

Abstract
In situ combination of diphosphinic amides and Zr(NMe2)4 results in the formation of chiral zirconium bis(amido) complexes. The complexes are competent catalysts for intramolecular asymmetric alkene hydroamintion, providing piperidines and pyrrolidines in up to 80% ee and high yield. This system utilizes an inexpensive zirconium precatalyst and readily prepared ligands and is the first asymmetric alkene hydroamination catalyst based upon a neutral zirconium bis(amido) complex.
The development of catalysts for intramolecular asymmetric alkene hydroamination has been the subject of intense investigation over the past 15 years.1,2,3,4,5 While considerable advances have been made using catalysts containing a variety of metals, no general solution has emerged. To date, catalysts based on Group 3 and lanthanide metals have shown the most promise for unactivated alkenes.1,2,3 However, even within this class only a small number (4) of highly enantioselective reactions (>90% ee) have been reported.3c Thus alkene hydroamination remains an open area of research.
Recently, Schafer6 (and subsequently Livinghouse7) reported that neutral Group IV bis(amido) complexes bearing achiral ligands are competent catalysts for intramolecular alkene hydroamination. Our group has previously reported that closely related catalysts containing chiral dialkoxide and diamide ligands are effective in intramolecular allene and alkyne hydroamination.8 We decided to explore the possibility that these types of chiral complexes could be applied to asymmetric alkene hydroamination. Herein, we report the first examples of asymmetric, intramolecular alkene hydroamination catalyzed by Group 4 bis(amido) complexes.9 The reported catalysts employ readily available chiral ligands and provide enantioselectivities of up to 80% ee.
Having previously demonstrated that in situ combination of various diamines or diols and Group IV tetrakis(dimethyl)amides provides competent hydroamination catalysts,8 we screened various combinations of these compounds as catalysts in the cyclization of 1 (eq 1).10,11 In an effort to develop a practical catalyst, we focused exclusively on commercially available or readily prepared diols, diamines and aminoalcohols (Table 1).12 Although enantioselective catalysts based on titanium and hafnium13 were also identified, the zirconium catalyst prepared by combination of diphosphinic amide 3c12c and Zr(NMe2)4 (entry 11) proved significantly more enantioselective than others that we studied. Under unoptimized conditions (see footnote, Table 1), pyrrolidine 2 was obtained in 67% ee and excellent yield. Given this initial promise, we elected to explore additional ligands of the same general structure.
Table 1.
Results from initial screen of ligands for conversion of 1 to 2.
| entry | ligand | M(NMe2)4, % conversion(% ee) | ||
|---|---|---|---|---|
| Ti | Zr | Hf | ||
| 1 | none | 97(0) | 80(0) | 88(0) |
| 2 | R,R-3a | <5(n/a) | 92(21) | 94(13) |
| 3 | R-4a | <5(n/a) | 89(18)a | 59(30)a |
| 4 | R-4b | <5(n/a) | 90(23)a | 24(26)a |
| 5 | S,S-3b | 97(9) | 94(20) | 94(13) |
| 6 | R-4c | 88(<5) | 87(<5) | 87(<5) |
| 5 | S-4d | <5(n/a) | 97(<5) | 63(19) |
| 6 | 5a | 59(13) | 97(24)a | 98(9)a |
| 7 | 5b | 52(6)a | 98(27)a | 99(12)a |
| 8 | 6 | 84(26) | 96(<5) | 98(7) |
| 9 | 7 | <5(n/a) | <5(n/a) | <5(n/a) |
| 10 | 8 | 95(15)a | 99(34)a | 73(18)a |
| 11 | R,R-3c | 8(<5) | 96(67) | 76(58) |
Conditions: 22 mol% ligand, 20 mol% M(NMe2)4, 135 °C, tol, 24 h; ligand and M(NMe2)4 were pre-heated 135 °C for 15 min prior to introduction of 1. Unless otherwise indicated, the (S)-enantiomer of product predominate. Conversion was determined using chiral GC by comparison of the sum of the enantiomers of product to the remaining starting material.
The (R)-enantiomer was predominate.
(1).
