

Abstract
Hybridization between distinct species may lead to introgression of genes across species boundaries, and this pattern can potentially persist for extended periods as long as selection at some loci or genomic regions prevents thorough mixing of gene pools. However, very few reliable estimates of long-term levels of effective migration are available between hybridizing species throughout their history. Accurate estimates of divergence dates and levels of gene flow require data from multiple unlinked loci as well as an analytical framework that can distinguish between lineage sorting and gene flow and incorporate the effects of demographic changes within each species. Here we use sequence data from 18 anonymous nuclear loci in two broadly sympatric sunflower species, Helianthus annuus and H. petiolaris, analyzed within an “isolation with migration” framework to make genome-wide estimates of the ages of these two species, long-term rates of gene flow between them, and effective population sizes and historical patterns of population growth. Our results indicate that H. annuus and H. petiolaris are approximately one million years old and have exchanged genes at a surprisingly high rate (long-term Nef m estimates of approximately 0.5 in each direction), with somewhat higher rates of introgression from H. annuus into H. petiolaris than vice versa. In addition, each species has undergone dramatic population expansion since divergence, and both species have among the highest levels of genetic diversity reported for flowering plants. Our results provide the most comprehensive estimate to date of long-term patterns of gene flow and historical demography in a nonmodel plant system, and they indicate that species integrity can be maintained even in the face of extensive gene flow over a prolonged period.
Keywords: Anonymous loci, effective population size, Helianthus, historical demography, hybridization, introgression
Natural hybridization is common in many different plant and animal taxa, and although the importance of hybridization in evolution has historically been a topic of some debate, it is becoming increasingly clear that it can be an important phenomenon in speciation and adaptation (Whitney et al. 2006; Mallet 2007). Although a common traditional (and still prevalent) view of species holds that widespread introgression between distinct species will be extremely rare if it occurs at all (Mayr 1963; Coyne and Orr 2004), recent evidence suggests that significant hybridization and introgression following species divergence may not be uncommon (Hey 2006), and theory suggests that if few genes or genomic regions maintain species differences and if there is sufficient opportunity for hybridization and recombination among genomes, much of the genome may be free to pass across species boundaries (Barton and Hewitt 1985; Wu 2001).
Numerous instances of introgression across species boundaries have been documented (Arnold 1992; Rieseberg and Wendel 1993), and recent studies provide further empirical evidence for the maintenance of species differences in the face of significant amounts of hybridization and introgression (Crow et al. 2007; de Casas et al. 2007; Yatabe et al. 2007). Although these studies document the occurrence of introgression, some questions remain about the amount and long-term dynamics of introgression, in part because studies are focused on populations in or near hybrid zones (but see Yatabe et al. 2007) or because the relative effects of introgression and sorting of ancestral polymorphisms are not explicitly taken into account (Nielsen and Wakeley 2001).
Models of strict isolation versus migration can be distinguished from each other using patterns of genetic variation within or among loci (Wakeley 1996; Nielsen and Wakeley 2001; Hey and Nielsen 2004). The past 20 years have seen a number of important theoretical (Felsenstein 1988; Geyer 1992; Nielsen and Wakeley 2001; Hudson 2002; Hey and Nielsen 2007) and computational advances, as well as a dramatic increase in the availability of sequence data from multiple loci, even in nonmodel organisms. These advances now allow investigators to not only distinguish between strict isolation and migration, but also to explicitly estimate a range of demographic quantities using parameter-rich models that can incorporate complex scenarios including population size changes, asymmetric rates of introgression between populations, and variation among loci in rates of introgression. These recently developed methods based on variation in coalescence patterns and shared genetic variation among multiple loci offer the best option currently available for sorting out the effects of incomplete lineage sorting and interspecific gene flow (Hey 2006). They have been successfully applied to primates (Hey 2005; Won and Hey 2005), Drosophila (Hey and Nielsen 2004; Machado et al. 2007b), Heliconius butterflies (Bull et al. 2006), and several other animal species. However, such studies remain very rare in plant systems, where there is a much richer history of exploring the roles hybridization and introgression can play in shaping species diversity.
