

Abstract
The ability of microbial populations to increase fitness through fixation of mutants with an increased growth rate has been well described. In experimental studies, this is often the only way fitness can be increased. In natural settings, however, fitness can also be improved by increasing the ability of the microbe to transmit from one host to the next. For many pathogens, transmission includes a phase outside the host during which they need to survive before the chance of reinfecting a new host occurs. In such a situation, a reduced death rate during this phase will lead to improved fitness. Here, we compute the fixation probability of mutants that better survive the transmission bottleneck during the evolution of microbial populations. We derive analytical results that show that transmission mutants are often likely to occur and that their importance relative to growth mutants increases as the population decline during the transmission phase increases. We confirm our theoretical results with numerical simulations and suggest specific experiments that can be done to test our predictions.
UNDERSTANDING the evolution of microbial populations is of both fundamental and applied interest. On a fundamental level, the short generation times of microbes allow the study of evolutionary processes in an experimental setting (Lenski et al. 1991; Elena and Lenski 2003; Manrubia and Lazaro 2006; Colegrave and Collins 2008). On an applied level, knowing what drives microbial evolution provides insights into the dynamics of pathogens in response to selective pressures created by factors such as antimicrobial drug use (Baquero and Blazquez 1997; Levin et al. 2000; Handel et al. 2006), host immunity (Kent et al. 2005; Nelson and Holmes 2007), or crossing a host–species barrier (Antia et al. 2003; Woolhouse et al. 2005).
In both experimental and real-world settings, many microbial populations alternate between a phase of population growth inside a flask or host followed by a population bottleneck created by serial passage or transmission. In serial passage experiments, the bottleneck is implemented as an essentially random sampling of the population, whereby a small aliquot of a culture is immediately placed into a flask with fresh nutrients. Even though it is known that bacteria sometimes die during the stationary phase, which usually follows the growth phase before the serial passage bottleneck, the possibility that differences in the survival during stationary phase might lead to fitness differences is usually ignored. Instead, most studies focus on the evolution of fitness as defined by an improved net growth rate [by means of a shorter generation time, increased fecundity, more efficient resource uptake, etc. (Wahl and Dehaan 2004; Novak et al. 2006)]. This is likely justified for most experimental setups. However, for many naturally spreading pathogens, the ability to survive outside a host, for instance on fomites or in an aquatic environment (Walther and Ewald 2004; Kramer et al. 2006), is crucial. Mutations that decrease the death rate of pathogens during this phase, and therefore increase the likelihood of transmission to a new host, can create microbes that have a greater overall fitness than the wild type, without increasing the within-host growth rate. Focusing only on fitness as determined by the pathogen's ability to grow inside a host will miss this important component.
A number of studies have considered trade-offs between pathogen growth inside and survival outside a host. These studies investigated how such trade-offs influence the evolution of virulence for different scenarios (Bonhoeffer et al. 1996; Gandon 1998; Day 2002; Kamo and Boots 2004; Caraco and Wang 2008). Here, we take a different, complementary approach. We use a framework that explicitly models the generation and growth of mutants with increased fitness, which allows us to compute the probability that such mutants invade microbial populations. We explore under what situations we expect to observe “growth mutants,” which have an improved net growth rate inside a host, vs. “transmission mutants,” which have an improved ability to survive outside the host. We derive analytical expressions and compare them with simulations. We find that transmission mutants are likely to arise and survive, especially in situations where the reduction in population size during the transmission phase is close to the increase in population size during the growth phase.
RESULTS
To approximate the process of within-host pathogen growth, followed by a transmission bottleneck, we study the simple model shown in Figure 1. A founder colony of size N0 grows exponentially and deterministically with a growth rate g for a time τg within a host to a size N1 > N0. At this time, a fraction F1 of the population, with size N2 ≤ N1, exits the host and enters the environment. While outside the host, the pathogen population declines at a rate d for a time τd down to a size N3 < N2. A fraction F2 of the pathogens that survive subsequently infect a new host with an inoculum of size N0 ≤ N3.
Figure 1.—

Serial passage with a transmission phase. A founder colony of size N0 grows exponentially at rate g for a time τg within a host (or flask) to a size 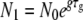 . At this time, a fraction F1 of the population, with size N2 ≤ N1, is transmitted into the environment. While outside the host, the population declines at a rate d for a time τd down to a size N3 < N2. A fraction F2 of the microbes that survive the transmission phase subsequently infect a new host with an inoculum of size N0 ≤ N3 and the next growth phase begins.
. At this time, a fraction F1 of the population, with size N2 ≤ N1, is transmitted into the environment. While outside the host, the population declines at a rate d for a time τd down to a size N3 < N2. A fraction F2 of the microbes that survive the transmission phase subsequently infect a new host with an inoculum of size N0 ≤ N3 and the next growth phase begins.