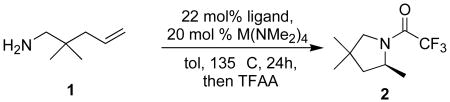
A series of diphosphinic amides was prepared using one of two short synthetic sequences.14 These ligands were investigated in the cyclization of 1 (Table 2). Ligands containing the cyclohexanediamine backbone proved to be the most selective (entries 1-5). Variation of the phosphorus substituent (R) had pronounced effects on catalytic activity. The ortho-tolyl-containing ligand 13a gave catalytically inactive complexes (entries 6), while the 3,5-bis(trifluromethyl)phenyl-substituted ligand 13b decomposed when exposed to the Zr(NMe2)4. Ligands containing other electron-deficient arenes proved more stable to the reaction conditions, but did not provide improved catalysts (entries 9 and 10). The addition of electron-donating groups did improve catalyst performance in the cyclization of 1. The para-methoxyphenyl-containing ligand 13e provided slightly higher enantioselectivity. The more sterically demanding ortho-methoxyphenyl group in 13f provided even higher selectivity, but at the cost of slightly diminished reactivity. Among the ligands screened, the best proved to be 3,5-dimethylphenyl-substituted ligand 13g (entry 12). This ligand provided the best combination of reactivity and enantioselectivity, providing 2 in 75% ee and 99% conversion under these unoptimized conditions. Larger meta substituents did not improve the catalyst performance (entry 13).
Table 2.
Results from the use of diphosphinic amide ligands with Zr(NMe2)4 in the conversion of 1 to 2.
| entry | ligand | R | % conversion | % ee |
|---|---|---|---|---|
| 1 | 3c | Ph | 98 | 67 |
| 2 | 9 | Ph | 96 | 50 |
| 3 | 10 | Ph | 95 | 55 |
| 4 | 11 | Ph | <1 | n/a |
| 5 | 12 | Ph | 46 | 16 |
| 6 | 13a | o-tol | <1 | n/a |
| 7 | 13b | 3,5- C6H3(CF3)2 | decomp | n/a |
| 8 | 13c | 3,5-C6H3F2 | 92 | 63 |
| 9 | 13d | 3,5-C6H3(OMe)2 | 99 | 68 |
| 10 | 13e | p-C6H4(OMe) | 99 | 70 |
| 11 | 13f | o-C6H4(OMe) | 95 | 76 |
| 12 | 13g | 3,5-C6H3Me2 | 99 | 75 |
| 13 | 13h | 3,5-C6H3(tBu)2 | 98 | 62 |
Conditions: 22 mol% ligand, 20 mol% Zr(NMe2)4, 135 °C, tol, 24 h; ligand and Zr(NMe2)4 were pre-heated 135 °C for 15 min prior to introduction of 1. In all cases, the ligand was the (R,R)-enantiomer and the (S)-enantiomer of 2 was predominate in the product. Conversion was determined using chiral GC by comparison of the sum of the enantiomers of product to the remaining starting material.
Having identified a promising ligand, we sought to further optimize the cyclization of 1 using the combination of 13g and Zr(NMe2)4 as catalyst. Neither variation of the solvent (PhH, Ph-F, Ph-Cl, 1,4-dioxane and pyridine), nor the addition of basic (LiNMe2)15 or acidic (HEt2O·B(C6F5)4) additives affected the yield or ee of the product obtained in the reaction. Lowering the reaction temperature did improve the ee, and under optimized conditions, 2 was produced in 95% yield and 80% ee in 24 h using 20 mol% each of 13g and Zr(NMe2)4 (Table 3). Lowering the catalyst loading to 10 mol% provided 2 in 91% yield and identical ee in 48 h.
Table 3.
Substrate scope under optimized conditions.