North American annual sunflower species Helianthus annuus and H. petiolaris are an excellent system for studying interspecific hybridization, introgression, and the maintenance of species differences. These two species, which are widespread throughout much of the central and western United States, differ by a minimum of 11 chromosomal rearrangements (Burke et al. 2004) and are morphologically and ecologically distinct (Gross et al. 2004; Rosenthal et al. 2005). They also show strong prezygotic (Rieseberg et al. 1995) and postzygotic (Lai et al. 2005b) reproductive isolation throughout their ranges (Buerkle and Rieseberg 2001). Nonetheless, they form numerous hybrid zones throughout their ranges and rates of F1 production in these zones are fairly high (Rieseberg et al. 1998), as are the frequencies of complex backcross genotypes (Rieseberg et al. 1999). This suggests that there is ample opportunity for recombination between the two species’ genomes to unlink neutral or even potentially adaptive genomic regions from regions that contribute to reproductive isolation (Rieseberg et al. 1999). Helianthus annuus and H. petiolaris are more similar to each other at anonymous nuclear markers than H. annuus is to its sister species, H. argophyllus (Yatabe et al. 2007). In addition, they have given rise to three stabilized homoploid hybrid species in the southwestern United States (Rieseberg 2006). Helianthus annuus also hybridizes with a number of other annual Helianthus species (Heiser 1951a, b; Dorado et al. 1992), and in at least one case adaptive introgression appears to have facilitated expansion into new habitat (Whitney et al. 2006). Hybridization has clearly played a central role in the evolution of this group.
Both species also have extremely high levels of genetic variation compared to other flowering plants (Lynch 2006). Reliable estimates of the long-term effective sizes, population growth patterns, and rates of gene flow between these species may help inform our understanding of these patterns of genetic diversity. Here we investigate the demographic histories of H. annuus and H. petiolaris within an “isolation with migration” framework using sequence data from 18 anonymous nuclear loci. We are particularly interested in the ages of these two species, their long-term patterns of historical gene flow and how they compare to data available for other flowering plant species, and their effective population sizes and patterns of historical population growth.
Materials and Methods
COLLECTIONS AND DNA ISOLATION
Leaves and/or achenes were collected from 10 populations of H. annuus and 10 populations of H. petiolaris in the western and southwestern United States (Fig. 1). None of these populations are known to be near hybrid zones between these two species. Achenes were germinated in greenhouses at Indiana University, and leaf tissue was sampled for genetic analyses. Individuals of two species in the closely related genus Bahiopsis (formerly Viguiera—Schilling and Paner 2002), B. lanata and B. reticulata, were used as outgroups. DNA was extracted from leaf tissue using a DNeasy Plant Mini Kit or DNeasy 96 Plant Kit (QIAGEN, Valencia, CA).
Figure 1.
Sampling localities for H. annuus and H. petiolaris individuals used in this study. Latitude/longitude data and sample sizes are given in Table 1.
DNA SEQUENCING
Primers were designed based on H. annuus EST sequence collected as part of the Compositae Genome Project (http://cgpdb.ucdavis.edu/) (Lai et al. 2005a). Where possible, primers were designed to anneal in conserved exon regions flanking one or more introns. Primer sequences and amplification conditions are described in online Supplementary Table S1. The 18 loci analyzed here represent at least 11 of 17 linkage groups in H. annuus. Because loci were developed based on an H. annuus EST library, they all contain portions of expressed genes. However, loci were chosen without regard to gene ID or homology to any other loci. Closest Arabidopsis thaliana BLAST hits and gene information are given in online Supplementary Table S2. Unincorporated primers and dNTPs were removed using ExoSAP-IT (USB, Cleveland, OH), and sequencing reactions using both forward and reverse primers were carried out on PCR products using ABI Big Dye Terminators version 3.1 and resolved using an ABI 3730 capillary sequencer (Applied Biosystems, Foster City, CA). For individuals heterozygous for a single indel, haplotypes were phased by comparing forward and reverse sequences at variable sites. For individuals heterozygous for multiple indels or for phasing haplotypes in individuals with no length heterozygosity, PCR products were cloned using a TOPO-TA cloning kit (INVITROGEN, Carlsbad, CA). Clone sequences were compared to sequences obtained through direct sequencing and to other clone sequences for the same individual to help identify polymerase errors and PCR-mediated recombination (Meyerhans et al. 1990) in clone sequences. Sequences were aligned using Sequencher version 4.7 (Gene Codes Corporation, Ann Arbor, MI), with minor adjustments made by eye. Ambiguous alignments, generally involving short regions of repetitive DNA, were removed prior to all analyses. Datasets are complete for both species (25 individuals = 50 haplotypes for H. annuus, 16 individuals = 32 haplotypes for H. petiolaris) except for two H. annuus individuals missing at locus JLS720a and one H. petiolaris individual missing at locus JLS244. All sequences have been submitted to GenBank; accession numbers are given in online Supplementary Table S3.
DATA ANALYSES
Coding regions and reading frames were identified by comparing sequences to Helianthus and Lactuca (lettuce) EST sequences retrieved from the Compositae Genome Project (http://compgenomics.ucdavis.edu/) as well as Arabidopsis sequences. For three loci, coding region/reading frame could not be reliably identified; in these cases, all sequence was considered noncoding.