Survival probability of transmission and growth mutants:
We are interested in the probability that a mutant that has a different growth rate inside the host (growth mutant) or a different death rate during the transmission phase (transmission mutant) will be able to survive and dominate the population. Ignoring stochastic drift, a mutant can invade only if its fitness is larger than the fitness of the existing clone. In our model, this can be achieved by either an increased growth rate (g′ = g(1 + s), s > 0) or a decreased death rate (d′ = d(1 − σ), σ > 0). Previous studies have studied the survival probability of mutants with an increased within-host growth rate (Wahl and Gerrish 2001; Wahl et al. 2002). Here, we allow mutants to change in both growth rate inside the host and death rate during the transmission phase. Let P(t, s, σ) be the probability that an individual mutant with growth mutation s and transmission mutation σ created at time t after the start of a growth phase (0 < t < τg) will not go extinct after all subsequent bottlenecks and therefore be able to take over the population. In the appendix, we show that for the situation illustrated in Figure 1, P(t, s, σ) is given by
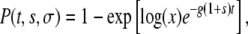 |
(1) |
where x is given by the implicit equation
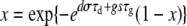 |
(2) |
and denotes the probability that a mutant with growth rate change s and death rate change σ, created at the beginning of the growth phase, will go extinct (Wahl and Gerrish 2001). If we make the—often biologically reasonable—approximation that fitness increases or decreases only by a small amount (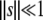 and
and 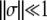 ), then we can approximate the nontrivial (x = 1) solution of Equation 2 as x ≈ 1 − 2gτgs − 2dτdσ, which leads us to
), then we can approximate the nontrivial (x = 1) solution of Equation 2 as x ≈ 1 − 2gτgs − 2dτdσ, which leads us to
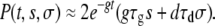 |
(3) |
This result agrees with a previous one for a scenario with no transmission mutants (Wahl and Gerrish 2001; Wahl et al. 2002). We refer the interested reader to these previous studies for some additional details and a discussion on how this fixation probability relates to the classical work by Haldane and others (Haldane 1927; Fisher 1930; Crow and Kimura 1970).
Having determined P(t, s, σ), we can calculate the expected number of mutations with growth advantage s and survival advantage σ that appear during the whole growth phase and survive subsequent bottlenecks, Λ(s, σ). Following Wahl et al. (2002), we can write
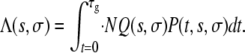 |
(4) |
The rate of creation of new microbes, 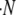 , is given by
, is given by 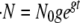 . The expression Q(s, σ) is the probability density per replication that a mutation occurs that has a change in growth rate of size s and a change in death rate of size σ. In other words, Q(s, σ)dsdσ is the probability that when a replication occurs, the new microbe will have a growth mutation between s and s + ds and a transmission mutation between σ and σ + dσ. We specify Q(s, σ) below.
. The expression Q(s, σ) is the probability density per replication that a mutation occurs that has a change in growth rate of size s and a change in death rate of size σ. In other words, Q(s, σ)dsdσ is the probability that when a replication occurs, the new microbe will have a growth mutation between s and s + ds and a transmission mutation between σ and σ + dσ. We specify Q(s, σ) below.
After substituting Equation 3 into Equation 4 we arrive at
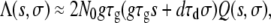 |
(5) |
which specifies the expected number of mutations with change s in growth rate and change σ in death rate that will arise and survive the bottlenecks. To complete Equation 5, we need to specify Q(s, σ), which we do in the next two sections.
Mutations that change a single trait:
Let us first assume that during the growth phase both types of mutations, growth and transmission, can occur but that a mutation in one phenotype does not affect the other. If a particular mutation improves the growth rate of the pathogen, it does not at the same time affect its death rate during the transmission phase, and vice versa (see Figure 2a).
Figure 2.—

Schematic of the mutation process. The possible mutations naturally partition into eight regions, depending on the relative signs of s and σ (s = 0 and σ = 0, which do not affect fitness, are not considered). Solid regions contain deleterious mutations, while mutations in open regions are always beneficial. Shaded regions, in which mutations are positive for one variable and negative for the other, may be either beneficial or deleterious. For each version of the model discussed, two types of mutation (“growth” or “transmission”) can occur, and the regions in which they exist are denoted by the circles. (a) Allowed mutations when pleiotropy is not present. Growth mutations increase growth, g, by s, while transmission mutations decrease death, d, by σ. (b) Allowed mutations with negative pleiotropic effects. Beneficial growth mutations lead to a decrease in d (σ < 0) while beneficial transmission mutations lead to a decrease in g (s < 0).