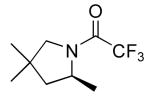
| entry | product | mol% cat.a | temp (°C)/time (h) | % yield (% ee) | |
|---|---|---|---|---|---|
| 1 |
|
2 | 20 | 115/24 | 95(80)c |
| 10 | 115/48 | 91(80)c | |||
| 2 |
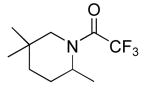
|
14 | 20b | 85/24 | 99(51)c |
| 10b | 85/48 | 91(51)c | |||
| 3 |
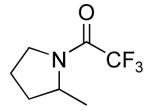
|
15 | 20 | 135/72 | 33(62)c,d |
| 4 |
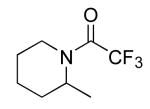
|
16 | 10 | 135/24 | 79(33)c |
| 5 |
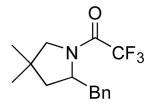
|
17 | 20 | 135/24 | 93(62)c |
| 6 |
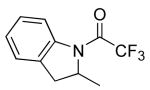
|
18 | 20 | 115/48 | 93(70)c |
| 20 | 115/48 | 85(70)e | |||
| 7 |
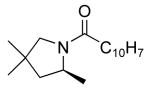
|
19 | 10 | 115/48 | 78(80)f |
| 8 |
|
20 | 20 | 135/24 | 75(55)f |
Reactions were performed in toluene.
Prepared in situ by combination of equimolar amounts of ligand and Zr(NMe2)4 in toluene and heating at the reaction temp for 15 min prior to introduction of substrate.
Ligand and Zr(NMe2)4 pre-heated at 105 °C for 15 min.
GC yield determined using C6Me6 as an internal standard; ee determined by chiral GC.
32% starting material remained in solution at the end of the reaction.
Isolated yield; ee determined by chiral GC.
Isolated yield as 2-napthoyl amide; ee determined by chiral HPLC.
Table 3 illustrates the substrate scope of the asymmetric hydroamination reaction using the combination of 13g and Zr(NMe2)4 as the catalyst. Formation of piperidine 14 (entry 2) proceeded in 99% yield and 51% ee using 20 mol% catalyst at 85 °C. Lowering the catalyst loading to 10 mol% provided 14 in slightly lower yield but without erosion of ee. Substrates lacking the geminal dimethyl group could also be cyclized, albeit in diminished yield and ee (entries 3 and 4). Substrates containing trans-disubstituted alkenes cyclized in high yield with moderate enantioselection (entry 5). Cyclization of 2-allylaniline provided 18 in 93% yield and 70% ee as determined by GC analysis (entry 5). On a larger scale, 18 could be isolated in 85% yield and identical ee. As a means of corroboration, we also wished to isolate other hydroamination products. 2-Napthoyl chloride proved the most convenient derivitizing agent for this propose, providing amides of sufficient molecular weight and stability for facile isolation. Isolated yields and enantioselectivities for amides prepared by this method (entries 7 and 8) are similar to those determined by the GC method.
Although we have not yet undertaken detailed mechanistic studies of this catalytic system, we have attempted to identify the catalytically active species generated from the combination of the diphosphinic amide ligands and Zr(NMe2)4. Upon combination of either 3c or 13g and an equimolar amount of Zr(NMe2)4 in d8-toluene at 25 °C, a complex mixture of compounds arose with 10 unassigned signals observed in the 31P NMR spectrum. Heating this mixture at 75 °C for 24 h led to the clean formation of the expected 1:1 Zr:ligand adduct 20 (Scheme 1), along with traces of the 2:1 adduct 21.14 The structure of 20 was assigned by the indicative dimethylamide signals in the 1H NMR spectrum at ca. 3.55 ppm and a single peak in the 31P NMR spectrum at ca. 33 ppm. Addition of substrate to in situ generated 20 and subsequent heating provided product at a rate similar to the rates of the reactions reported in Table 1. Heating the solution of 20 at 135 °C (with or without substrate present) resulted in the slow formation of 21. At 150 °C, in the absence of substrate, 20 was fully converted to 21 within 24 h. The structure of 21a has been determined by X-ray crystallographic analysis (Fig 2). Both 21a and 21g were catalytically inactive.
Scheme 1.

Products from the reaction of diphosphinic amide ligands and Zr(NMe2)4.
Figure 2.