Sequence diversity (π) using the Jukes-Cantor (1969) correction and Watterson’s (1975) θ were calculated for entire sequences as well as silent/noncoding and replacement partitions using DnaSP version 4.10.9 (Rozas et al. 2003). Interspecific gross and net sequence divergence (net divergence = gross divergence – average diversity within each species) were calculated using the computer program SITES (Hey and Wakeley 1997). Summary statistics for four categories of segregating sites described by Wakeley and Hey (1997) were also calculated using SITES. Neighbor joining trees were constructed using PAUP 4.0b10 (Swofford 1999). AMOVAs were run using the program Arlequin version 3.11 (Excoffier et al. 2005). Estimates of current and ancestral inbreeding effective population sizes, divergence time, long-term rates of effective migration, and the ancestral population splitting parameter (measuring the proportion of the ancestral population that gave rise to H. annuus) were made using the computer program IM (written by Jody Hey; available at http://lifesci.rutgers.edu/~heylab/HeylabSoftware.htm%IM). Analyses in IM were run using the HKY (Hasegawa et al. 1985) mutation model. Three independent (different random number seeds) runs of at least 2–3 million steps following a 100,000 step burn-in were used to ensure convergence; all runs involved 10 independent chains, and the lowest effective sample size (ESS) among the seven parameters was at least 50 in each case, as recommended in the IM documentation (in most cases ESS values ranged from several hundred to >10,000). Upper bounds of the prior distributions for each parameter were set based on the results of a preliminary run. Maxima of posterior distributions for each parameter were well under the upper bounds of the prior distributions for all three runs. Results were highly consistent across runs, and a single representative run is presented here. Maximum-likelihood estimates (MLEs) and 90% highest posterior density (HPD) ranges for each parameter were converted to meaningful biological quantities using a mutation rate estimate of 1 × 10−8 substitutions/site/year, derived from a range of EST sequence comparisons and fossil calibrations in flowering plants, with an emphasis on Asteraceae and closely related taxa (M. S. Barker and L. H. Rieseberg, unpubl. ms.), and a one-year generation time (these sunflowers are annuals). MLEs and HPD ranges were very consistent across IM runs.
The “isolation with migration” model implemented in IM assumes no recombination within loci and free recombination among loci. There is no significant linkage between any pair of loci (data not shown); in fact, most of the loci used here are on different chromosomes (see Table 2). The program SITES was used to infer apparently nonrecombining blocks of sequence based on the algorithm of Hudson and Kaplan (1985), with sites containing indels and sites with more than two DNA character states excluded. The largest nonrecombining block of sequence for each locus was included for analyses using IM. Sequences from six Helianthus species involved in a larger study (H. annuus and H. petiolaris as well as three species resulting from hybridization between them—Rieseberg 2006—plus H. annuus’ sister species, H. argophyllus) were used for recombination analyses. In one case—locus (3724)—all sequence was included because there was no evidence of recombination. The method implemented in IM also assumes that all sequences have evolved neutrally. To test this, Tajima’s (1989) D and Fu’s (1997) Fs statistics and P-values were calculated in Arlequin version 3.11 using 10,000 simulations to assess significance. In addition, Hudson-Kreitman-Aguade (HKA) tests (Hudson et al. 1987) were conducted using Jody Hey’s HKA program (available at http://lifesci.rutgers.edu/~heylab/HeylabSoftware.htm%HKA) with significance determined based on 10,000 coalescent simulations; and McDonald–Kreitman (MK) tests (McDonald and Kreitman 1991) comparing H. annuus and H. petiolaris sequences to B. lanata and B. reticulata sequences were conducted using DnaSP version 4.10.9.
Table 2.
Basic information and summary statistics for all 18 loci. BLAST E values for Arabidopsis thaliana genes and GenBank accession numbers for H. annuus and H. petiolaris sequences are given in online Supplementary Tables S2 and S3, respectively.