The function Q(s, σ) for these mutations can then be written as
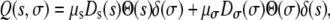 |
(6) |
where δ(x) is the Dirac delta function and Θ(x) is the Heaviside step function. This choice for Q(s, σ) ensures that only either pure growth mutants (s > 0, σ = 0) or transmission mutants (s = 0, σ > 0) occur. The constants μs and μσ are the rates at which either a growth or a transmission mutant is generated (essentially the probability of a mutation occurring during a single replication). The distributions Ds(s) and Dσ(σ) describe the probability distribution for a growth mutation of size s or transmission mutation of size σ (s, σ > 0). For instance, the first term in Equation 6 says that when a growth mutant occurs (with frequency μs), the change in the growth rate, s is ensured to be positive [since Θ(s) = 0 for s < 0] and distributed according to the function Ds(s). Furthermore, when the growth mutation occurs, the delta function, δ(σ), ensures that there will be no corresponding change in the death rate.
On the basis of theoretical arguments and previous studies (Gillespie 1984; Imhof and Schlotterer 2001; Rozen et al. 2002; Orr 2005), we choose the distributions Ds(s) and Dσ(σ) to be exponentially decaying functions as the strength of the mutation increases; i.e., Ds(s) = αsexp(−αss) and Dσ(σ) = ασexp(−ασσ). Substituting Equation 6 into Equation 5 and integrating over s and σ (see appendix) gives
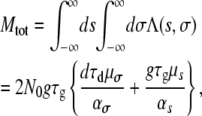 |
(7) |
which is the expected number of mutants that survive the bottlenecks and reach fixation. Note that Equation 7 naturally partitions into two contributions, Mtot = Mσ + Ms, giving the total number of fixed transmission and growth mutants, respectively. The ratio R of expected transmission mutants to growth mutants that reach fixation is given by
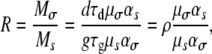 |
(8) |
where we defined
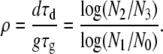 |
(9) |
Equations 8 and 9 show that the ratio of transmission to growth mutants, R, depends on the mutation rates and the distribution of fitness effects, μs, μσ, αs, ασ. While these quantities are likely difficult to measure, it is not unreasonable to assume that in many cases, there are a similar number of ways to improve growth inside a host as there are ways to improve survival in the environment; i.e., these parameters might be of similar magnitude. If this is the case, then R is most strongly influenced by the sizes of the different populations at the beginning and the end of the growth and transmission phases. As ρ varies from zero to one, the scenario changes from one where the transmission phase is negligible to one where contraction during the transmission phase equals the amount of expansion during the growth phase. For a situation where the pathogen population does not contract much during the transmission phase (i.e., N3 ≈ N2), a reduced death rate is obviously of little advantage and we have ρ ≈ 0. Such a situation likely applies to pathogens that use the respiratory route and directly transmit from person to person. For a situation where almost all pathogens are released into the environment and die down to the inoculum size, i.e., N1 ≳ N2 and N3 ≳ N0, we find ρ ≲ 1 (the symbols ≳ and ≲ stand for “greater/less than but very close to”). Such a scenario might be more likely for gastrointestinal infections that use fecal–oral transmission routes or for pathogens that have a long fomite phase. Note that due to the constraint of continued survival of the population, values for ρ are between 0 and 1. Importantly, if we know the inoculum size, N0, the pathogen load at the time of shedding, N1, the amount of pathogens released into the environment, N2, and the number of pathogens at the end of the transmission phase, N3, it is not necessary to know the growth or death rates and duration of the growth and transmission phases.
Mutations that change both traits:
Trade-offs and pleiotropic effects are well-known mechanisms that can influence evolution (Taylor and Higgs 2000; Ostrowski et al. 2005; De Paepe and Taddei 2006; Knies et al. 2006; Sleight et al. 2006). We now account for them by assuming that mutations occur that simultaneously change growth rate, g, and death rate, d. We assume that a mutation that improves fitness in one of the traits causes a pleiotropic negative effect in the other, as shown in Figure 2b. For instance, if a mutation allows a pathogen to survive better in the environment (perhaps by increasing cell wall density), there will be a related decrease in the growth rate (perhaps because building a stronger cell wall slows down the rate of division). We can model this by modifying Q(s, σ), such that a positive effect in one trait is now accompanied by a negative contribution in the other. This leads to
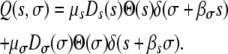 |
(10) |
This equation says that if a growth mutation of magnitude s occurs, there is a related decrease in the survival, with σ = −βσs. Similarly, if there is a positive transmission mutation of size σ, the resulting change in the growth will be s = −βsσ.
