20348. [DOI] [PubMed] [Google Scholar]
- 40.Mohamed ZA. Water Res. 2001;35:4405–4409. doi: 10.1016/s0043-1354(01)00160-9. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa T, Bao DH, Katoh H, Shibata M, Pakrasi HB, Bhattacharyya-Pakrasi M. J Biol Chem. 2002;277:28981–28986. doi: 10.1074/jbc.M204175200. [DOI] [PubMed] [Google Scholar]
- 42.Keren N, Kidd MJ, Penner-Hahn JE, Pakrasi HB. Biochemistry. 2002;41:15085–15092. doi: 10.1021/bi026892s. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharyya-Pakrasi M, Pakrasi HB, Ogawa T, Aurora R. Biochem Soc Trans. 2002;30:768–770. doi: 10.1042/bst0300768. [DOI] [PubMed] [Google Scholar]
- 44.Smith RM, Martell AE. Critical Stability Constants. Plenum; New York: 1976. [Google Scholar]
- 45.Ananyev GM, Dismukes GC. Biochemistry. 1996;35:4102–4109. doi: 10.1021/bi952667h. [DOI] [PubMed] [Google Scholar]
- 46.Schroda M. Photosynth Res. 2004;82:221–240. doi: 10.1007/s11120-004-2216-y. [DOI] [PubMed] [Google Scholar]
- 47.Schroda M, Kropat J, Oster U, Rudiger W, Vallon O, Wollman FA, Beck CF. Biochem Soc Trans. 2001;29:413–418. doi: 10.1042/bst0290413. [DOI] [PubMed] [Google Scholar]
- 48.Cheniae G, Martin IF. Plant Physiol. 1976;57:25. [Google Scholar]
- 49.Baranov S, Tyryshkin A, Katz D, Dismukes G, Ananyev G, Klimov V. Biochemistry. 2004;43:2070–2079. doi: 10.1021/bi034858n. [DOI] [PubMed] [Google Scholar]
- 50.Baranov SV, Ananyev GM, Klimov VV, Dismukes GC. Biochemistry. 2000;39:6060–6065. doi: 10.1021/bi992682c. [DOI] [PubMed] [Google Scholar]
- 51.Hunziker D, Abramowicz DA, Damoder R, Dismukes GC. Biochim Biophys Acta. 1987;890:6–14. [Google Scholar]
- 52.Ananyev GM, Dismukes GC. Biochemistry. 1996;35:14608–14617. doi: 10.1021/bi960894t. [DOI] [PubMed] [Google Scholar]
- 53.Tamura N, Cheniae GM. Biochim Biophys Acta. 1987;890:179–194. [Google Scholar]
- 54.Miller AF, Brudvig G. Biochemistry. 1989;28:8181–8190. doi: 10.1021/bi00446a033. [DOI] [PubMed] [Google Scholar]
- 55.Zaltsman L, Ananyev G, Bruntrager E, Dismukes GC. Biochemistry. 1997;36:8914–8922. doi: 10.1021/bi970187f. [DOI] [PubMed] [Google Scholar]
- 56.Bersuker IB. Electronic structure and properties of transitioin metal compounds. J. Wiley; New York: 1996. [Google Scholar]
- 57.Dismukes GC, Klimov VV, Baranov SV, Kozlov YN, Dasgupta J, Tyryshkin A. Proc Natl Acad Sci U S A. 2001;98:2170–2175. doi: 10.1073/pnas.061514798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen GX, Blubaugh DJ, Homann PH, Golbeck JH, Cheniae GM. Biochemistry. 1995;34:2317–2332. doi: 10.1021/bi00007a028. [DOI] [PubMed] [Google Scholar]
- 59.Ananyev GM, Murphy A, Abe Y, Dismukes GC. Biochemistry. 1999;38:7200–7209. doi: 10.1021/bi990023u. [DOI] [PubMed] [Google Scholar]