| H. annuus | H. petiolaris | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Linkage group | Protein homology (based on closest Arabidopsis thaliana BLAST hit) | Aligned size (bp) | Largest nonrecombining block (bp) | No. of seqs. | π (silent) | θ (silent) | No. of seqs | π (silent) | θ (silent) | % Gross divergence | % Net divergence |
| (2213) | 6 | scarecrow transcription factor family protein | 656 | 594 | 50 | 0.01018 | 0.01392 | 32 | 0.01884 | 0.02246 | 3.02 | 1.31 |
| (2352) | 7 | protein kinase family protein | 776 | 620 | 50 | 0.03344 | 0.05606 | 32 | 0.04136 | 0.06043 | 3.36 | 0.49 |
| (3242) | 6 | short-chain dehydrogenase/reductase (SDR) family protein | 869 | 285 | 50 | 0.01647 | 0.02486 | 32 | 0.02501 | 0.03851 | 2.43 | 0.50 |
| (3724) | unknown | unknown protein | 1059 | 1059 | 50 | 0.00173 | 0.00291 | 32 | 0.00621 | 0.00803 | 1.85 | 1.32 |
| 724 | unknown | indoleacetic acid-induced protein 16 | 581 | 368 | 50 | 0.02016 | 0.03158 | 32 | 0.02465 | 0.03187 | 2.39 | 0.12 |
| 2695 | 5 | NAC domain containing protein 2; transcription factor | 591 | 330 | 50 | 0.03656 | 0.05864 | 32 | 0.06393 | 0.06907 | 4.08 | 1.19 |
| 2725 | 8 | adenosylmethionine decarboxylase family protein | 297 | 65 | 50 | 0.02545 | 0.03417 | 32 | 0.02085 | 0.03674 | 1.88 | 0.51 |
| JLS244 | 9 | proton-dependent oligopeptide transport (POT) family protein | 562 | 315 | 50 | 0.02757 | 0.02729 | 30 | 0.02404 | 0.03586 | 2.33 | 0.98 |
| JLS720a | 16 | cellulose synthase-related protein | 687 | 561 | 46 | 0.01326 | 0.02665 | 32 | 0.02834 | 0.03718 | 2.48 | 1.34 |
| JLS810R1 | 13 | auxin efflux carrier protein family | 740 | 375 | 50 | 0.04154 | 0.03369 | 32 | 0.05294 | 0.03939 | 5.18 | 1.91 |
| JLS836F2 | 8 | glyceraldehyde-3-phosphate dehydrogenase C subunit | 377 | 214 | 50 | 0.02861 | 0.02996 | 32 | 0.02426 | 0.02532 | 3.43 | 0.91 |
| JLS925R2 | 9 | histone deacetylase 15 | 790 | 411 | 50 | 0.02299 | 0.03434 | 32 | 0.03541 | 0.05299 | 2.61 | 0.54 |
| JLS1040R2 | 10 | serine/threonine phosphatase | 653 | 445 | 50 | 0.02164 | 0.03025 | 32 | 0.03299 | 0.04155 | 2.13 | 0.39 |
| JLS1615a | 14 | unknown protein | 572 | 247 | 50 | 0.03715 | 0.06132 | 32 | 0.0475 | 0.07640 | 1.10 | 0.07 |
| JLS1689a | 10 | unknown protein | 708 | 121 | 50 | 0.03692 | 0.04554 | 32 | 0.04512 | 0.06358 | 3.25 | 1.49 |
| JLS1747R2 | 17 | amino acid permease family protein | 785 | 100 | 50 | 0.04536 | 0.05024 | 32 | 0.05802 | 0.07068 | 2.29 | 0.20 |
| JLS1953a | 17 | galacturonosyltransferase 4 | 450 | 192 | 50 | 0.00463 | 0.00794 | 32 | 0.01550 | 0.02379 | 2.45 | 1.46 |
| JLS2899 | 14 | unknown protein | 774 | 517 | 50 | 0.02950 | 0.02853 | 32 | 0.03220 | 0.05705 | 1.26 | 0.23 |
| Average | 663 | 379 | 0.02518 | 0.03322 | 0.03318 | 0.04394 | 2.64 | 0.83 | ||||
Results
Eighteen anonymous nuclear loci representing at least 11 of 17 linkage groups were sequenced; aligned sizes range from 297 to 1059 bp, with an average size of 663 bp (11,927 bp total). Basic summary statistics for all loci are shown in Table 2. Average silent site nucleotide diversity across loci within H. annuus and H. petiolaris is 2.5% and 3.3%, respectively, and average pairwise distance across loci between species is 2.6%. As is evident from Table 2, although there is a large amount of variation within each species, differentiation between species is fairly low (mean ratio of net to gross divergence across loci is 0.30). In neighbor joining trees using B. lanata and B. reticulata as outgroups, H. annuus and H. petiolaris haplotypes are reciprocally monophyletic for only one locus, JLS244 (data not shown). Helianthus annuus haplotypes are paraphyletic with respect to H. petiolaris haplotypes for one locus, the reverse is true for four loci, and for 12 loci haplotypes sampled from the two species have a polyphyletic relationship.
Consistent with previous data on the decay of linkage disequilibrium in H. annuus (Liu and Burke 2006), intralocus recombination appears common in our dataset; only one locus showed no evidence of recombination, and the average size of the largest nonrecombining block is almost 300 bp smaller than the average size of the full aligned sequence (379 bp vs. 663 bp).