Substituting Equation 10 into Equation 5 again gives us two terms representing the number of successful mutations of each type. After integration we arrive at
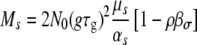 |
(11) |
and
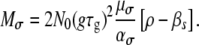 |
(12) |
Note that now a mutant has changed both growth and death rates. We define a transmission mutant as a mutant with reduced death rate, while a growth mutant is a mutant with increased growth rate, as previously. The ratio of expected transmission to growth mutants is found to be
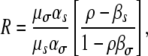 |
(13) |
which reduces to the previous result (Equation 8) in the absence of pleiotropic effects (βs = βσ = 0). As Equation 11 shows, a growth mutation can be successful only if the pleiotropic decrease in σ satisfies βσ < 1/ρ. Otherwise, the mutant would not survive the bottlenecks. Similarly, for transmission mutants to survive we must have βs < ρ. Since ρ ∈ [0, 1], transmission mutants become impossible for βs ≥ 1, i.e., if a decrease in the death rate leads to a comparably strong decrease in the growth rate. Therefore, if the population decline during the transmission phase is small, even small pleiotropic effects on the growth rate make transmission mutants impossible, something that is intuitively obvious. However, for a larger contraction phase, transmission mutants become more likely, and the ratio of transmission to growth mutants increases faster compared to the situation without pleiotropy (Figure 3). While we assumed for simplicity that the pleiotropic relation between growth and survival is linear, qualitatively similar results can be obtained if one considers nonlinear pleiotropic effects.
Figure 3.—

Ratio of transmission to growth mutants, R, with and without pleiotropic effects. We show a situation where a growth mutation leads to a reduction in survival (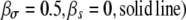 , a transmission mutation leads to a reduction in growth rate
, a transmission mutation leads to a reduction in growth rate 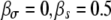 , dashed line), and for both growth and transmission mutations, a reduction in the other trait occurs
, dashed line), and for both growth and transmission mutations, a reduction in the other trait occurs 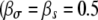 , dashed-dotted line). For comparison, we also plot the case of no pleiotropy (
, dashed-dotted line). For comparison, we also plot the case of no pleiotropy (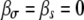 , shaded line). In all cases,
, shaded line). In all cases, 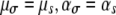 . Changing either
. Changing either 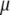 or
or 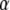 decreases or increases the slope of R, as seen from Equations 8 and 13.
decreases or increases the slope of R, as seen from Equations 8 and 13.
Comparison with simulations:
To test the analytically derived results, we decided to compare them to a simplified version of a previously developed simulation of bacterial growth and dilution cycles (Rozen et al. 2008). At the start of the simulation, the population consists of N0 identical clones. The bacteria divide at intervals given by the inverse of their growth rate. Whenever a clone divides, there is a certain probability that it creates a mutant offspring with either changed within-host growth rate or changed death rate during transmission. Growth continues until the population has reached final size, N1. At this point, multinomial sampling reduces the population size to N2. This is followed by another multinomial sampling, this time weighted by the death rate of each clone, to a population size N3, and one more (unweighted) sampling to population size N0, which then starts a new growth cycle. As in the mathematical model, death during the within-host growth phase is ignored. The only way a clone gets eliminated is by not surviving the transmission bottlenecks. The simulation is implemented in Matlab R2007a (The Mathworks), and the code is available from the authors upon request.
We ran simulations in two ways. In the first scenario, growth or transmission mutants were created during the first growth cycle, and then we recorded how often a mutant reached fixation. This number, divided by the total number of simulations (in most of which no mutant invades), corresponds to Ms and Mσ obtained analytically. From these values, we can compute R. The squares and the solid line in Figure 4 show R obtained from this simulation and the analytical result, respectively. Additionally, we ran simulations for the more realistic situation where mutants can arise during all growth cycles. The simulation is run until either a growth or a transmission mutant has reached fixation. This approach does not lead to values for Ms or Mσ; however, we can still determine the ratio of transmission to growth mutants (circles in Figure 4). Reassuringly, this more realistic scenario leads to results that also agree well with the theory. Since the implementation of the simulation differs somewhat from the model on which our analytics are based, the analytical and simulation results are not in perfect agreement, but nevertheless overall agreement is good, suggesting that the analytical results hold in more realistic situations.
Figure 4.—

Ratio of transmission to growth mutants, no pleiotropy. Solid line: analytics. Squares: mutants can arise only during the first growth cycle, corresponding to the theoretical model. Circles: mutants can arise at any time during the evolution. See text for more details on the simulations. Parameters are N0 = 103, N1 = 107, μs = μσ = 10−7 (Rozen et al. 2002; Perfeito et al. 2007), αs = ασ = 40 (Rozen et al. 2002). For each data point, 5 × 106 (squares) or 103 (circles) simulations were run.