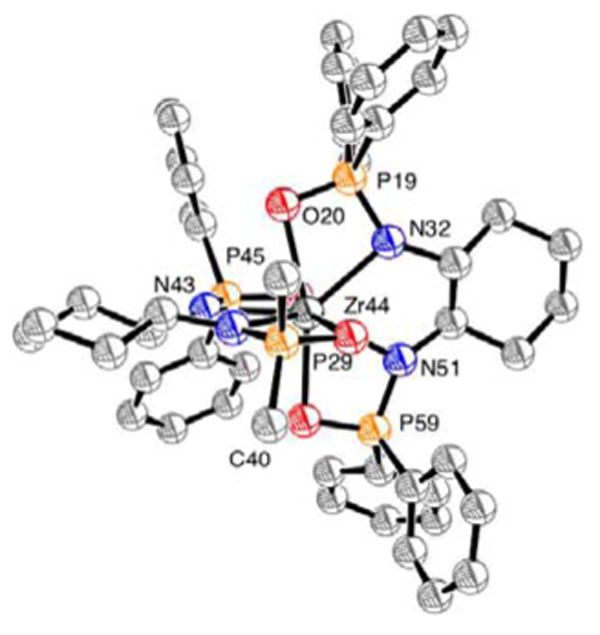
ORTEP representation of the solid state structure of 21a. Arenes connected to P29 have been truncated for clarity. Thermal ellipsoids are shown at 50% probability.
Based upon these studies, and those previously reported by our group and others,6,7,8 this reaction likely proceeds via a transient imidozirconium species according to the general mechanism presented in Scheme 2. Combination of 13 and Zr(NMe2)4 results in the formation of 20. Addition of substrate allows the reversible loss of dimethylamine and the formation of the imidozirconium species 22. Subsequent [2+2] cycloaddition and protonation of the resulting azametallocyclobutane 23 by substrate regenerates the imido species and delivers the cyclic amine product. Slow decomposition of 20 to 21 represents a pathway for catalyst decomposition. Further research designed to inhibit the latter process represents a potential avenue for further optimization of this catalyst system.
Scheme 2.

Proposed catalytic cycle and catalyst decomposition.
In summary, we have developed the first asymmetric alkene hydroamination catalyst based upon a neutral zirconium bis(amido) complex. This system utilizes an inexpensive zirconium precatalyst and a readily prepared diphosphinic amide ligand and provides cyclic amines in high yields and up to 80% ee. In situ preparation of the active catalyst obviates the need to isolate moisture-sensitive compounds. Studies aimed at further optimization of the ligand architecture and full elucidation of the mechanism of this transformation are ongoing.
Supplementary Material
Complete experimental details for the preparation of diphosphinic amide ligands and cyclization experiments, copies of the 1H and 13C NMR spectra for 3c, 9-12, 13a-13g, 17-21, and chromatograms for ee analysis.
Figure 1.

Structures of ligands examined.
Acknowledgments
We thank Mr. Jared Lewis for helpful discussions regarding experimental design. This work was supported by NIH National Institute of General Medical Science (GM-25459), a NIH-NRSA postdoctoral fellowship (GM068266) to DAW and a NSF predoctoral fellowship to MC. We are grateful to Prof. Laurel Schafer (Univ. of British Columbia) for a mutual exchange of information regarding an analogous project proceeding in her laboratory.
References
- 1.Gagne MR, Brard L, Conticello VP, Giardello MA, Stern CL, Marks TJ. Organometallics. 1992;11:2003–2005. [Google Scholar]
- 2.For reviews of asymmetric hydroamination, see: (a) Togni, A.; Dorta, R.; Kollner, C.; Pioda, G. 1998, 70, 1477–1485.; a Togni A, Dorta R, Kollner C, Pioda G. 1998;70:1477–1485. [Google Scholar]; b Roesky PW, Muller TE. Angew Chem Int Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637. [DOI] [PubMed] [Google Scholar]; c Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]; d Hultzsch KC. Org Biomol Chem. 2005;3:1819–1824. doi: 10.1039/b418521h. [DOI] [PubMed] [Google Scholar]; e Hultzsch KC. Adv Synth Catal. 2005;347:367–391. [Google Scholar]; f Hii KK. Pure Appl Chem. 2006;78:341–349. [Google Scholar]
- 3.For recent reports of hydroaminations using lanthanum and Group 3 metal complexes, see:; a Kim JY, Livinghouse T. Org Lett. 2005;7:1737–1739. doi: 10.1021/ol050294z. [DOI] [PubMed] [Google Scholar]; b Collin J, Daran J, Jacquet O, Schulz E, Trifonov A. Chem Eur J. 2005;11:3455–3462. doi: 10.1002/chem.200401203. [DOI] [PubMed] [Google Scholar]; c Birkov DV, Hultzsch KC, Hampel F. J Am Chem Soc. 2006;128:3748–3759. doi: 10.1021/ja058287t. [DOI] [PubMed] [Google Scholar]; d Riegert D, Collin J, Meddour A, Schulz E, Trifonov A. J Org Chem. 2006;71:2514–2517. doi: 10.1021/jo052322x. [DOI] [PubMed] [Google Scholar]