Tajima’s D and Fu’s Fs statistics for each locus are given in Table 3. Fs is considered more sensitive to population growth, whereas D is considered more sensitive to selection (Fu 1997). For H. annuus and H. petiolaris, zero and two loci are significantly negative for D, and 15 and 10 loci are significantly negative for Fs, respectively. To test whether recombination or population structure within each species had a significant effect on D and Fs, both statistics were calculated using the largest nonrecombining sequence blocks for each locus (the same blocks used for IM analyses) and using a single randomly selected haplotype from each sampling locality (10 haplotypes for each species; see Table 1), as recommended by Ramos-Onsins et al. (2004). Results are shown in online Supplementary Tables S4 and S5, and are qualitatively very similar to those in Table 3. For nonrecombining blocks, five of 36 (18 loci × two species) D values are significantly negative, and 23 of 36 Fs values are significantly negative. For single haplotypes from each population, zero of 36 and 12 of 36 values are significantly negative for D and Fs, respectively. In addition, HKA tests were not significant between H. annuus and H. petiolaris across all loci. For MK tests, no comparisons between H. annuus or H. petiolaris and B. lanata or B. reticulata were significant following Bonferroni correction. Taken overall, the results of D and Fs calculations and HKA and MK tests are most consistent with population growth and selective neutrality.
Table 3.
Results of tests of selection/demographic changes for all 18 loci individually, as well as for all loci concatenated together. Tests in bold italics are significant at the 0.05 level, and those marked with asterisks are significant after Bonferroni correction.
| H. annuus | H. petiolaris | |||||||
|---|---|---|---|---|---|---|---|---|
| Locus | Tajima’s D | Fu’s Fs | Tajima’s D | Fu’s Fs | ||||
| D | P | Fs | P | D | P | Fs | P | |
| (2213) | −0.934 | 0.185 | −13.753 | <0.001* | −0.534 | 0.330 | −5.588 | 0.032 |
| (2352) | −1.392 | 0.059 | −3.236 | 0.181 | −1.215 | 0.100 | 0.167 | 0.569 |
| (3242) | −0.960 | 0.171 | −12.765 | 0.001* | −1.026 | 0.155 | −5.105 | 0.048 |
| (3724) | −1.233 | 0.100 | −2.631 | 0.060 | −1.137 | 0.121 | −4.745 | 0.019 |
| 724 | −1.278 | 0.082 | −21.960 | <0.001* | −0.539 | 0.331 | −3.685 | 0.098 |
| 2695 | −1.382 | 0.061 | −7.577 | 0.027 | −0.380 | 0.399 | −0.640 | 0.423 |
| 2725 | −1.248 | 0.093 | −24.526 | <0.001* | −1.337 | 0.072 | −15.396 | <0.001* |
| JLS244 | −0.273 | 0.454 | −8.723 | 0.005 | −0.857 | 0.211 | −3.330 | 0.104 |
| JLS720a | −1.281 | 0.087 | −5.553 | 0.042 | −1.008 | 0.156 | −8.992 | 0.004 |
| JLS810R1 | 0.416 | 0.735 | −5.363 | 0.080 | 1.375 | 0.941 | 2.009 | 0.799 |
| JLS836F2 | −0.234 | 0.465 | −6.655 | 0.035 | −0.296 | 0.441 | −9.635 | 0.003 |
| JLS925R2 | −1.308 | 0.077 | −10.596 | 0.008 | −1.328 | 0.068 | −5.024 | 0.053 |
| JLS1040R2 | −1.293 | 0.076 | −23.073 | <0.001* | −0.778 | 0.235 | −2.280 | 0.215 |
| JLS1615a | −1.344 | 0.070 | −23.025 | <0.001* | −1.477 | 0.046 | −7.992 | 0.005 |
| JLS1689a | −0.685 | 0.277 | −16.556 | <0.001* | −0.931 | 0.174 | −6.270 | 0.026 |
| JLS1747R2 | −0.386 | 0.408 | −7.277 | 0.033 | −0.818 | 0.217 | −6.118 | 0.031 |
| JLS1953a | −1.295 | 0.087 | −8.116 | <0.001* | −1.326 | 0.076 | −0.372 | 0.477 |
| JLS2899 | −0.213 | 0.476 | −8.976 | 0.007 | −1.697 | 0.025 | −6.035 | 0.024 |
| All Loci | −1.034 | >0.10 | −8.930 | <0.001* | −1.065 | >0.10 | −2.607 | 0.069 |
Table 1.