DISCUSSION
A large number of both theoretical and experimental studies have contributed toward our understanding of microbial evolution. Most theoretical results apply to the evolution of populations of fixed sizes (Crow and Kimura 1970; Woodcock and Higgs 1996; Orr 2005; Cowperthwaite et al. 2006). The realization that microbial population sizes often change rapidly and widely led to the study of evolution of populations that vary in size (Otto and Whitlock 1997; Pollak 2000; Wahl and Gerrish 2001; Wilke et al. 2001; Wahl et al. 2002). Transmission bottlenecks are one important case in which the size of microbial populations undergoes extreme changes. A number of studies have considered the impact of bottlenecks for the evolution of pathogens (Bergstrom et al. 1999; Wahl and Gerrish 2001; Wahl et al. 2002; Manrubia et al. 2005; Escarmis et al. 2006). Those studies focus on mutants that have a different fitness as measured by the effective growth rate. However, successful spread of a pathogen in a population depends on its ability to transmit from host to host. While increasing population numbers due to an increase in the growth rate might lead to increased transmission, so does the ability to better survive during the transmission phase (Bonhoeffer et al. 1996; Gandon 1998; Day 2002; Kamo and Boots 2004; Caraco and Wang 2008).
The results obtained from our study suggest that for pathogens that spend time in the environment as part of their transmission strategy, the emergence of mutants that better survive the transmission bottleneck is likely if the transmission phase is an important part of the dynamics of the population. Our study shows that the impact of the transmission phase does not depend on its duration, per se. Instead, the important factor is the amount by which the population is reduced during the transmission phase. Thus a fast death rate with a short transmission phase can have the same effect as a long transmission phase, coupled with a slow death rate. On the basis of our results, we predict that for infections that involve shedding of a large number of pathogens into the environment, followed by strong contraction of the population, the evolution of transmission mutants is likely. For other pathogens that do not undergo strong population contraction during the transmission phase, for instance respiratory pathogens that directly transmit from person to person, mutants that increase survival seem to be less likely to evolve.
Our theoretical predictions should be easily amenable to experimental testing. One might, for instance, alter the standard serial dilution protocol by including an artificial transmission phase. After reaching the end of a growth phase, one can transfer a fraction of the microbes under study into a hostile medium to induce death in some of the remaining population. Then, after some time, a subpopulation from the hostile medium is used to found a new colony and begin a new cycle. Standard competition experiments would detect an increase in growth rate, while “inverse competition experiments” in the hostile medium would be able to detect reduced death rates. On the basis of our results, experiments that had equal growth and decline phases would lead—in the absence of pleiotropy—to equal rates of fixation of transmission or growth mutants. By altering the transmission/decline phase, one should be able to scan the range of R as a function of ρ and obtain results similar to the ones shown in Figures 3 and 4. Additionally, by determining how R changes with ρ and comparing it with the theory, one might be able to obtain indirect experimental evidence for the existence of pleiotropic effects and trade-offs between growth and transmission adaptations. Without pleiotropy, one expects a linear relationship between R and ρ (Equation 8), whereas the relationship will be nonlinear when pleiotropic effects are prominent. The caveat to this is that experimental noise might be too high to discern if there are systematic deviations from the linear slope that might indicate pleiotropy.
Additionally, other effects that occur in real systems but are not part of our model might influence experimental results. For instance, we assumed that extinction of mutants occurs only during bottlenecks. It might be possible to extend the results to a situation where stochastic extinction during the growth phase can occur (Heffernan and Wahl 2002). Further, the analytical part focused on the probability of extinction or fixation of an individual mutation that arises during a growth cycle. While the latter equations allow for multiple mutants, they are all considered to evolve independently; i.e., the model cannot account for features such as clonal interference (Gerrish and Lenski 1998; Orr 2000; Wilke 2004). Nevertheless, we found good agreement with more realistic numerical simulations (which allowed for multiple mutants to compete), and we expect that the theory developed here can help to guide experiments to establish the importance of either growth or transmission mutants in biological systems.
In summary, our study shows that transmission mutants can gain a significant advantage and are quite likely to survive and emerge. The current approach of measuring pathogen fitness by in vitro or in vivo growth rates is useful to answer basic evolutionary questions. However, it cannot be directly applied to the spread of pathogens in populations. To better quantify fitness in these situations, one needs to consider both within-host growth and the ability to transmit. Therefore, it will be important in future experiments to consider transmission mutants as well as growth mutants. We hope the present study will trigger experimental studies of this important aspect of pathogen evolution.
Acknowledgments
We thank Omar Cornejo, Daniel Rozen, and Lev Tsimring for comments on an earlier version of this manuscript. M.R.B. was supported by National Institutes of Health grant GM082168-01.
APPENDIX
Here we present the mathematical derivation of the results discussed in the main text. We first note some useful relations between the populations at the start and the end of each phase. As can be seen in Figure 1, we have 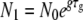 , N2 = F1N1,
, N2 = F1N1, 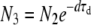 , and N0 = F2N3, leading to
, and N0 = F2N3, leading to 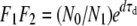 and
and 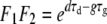 . Note that since 0 < F1F2 < 1, it means that the inequality dτd < gτg must hold, to ensure colony survival. This is just another way of stating the obvious fact that the final size of the microbial population at the end of the transmission bottleneck must be at least as large as the new inoculum.