- 60.Vrettos JS, Stone DA, Brudvig GW. Biochemistry. 2001;40:7937–7945. doi: 10.1021/bi010679z. [DOI] [PubMed] [Google Scholar]
- 61.Tyryshkin AM, Watt RK, Baranov SV, Dasgupta J, Hendrich MP, Dismukes GC. Biochemistry. 2006;45:12876–12889. doi: 10.1021/bi061495t. [DOI] [PubMed] [Google Scholar]
- 62.Boussac A, Rutherford AW. Biochemistry. 1988;27:3476–3483. [Google Scholar]
- 63.Westphal KL, Lydakis-Simantiris N, Cukier RI, Babcock GT. Biochemistry. 2000;39:16220–16229. doi: 10.1021/bi0018077. [DOI] [PubMed] [Google Scholar]
- 64.Badger MR, Price GD. J Exp Bot. 2003;54:609–622. doi: 10.1093/jxb/erg076. [DOI] [PubMed] [Google Scholar]
- 65.Rudenko NN, Ignatova LK, Ivanov BN. Photosyn Res. 2007;91:81–89. doi: 10.1007/s11120-007-9148-2. [DOI] [PubMed] [Google Scholar]
- 66.Hillier W, McConnell I, Badger MR, Boussac A, Klimov VV, Dismukes GC, Wydrzynski T. Biochemistry. 2006;45:2094–2102. doi: 10.1021/bi051892o. [DOI] [PubMed] [Google Scholar]
- 67.Carrieri D, Ananyev GM, Brown T, Dismukes GC. submitted. [Google Scholar]
- 68.Ananyev G, Nguyen T, Putnam-Evans C, Dismukes GC. Photochem Photobiol Sci. 2005;4:991–998. doi: 10.1039/b507519j. [DOI] [PubMed] [Google Scholar]
- 69.Lu YK, Theg SM, Stemler AJ. Plant Cell Physiol. 2005;46:1944–1953. doi: 10.1093/pcp/pci209. [DOI] [PubMed] [Google Scholar]
- 70.Lu YK, Stemler AJ. Plant Physiol. 2002;128:643–649. doi: 10.1104/pp.010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hunziker D, Abramowicz DA, Damoder R, Dismukes GC. In: Progress in Photosynthesis Research. Biggins J, editor. I. Martinus-Nijhoff; Dordrecht: 1987. pp. 5.597–5.600. [Google Scholar]
- 72.Moskvin OV, Shutova TV, Khristin MS, Ignatova LK, Villarejo A, Samuelsson G, Klimov VV, Ivanov BN. Photosynth Res. 2004;79:93–100. doi: 10.1023/B:PRES.0000011925.93313.db. [DOI] [PubMed] [Google Scholar]
- 73.Dasgupta J, Tyryshkin AM, Kozlov YN, Klimov VV, Dismukes GC. J Phys Chem B. 2006;110:5099–5111. doi: 10.1021/jp055213v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalman L, LoBrutto R, Allen JP, Williams JC. Biochemistry. 2003;42:11016–11022. doi: 10.1021/bi034747o. [DOI] [PubMed] [Google Scholar]
- 75.Dasgupta J. PhD Thesis. Princeton University; Princeton: 2006. [Google Scholar]
- 76.Yu H, Aznar CP, Xu XZ, Britt RD. Biochemistry. 2005;44:12022–12029. doi: 10.1021/bi0505767. [DOI] [PubMed] [Google Scholar]
- 77.Hasegawa K, Kimura Y, Ono T. Biophys J. 2004;86:1042–1050. doi: 10.1016/S0006-3495(04)74179-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olesen K, Andréasson LE. Biochemistry. 2003;42:2025–35. doi: 10.1021/bi026175y. [DOI] [PubMed] [Google Scholar]
- 79.Dasgupta J, Ananyev GM, Dismukes GC. submitted. [Google Scholar]