Sampling localities. Sampling sites are shown in Figure 1.
| Species | Population | Location | Latitude (N) | Longitude (W) | No. of individuals |
|---|---|---|---|---|---|
| H. annuus (n= 25) | Ann-NM1 | North of Capitan, NM | 33.57 | 105.57 | 3 |
| Ann-NM2 | West of Roswell, NM | 33.40 | 104.73 | 3 | |
| Ann-AZ1 | Chinle, AZ | 36.15 | 109.53 | 2 | |
| Ann-UT1 | Washington Co., UT | 37.26 | 113.28 | 2 | |
| Ann-NV1 | Clark Co., NV | 36.78 | 114.25 | 2 | |
| Ann-UT2 | Juab Co., UT | 39.72 | 112.23 | 2 | |
| Ann-NV2 | Washoe Co., NV | 39.63 | 119.18 | 3 | |
| Ann-UT3 | Carbon Co., UT | 39.58 | 110.78 | 2 | |
| Ann-NM3 | Chaves Co., NM | 33.19 | 104.34 | 2 | |
| Ann-TX1 | Ft. Stockton, TX | 30.90 | 102.85 | 4 | |
| H. petiolaris (n= 16) | Pet-UT1 | Kane Co., UT | 37.03 | 112.48 | 2 |
| Pet-NM1 | Lincoln Co., NM | 33.75 | 105.45 | 2 | |
| Pet-NM2 | Tularosa Basin, NM | 32.75 | 106.00 | 2 | |
| Pet-AZ1 | Coconino Co., AZ | 36.78 | 111.55 | 2 | |
| Pet-AZ2 | Navajo Co., AZ | 35.75 | 109.91 | 2 | |
| Pet-UT2 | Wayne Co., UT | 38.35 | 110.69 | 2 | |
| Pet-UT4 | Kane Co., UT | 37.52 | 111.98 | 1 | |
| Pet-UT5 | San Juan Co., UT | 37.03 | 110.13 | 1 | |
| Pet-UT3 | Iron Co., UT | 38.08 | 112.69 | 1 | |
| Pet-UT6 | Iron Co., UT | 38.08 | 112.68 | 1 | |
| Bahiopsis lanata | Bah-1 | 1 | |||
| B. reticulata | Bah-2 | 1 |
Another useful way to describe the distribution of genetic variation between H. annuus or H. petiolaris is using the four mutually exclusive categories of segregating sites that Wakeley and Hey (1997) introduced to estimate current and ancestral effective sizes and divergence time. These are the numbers of sites polymorphic in the first population but fixed in the second and vice versa, the number of shared polymorphisms, and the number of fixed differences. As can be seen in Table 4, there is a large amount of variation within each species, with an average of 51 and 59 variable sites per locus in H. annuus and H. petiolaris, respectively; in contrast, across all loci there are a total of 15 fixed differences between the two species.
Table 4.
Counts of four polymorphism categories for all sites that are variable within or between H. annuus and H. petiolaris (see Wakeley and Hey 1997).
| Locus | Polymorphic in H. annuus only | Polymorphic in H. petiolaris only | Polymorphic in both species | Fixed differences between species |
|---|---|---|---|---|
| (2213) | 22 | 41 | 6 | 1 |
| (2352) | 100 | 89 | 27 | 0 |
| (3242) | 30 | 51 | 16 | 0 |
| (3724) | 7 | 19 | 0 | 6 |
| 724 | 44 | 33 | 24 | 0 |
| 2695 | 54 | 65 | 25 | 1 |
| 2725 | 20 | 16 | 9 | 0 |
| JLS244 | 29 | 37 | 0 | 1 |
| JLS720a | 43 | 49 | 1 | 2 |
| JLS810R1 | 46 | 47 | 19 | 1 |
| JLS836F2 | 37 | 26 | 6 | 0 |
| JLS925R2 | 58 | 81 | 22 | 0 |
| JLS1040R2 | 45 | 52 | 12 | 0 |
| JLS1615a | 25 | 28 | 15 | 0 |
| JLS1689a | 51 | 53 | 13 | 2 |
| JLS1747R2 | 33 | 52 | 36 | 0 |
| JLS1953a | 16 | 41 | 0 | 1 |
| JLS2899 | 20 | 45 | 14 | 0 |
Results from IM analysis are shown in Table 5 and Figure 2. Estimates of effective population sizes for each species are 1.8 million for H. annuus and 2.4 million for H. petiolaris. These sizes are four to six times larger than the effective size of the ancestral population that gave rise to these species, indicating that both H. annuus and H. petiolaris have undergone dramatic population growth following formation. The MLE of the splitting parameter is 0.21, indicating that H. annuus was initially considerably smaller than H. petiolaris and has subsequently undergone much greater expansion (approximately 21× vs. 8×).