. Note that since 0 < F1F2 < 1, it means that the inequality dτd < gτg must hold, to ensure colony survival. This is just another way of stating the obvious fact that the final size of the microbial population at the end of the transmission bottleneck must be at least as large as the new inoculum.
Now consider a mutant that has either an increased growth rate, g′ = g(1 + s), or a decreased death rate during the transmission phase d′ = d(1 − σ). We now wish to calculate the probability, x, that the descendants of a single mutant present at the start of the growth phase will eventually go extinct due to the bottleneck. First, if we assume that the growth phase is long enough such that 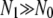 , then the probability Pg(n) that n mutants survive the first dilution and enter the environment is well approximated by
, then the probability Pg(n) that n mutants survive the first dilution and enter the environment is well approximated by
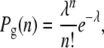 |
(A1) |
where  is the expected number of mutants to exit the host and reach the transmission phase. Once in the environment, the mutant cells die at a rate d′, meaning that the probability for a single mutant to survive the transmission phase is given by
is the expected number of mutants to exit the host and reach the transmission phase. Once in the environment, the mutant cells die at a rate d′, meaning that the probability for a single mutant to survive the transmission phase is given by 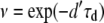 . Therefore, the conditional probability, Pd(m | n) that m mutants survive the transmission phase given that n entered is given by a binomial distribution. In particular, we have
. Therefore, the conditional probability, Pd(m | n) that m mutants survive the transmission phase given that n entered is given by a binomial distribution. In particular, we have
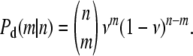 |
(A2) |
After surviving the transmission phase each mutant has a probability F2 to reach the new host. Therefore, the probability, Pnh(l |m), that l mutants reach the new host given that there are m mutants at the end of the transmission phase is once again given by the binomial distribution,
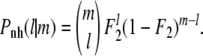 |
(A3) |
Finally, the probability, Pse(l), that l mutants reaching a new host face subsequent extinction is simply given by
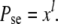 |
(A4) |
If we combine Equations A1–A4 and sum over all possible outcomes, we arrive at an implicit equation for x, namely
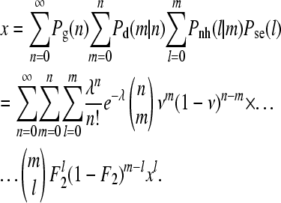 |
(A5) |
These sums can be done with result
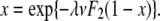 |
(A6) |
Substituting in for λ and ν, using  and our definitions of d′ and g′ allows us to reduce this to
and our definitions of d′ and g′ allows us to reduce this to
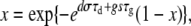 |
(A7) |
which is the probability that a mutant with growth rate change s and death rate change σ will go extinct if there was exactly one mutant at time t = 0 (Wahl and Gerrish 2001). Next, we calculate the extinction probability, V(t, s, σ), of a mutant with a death rate changed by σ and a growth rate changed by s appearing at time t < τg. Let γ = F1exp[g′(τg − t)] be the expected number of mutants after the first dilution. Then, using the same logic as above, we have
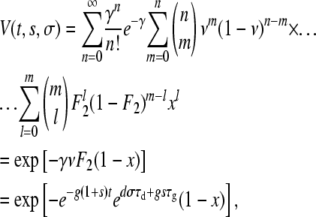 |
(A8) |
where x is the first real root of Equation A7. The probability P(t, s, σ) that a created mutant will survive is then simply
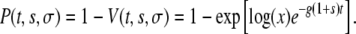 |
(A9) |
Note that we assumed in the derivation that population sizes and growth and decay times stay the same. Strictly speaking, once a mutant with a different growth or death rate is created, either the values for the initial and final populations, i.e., N0, N1, N2, N3, or the growth and death intervals, τg or τd, need to be adjusted. However, since we consider only small changes in either growth rate or survival, and mutant strains are most likely to go extinct when their overall numbers are small in comparison to the entire culture, the adjusted growth or death rate during these times is nearly identical to the mutant-free culture and we can therefore assume the remaining parameters to stay constant. Also note that even though transmission mutations are important only during the exponential decline in the fomite phase, they are created during the exponential growth phase, just like the growth mutations. This scenario should not be confused with a previous study that showed that the fixation probability is different for exponentially growing or declining populations (Otto and Whitlock 1997).