- 4.For asymmetric hydroaminations using late metal catalysts, see:; a Löber O, Kawatsura M, Hartwig JF. J Am Chem Soc. 2001;123:4366–4367. doi: 10.1021/ja005881o. [DOI] [PubMed] [Google Scholar]; b Patil NT, Lutete LM, Wu H, Pahadi NK, Gridnev ID, Yamamoto Y. J Org Chem. 2006;71:4270–4279. doi: 10.1021/jo0603835. [DOI] [PubMed] [Google Scholar]
- 5.For examples of asymmetric hydroaminations using other metals, including cationic zirconium complexes, see:; a Knight PD, Munslow I, O'Shaughnessy PN, Scott P. Chem Commun. 2004:894–895. doi: 10.1039/b401493f. [DOI] [PubMed] [Google Scholar]; b Gribkov DV, Hultzcsh KC. Angew Chem Int Ed. 2004;43:5542–5546. doi: 10.1002/anie.200460880. [DOI] [PubMed] [Google Scholar]; c Martinez PH, Hultzsch KC, Hampel F. Chem Commun. 2006:2221–2223. doi: 10.1039/b518360j. [DOI] [PubMed] [Google Scholar]
- 6.Bexrud JA, Beard JD, Leitch DC, Schafer LL. Org Lett. 2005;7:1959–1962. doi: 10.1021/ol0503992. [DOI] [PubMed] [Google Scholar]
- 7.Kim H, Lee PH, Livinghouse T. Chem Commun. 2005:5205–5207. doi: 10.1039/b505738h. [DOI] [PubMed] [Google Scholar]
- 8.a Ackermann L, Bergman RG. Org Lett. 2002;4:1475–1478. doi: 10.1021/ol0256724. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ackermann L, Bergman RG, Loy RN. J Am Chem Soc. 2003;125:11956–11963. doi: 10.1021/ja0361547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.There has been one report of an asymmetric intramolecular allene hydroamination catalyzed by neutral titanium bis(amido) complexes that employed chiral aminoalcohol ligands. The maximum reported ee was 16%. See:; Hoover JM, Petersen JR, Pikul JH, Johnson AR. Organometallics. 2004;23:4614–4620. [Google Scholar]
- 10.In screening efforts, ligand (22 mol%) and Group IV tetrakis(dimethylamide) (20 mol%) were combined in toluene and heated to 135 °C for 15 min to insure formation of ligand/metal complex. Aminoalkene 1 was then introduced, and the reaction mixture was heated with stirring for 24 h. After acylation with trifluoroacetyl anhydride (TFAA), the product was analyzed by chiral GC.
- 11.The absolute stereochemistry of amide 2 was established by determination of the des-acyl piperdine using the method reported by Livinghouse using O-acetylmandelatic acid; see reference 3a. The absolute stereochemistry of the remaining products have not been established.
- 12.a Jamieson JY, Schrock RR, Davis WM, Bonitatebus PJ, Zhu SS, Hoveyda AH. Organometallics. 2000;19:925–930. [Google Scholar]; b Ghosh AK, Chen Y. Tetrahedron Lett. 1995;36:6811–6814. doi: 10.1016/0040-4039(95)01385-U. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shi M, Sui W. Tetrahedron: Asymmetry. 1999;10:3319–3325. [Google Scholar]; d Shi M, Sui W. Chirality. 2000;12:574–580. doi: 10.1002/1520-636X(2000)12:7<574::AID-CHIR5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 13.To our knowledge, these are the first examples of hafnium-catalyzed hydroaminations.
- 14.See Supporting Information for details.
- 15.The fact that LiNMe2 does not inhibit the reaction excludes the possibility that catalysis is due to trace acidic impurities.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental details for the preparation of diphosphinic amide ligands and cyclization experiments, copies of the 1H and 13C NMR spectra for 3c, 9-12, 13a-13g, 17-21, and chromatograms for ee analysis.