Table 5.
Model parameter and biological quantity estimates from IM analysis of largest nonrecombining sequence blocks. Conversions are based on a mutation rate estimate of 1×10−8 substitutions/site/generation and one-year generation time. Sizes of the initial populations that founded the two current species are based on the high point estimate for Nef of the common ancestor and the confidence interval for s, the proportion of the common ancestral population that contributed to H. annuus. Nef m estimates are based on the high point estimate for the relevant Nef and the confidence interval for m. HPD90LO and HPD90HI are the lower and upper limits, respectively, of the 90% highest posterior density interval.
| Model Parameter | High point | HPD90Lo | HPD90Hi |
|---|---|---|---|
| θannuus | 15.24 | 12.65 | 18.32 |
| θpetiolaris | 21.21 | 17.29 | 25.91 |
| θancestor | 3.50 | 2.37 | 5.11 |
| T | 2.28 | 1.86 | 2.71 |
| m1 | 0.08 | 0.04 | 0.14 |
| m2 | 0.11 | 0.04 | 0.18 |
| S | 0.21 | 0.12 | 0.34 |
| Demographic quantity | High point | HPD90Lo | HPD90Hi |
| Nef —H. annuus | 1.75 × 106 | 1.45 × 106 | 2.10 × 106 |
| Nef — H. petiolaris | 2.43 × 106 | 1.98 × 106 | 2.97 × 106 |
| Nef — common ancestor | 4.01 × 105 | 2.72 × 105 | 5.86 × 105 |
| Nef — H. annuus initial | 8.31 × 104 | 4.87 × 104 | 1.38 × 105 |
| Nef — H. petiolaris initial | 3.18 × 105 | 2.63 × 105 | 3.52 × 105 |
| Divergence time | 1.05 × 106 | 8.53 × 105 | 1.24 × 106 |
| Nef m: H. petiolaris → H. annuus | 0.31 | 0.14 | 0.54 |
| Nef m: H. annuus → H. petiolaris | 0.60 | 0.19 | 0.96 |
Figure 2.
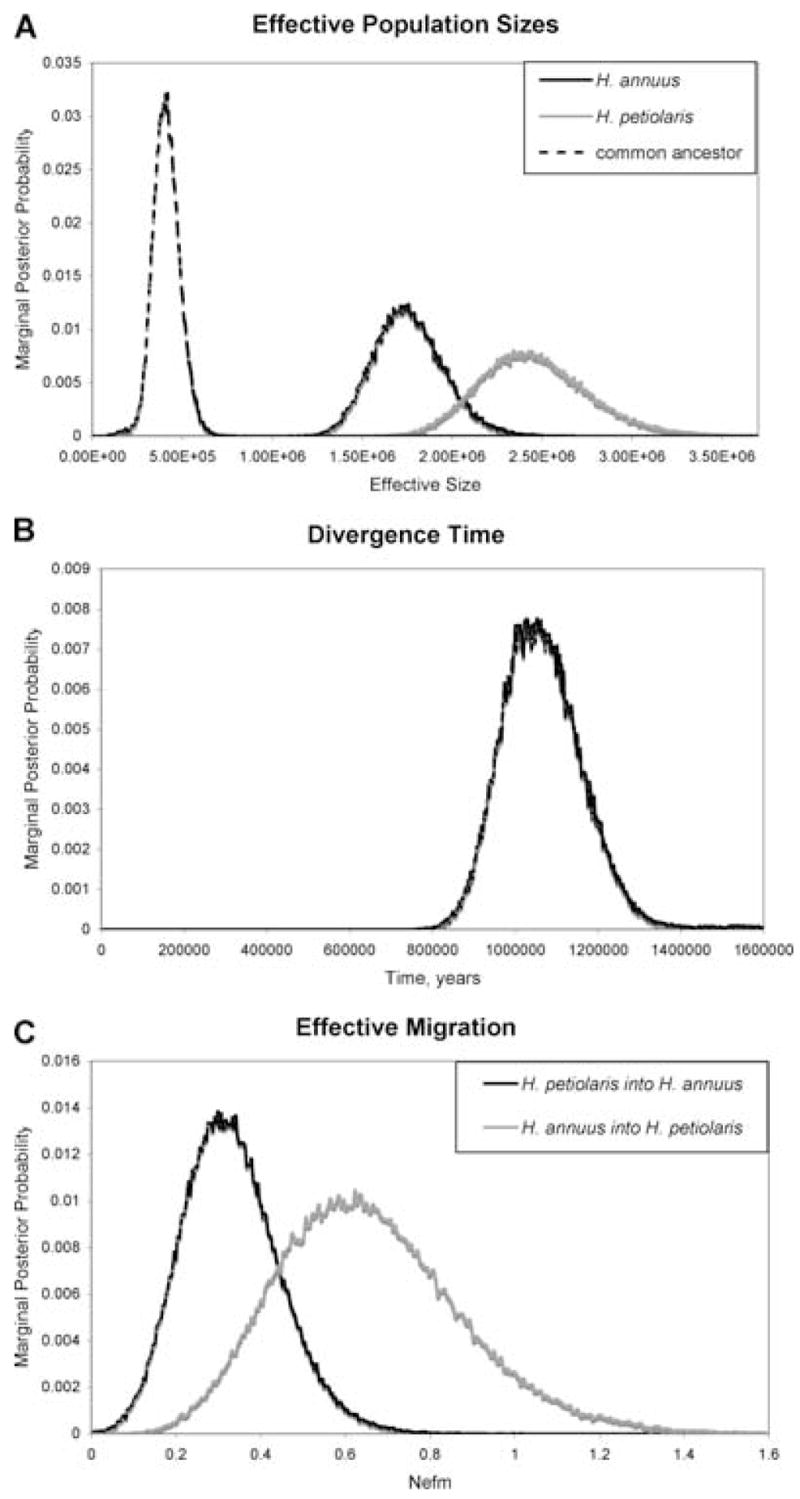
Marginal posterior probability densities for various demographic parameters. (A) inbreeding effective population sizes for H. annuus, H. petiolaris, and their common ancestor at time of divergence; (B) divergence time; (C) effective migration rates (Nef m) between the two species.
Our analysis indicates that H. annuus and H. petiolaris are approximately one million years old (90% HPD range 0.85–1.24 million years). This estimate is fairly concordant with previous estimates based on cpDNA (Rieseberg et al. 1991a). Long-term estimates of effective migration between H. annuus and H. petiolaris are exceptionally high for two species that diverged one million years ago. MLEs of Nef m for H. petiolaris into H. annuus and H. annuus into H. petiolaris are 0.31 and 0.6, respectively. The upper limit of the 90% HPD range for the latter value is approximately one, a level of effective gene flow often cited as high enough to prevent differentiation through genetic drift among populations within a single species (Templeton 2006).
Discussion