To find the expected number of mutants, we need to integrate Λ(s, σ) (Equation 5) over all possible values of s and σ. This integral can be split into two parts, such that Mtot = Ms + Mσ, representing the total number of growth mutants (Ms) and transmission mutants (Mσ). To do this, let us first examine the function Q(s, σ). In general, we can write Q(s, σ) as Q(s, σ) = Qs + Qσ, where Qs and Qσ represent the contributions of the growth mutations and transmission mutations, respectively. For instance, we can write Qs as
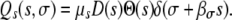 |
(A10) |
Note that we use the convention that the mutation is always positive in the main trait (here growth rate), since negative mutations lead to decreased fitness. This is ensured by the Heaviside step function, which is unity for positive arguments and zero otherwise. On the other hand, we assume that the corresponding pleiotropic effect is always negative or zero. Here, this is governed by the Dirac delta function. Provided βσ ≥ 0, σ is negative (or zero) for all positive s. We can now integrate Λ(s, σ) to find the expected number of growth mutants,
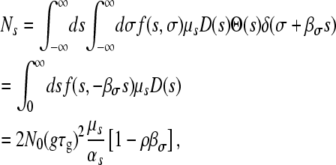 |
(A11) |
where we used, for convenience, 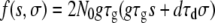 . Mσ can be computed in a similar manner. The results for the nonpleiotropic case are easily derived by setting βs = βσ = 0.
. Mσ can be computed in a similar manner. The results for the nonpleiotropic case are easily derived by setting βs = βσ = 0.
References
- Antia, R., R. R. Regoes, J. C. Koella and C. T. Bergstrom, 2003. The role of evolution in the emergence of infectious diseases. Nature 426(6967): 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero, F., and J. Blazquez, 1997. Evolution of antibiotic resistance. Trends Ecol. Evol. 12(12): 482–487. [DOI] [PubMed] [Google Scholar]
- Bergstrom, C. T., P. McElhany and L. A. Real, 1999. Transmission bottlenecks as determinants of virulence in rapidly evolving pathogens. Proc. Natl. Acad. Sci. USA 96(9): 5095–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer, S., R. E. Lenski and D. Ebert, 1996. The curse of the pharaoh: the evolution of virulence in pathogens with long living propagules. Proc. Biol. Sci. 263(1371): 715–721. [DOI] [PubMed] [Google Scholar]
- Caraco, T., and I.-N. Wang, 2008. Free-living pathogens: life-history constraints and strain competition. J. Theor. Biol. 250(3): 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrave, N., and S. Collins, 2008. Experimental evolution: experimental evolution and evolvability. Heredity 100(5): 464–470. [DOI] [PubMed] [Google Scholar]
- Cowperthwaite, M. C., J. J. Bull and L. Ancel Meyers, 2006. From bad to good: fitness reversals and the ascent of deleterious mutations. PLoS Comput. Biol. 2(10):e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow, J. F., and M. Kimura, 1970. Introduction to Population Genetics Theory. Harper & Row, New York.
- Day, T., 2002. Virulence evolution via host exploitation and toxin production in spore-producing pathogens. Ecol. Lett. 5(4): 471–476. [Google Scholar]
- De Paepe, M., and F. Taddei, 2006. Viruses' life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4(7): e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena, S. F., and R. E. Lenski, 2003. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4(6): 457–469. [DOI] [PubMed] [Google Scholar]
- Escarmis, C., E. Lazaro and S. C. Manrubia, 2006. Population bottlenecks in quasispecies dynamics. Curr. Top. Microbiol. Immunol. 299 141–170. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Oxford University Press, London/New York/Oxford.
- Gandon, S., 1998. The curse of the pharaoh hypothesis. Proc. Biol. Sci. 265(1405): 1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish, P. J., and R. E. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102/103(1–6): 127–144. [PubMed] [Google Scholar]
- Gillespie, J. H., 1984. Molecular evolution over the mutational landscape. Evolution 38(5): 1116–1129. [DOI] [PubMed] [Google Scholar]
- Haldane, J. B. S., 1927. A mathematical theory of natural and artificial selection, part v: selection and mutation. Proc. Camb. Philos. Soc. 23 838–844. [Google Scholar]
- Handel, A., R. R. Regoes and R. Antia, 2006. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput. Biol. 2(10): e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan, J. M., and L. M. Wahl, 2002. The effects of genetic drift in experimental evolution. Theor. Popul. Biol. 62(4): 349–356. [DOI] [PubMed] [Google Scholar]
- Imhof, M., and C. Schlotterer, 2001. Fitness effects of advantageous mutations in evolving Escherichia coli populations. Proc. Natl. Acad. Sci. USA 98(3): 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamo, M., and M. Boots, 2004. The curse of the pharaoh in space: free-living infectious stages and the evolution of virulence in spatially explicit populations. J. Theor. Biol. 231(3): 435–441. [DOI] [PubMed] [Google Scholar]