- 80.Matysik J, Alia A, Nachtegaal G, van Gorkom H, Hoff A, de Groot H. Biochemistry. 2000;39:6751–6755. doi: 10.1021/bi0004145. [DOI] [PubMed] [Google Scholar]
- 81.Bartlett JE, Baranov SV, Ananyev GM, Dismukes GC. Phil Trans Roy Soc. In Press. [Google Scholar]
- 82.Rögner M, Chisholm DA, Diner BA. Biochemistry. 1991;30:5387–5395. doi: 10.1021/bi00236a009. [DOI] [PubMed] [Google Scholar]
- 83.Burnap RL, Qian M, Pierce C. Biochemistry. 1996;35:874–882. doi: 10.1021/bi951964j. [DOI] [PubMed] [Google Scholar]
- 84.Qian M, AlKhaldi SF, PutnamEvans C, Bricker TM, Burnap RL. Biochemistry. 1997;36:15244–15252. doi: 10.1021/bi9713198. [DOI] [PubMed] [Google Scholar]
- 85.Qian M, Al-Khaldi SF, Putnam-Evans C, Bricker TM, Burnap RL. Biochemistry. 1997;36:15244–15252. doi: 10.1021/bi9713198. [DOI] [PubMed] [Google Scholar]
- 86.Hwang HJ, Burnap RL. Biochemistry. 2005;44:9766–9774. doi: 10.1021/bi050069p. [DOI] [PubMed] [Google Scholar]
- 87.Campbell KA, Force DA, Nixon PJ, Dole F, Diner BA, Britt RD. J Am Chem Soc. 2000;122:3754–3761. [Google Scholar]
- 88.Debus RJ. Biochim Biophys Acta. 1992;1102:269–352. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 89.Hsu BD, Lee JY, Pan RL. Biochim Biophys Acta. 1987;890:89–96. [Google Scholar]
- 90.Tamura N, Cheniae GM. FEBS Lett. 1986;200:231–236. [Google Scholar]
- 91.Kozlov YN, Zharmukhamedov SK, Tikhonov KG, Dasgupta J, Kazakova AA, Dismukes GC, Klimov VV. Phys Chem Chem Phys. 2004;6:4905–4911. [Google Scholar]
- 92.Dasgupta J, Tyryshkin AM, Baranov SV, Dismukes GC. submitted. [Google Scholar]
- 93.Dasgupta J, Tyryshkin AM, Dismukes GC. Angew Chem Int Ed. In press. [Google Scholar]
- 94.Barra M, Haumann M, Loja P, Krivanek R, Grundmeier A, Dau H. Biochemistry. 2006;45:14523–14532. doi: 10.1021/bi061842z. [DOI] [PubMed] [Google Scholar]
- 95.Li SQ, Chen GY, Han GY, Ling L, Huang DG, Khorobrykh AA, Zharmukhamedov SK, Liu QT, Klimov VV, Kuang TY. J Biol Inorg Chem. 2006;11:783–790. doi: 10.1007/s00775-006-0126-5. [DOI] [PubMed] [Google Scholar]
- 96.Han GY, Li J, Chen GY, Ling L, Li SQ, Khorobrykh AA, Zharmukhamedov SK, Klimov VV, Kuang TY. J Photochem Photobiol B. 2005;81:114–120. doi: 10.1016/j.jphotobiol.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 97.Liu B, Shen PP, Shi W, Song YG, Li W, Nie Z, Liu Y. J Biol Inorg Chem. 2006;11:626–632. doi: 10.1007/s00775-006-0111-z. [DOI] [PubMed] [Google Scholar]
- 98.Thielges M, Uyeda G, Camara-Artigas A, Kalman L, Williams JC, Allen JP. Biochemistry. 2005;44:7389–7394. doi: 10.1021/bi050377n. [DOI] [PubMed] [Google Scholar]
- 99.Koder RL, Dutton PL. Dalton Trans. 2006:3045–3051. doi: 10.1039/b514972j. [DOI] [PubMed] [Google Scholar]