- Kent, S. J., C. S. Fernandez, C. J. Dale and M. P. Davenport, 2005. Reversion of immune escape HIV variants upon transmission: insights into effective viral immunity. Trends Microbiol. 13(6): 243–246. [DOI] [PubMed] [Google Scholar]
- Kramer, A., I. Schwebke and G. Kampf, 2006. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 6 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knies, J. L., R. Izem, K. L. Supler, J. G. Kingsolver and C. L. Burch, 2006. The genetic basis of thermal reaction norm evolution in lab and natural phage populations. PLoS Biol. 4(7): e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski, R. E., M. R. Rose, S. C. Simpson and S. C. Tadler, 1991. Long-term experimental evolution in Escherichia-coli. 1. Adaptation and divergence during 2,000 generations. Am. Nat. 138(6): 1315–1341. [Google Scholar]
- Levin, B. R., V. Perrot and N. Walker, 2000. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrubia, S. C., and E. Lazaro, 2006. Viral evolution. Phys. Life Rev. 3(2): 65–92. [Google Scholar]
- Manrubia, S. C., C. Escarmis, E. Domingo and E. Lazaro, 2005. High mutation rates, bottlenecks, and robustness of RNA viral quasispecies. Gene 347(2): 273–282. [DOI] [PubMed] [Google Scholar]
- Nelson, M. I., and E. C. Holmes, 2007. The evolution of epidemic influenza. Nat. Rev. Genet. 8(3): 196–205. [DOI] [PubMed] [Google Scholar]
- Novak, M., T. Pfeiffer, R. E. Lenski, U. Sauer and S. Bonhoeffer, 2006. Experimental tests for an evolutionary trade-off between growth rate and yield in E. coli. Am. Nat. 168(2): 242–251. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2000. The rate of adaptation in asexuals. Genetics 155 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2005. The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6(2): 119–127. [DOI] [PubMed] [Google Scholar]
- Ostrowski, E. A., D. E. Rozen and R. E. Lenski, 2005. Pleiotropic effects of beneficial mutations in Escherichia coli. Evol. Int. J. Org. Evol. 59(11): 2343–2352. [PubMed] [Google Scholar]
- Otto, S. P., and M. C. Whitlock, 1997. The probability of fixation in populations of changing size. Genetics 146 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito, L., L. Fernandes, C. Mota and I. Gordo, 2007. Adaptive mutations in bacteria: high rate and small effects. Science 317(5839): 813–815. [DOI] [PubMed] [Google Scholar]
- Pollak, E., 2000. Fixation probabilities when the population size undergoes cyclic fluctuations. Theor. Popul. Biol. 57(1): 51–58. [DOI] [PubMed] [Google Scholar]
- Rozen, D. E., J. A. G. M. de Visser and P. J. Gerrish, 2002. Fitness effects of fixed beneficial mutations in microbial populations. Curr. Biol. 12(12): 1040–1045. [DOI] [PubMed] [Google Scholar]
- Rozen, D. E., M. G. J. L. Habets, A. Handel and J. A. G. M. de Visser, 2008. Heterogeneous adaptive trajectories of small populations on complex fitness landscapes. PLoS ONE 3(3):e1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleight, S. C., N. S. Wigginton and R. E. Lenski, 2006. Increased susceptibility to repeated freeze-thaw cycles in Escherichia coli following long-term evolution in a benign environment. BMC Evol. Biol. 6 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, C. F., and P. G. Higgs, 2000. A population genetics model for multiple quantitative traits exhibiting pleiotropy and epistasis. J. Theor. Biol. 203(4): 419–437. [DOI] [PubMed] [Google Scholar]
- Wahl, L. M., and C. S. DeHaan, 2004. Fixation probability favors increased fecundity over reduced generation time. Genetics 168 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, L. M., and P. J. Gerrish, 2001. The probability that beneficial mutations are lost in populations with periodic bottlenecks. Evol. Int. J. Org. Evol. 55(12): 2606–2610. [DOI] [PubMed] [Google Scholar]
- Wahl, L. M., P. J. Gerrish and I. Saika-Voivod, 2002. Evaluating the impact of population bottlenecks in experimental evolution. Genetics 162 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther, B. A., and P. W. Ewald, 2004. Pathogen survival in the external environment and the evolution of virulence. Biol. Rev. Camb. Philos. Soc. 79(4): 849–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke, C. O., 2004. The speed of adaptation in large asexual populations. Genetics 167 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke, C. O., C. Ronnewinkel and T. Martinetz, 2001. Dynamic fitness landscapes in molecular evolution. Phys. Rep. 349 395–446. [Google Scholar]
- Woodcock, G., and P. G. Higgs, 1996. Population evolution on a multiplicative single-peak fitness landscape. J. Theor. Biol. 179(1): 61–73. [DOI] [PubMed] [Google Scholar]
- Woolhouse, M. E. J., T. H. Daniel and R. Antia, 2005. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol. Evol. 20(5): 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]