To date the “isolation with migration” model implemented in IM and closely related methods has been mainly applied to animals, most notably (although certainly not exclusively) primates and Drosophila. To the extent that a general pattern has emerged, it suggests that low, often asymmetric levels of gene flow are not uncommon between recently formed species (Hey 2006). In addition, inferred rates of gene flow sometimes vary widely among loci, reinforcing the need for multilocus approaches to the study of divergence, gene flow, and species relationships. Variation across the genome in rates of introgression can also provide clues to the genetic or genomic basis of species differences and reproductive isolation (Noor et al. 2001; Rieseberg 2001; Machado et al. 2007a; Turner and Hahn 2007). Hey and Nielsen (2004), in introducing the IM methodology and computer program, found an average Nm of approximately 0.05 in both directions between Drosophila pseudoobscura and D. persimilis, but with considerable variation across 14 nuclear loci, consistent with previous work indicating gene flow is limited by natural selection at some loci (Wang et al. 1997; Machado et al. 2002). Interestingly, although average rates of gene flow were comparable in each direction, most loci showed some evidence of asymmetric gene flow; in the most extreme case, mitochondrial introgression from D. pseudoobscura into D. persimilis was two orders of magnitude higher than in the reverse direction. Other results from Drosophila have been mixed. Counterman and Noor (2006) found no evidence for introgression between Drosophila mohavensis and D. arizonae, whereas Llopart et al. (2005) found introgression of mtDNA and two nuclear loci between D. yakuba and D. santomea (the authors did not make a quantitative estimate of the population migration rate at these loci). Bull et al. (2006) found evidence for gene flow on the order of Nm = 1 for one locus (calculated by us from the estimate of m in Table 6 and effective population sizes on page 6 of that paper), but no evidence of gene flow at three other loci. Two other illustrative examples involve chimpanzees (Won and Hey 2005), in which substantial unidirectional gene flow occurs between two subspecies of the common chimpanzee, but little exchange was found between common chimpanzees and bonobos; and Lake Malawi cichlids (Hey et al. 2004), where two very recently diverged species are exchanging genes at reciprocal rates of Nm = 0.16 and Nm = 0.31.
Table 6.
Model parameter and biological quantity estimates from IM analysis of entire sequences, including inferred recombinants. Due to computational constraints, a single haplotype was randomly chosen from each individual. See Table 5 legend for additional details.
| Model parameter | High point | HPD90Lo | HPD90Hi |
|---|---|---|---|
| θannuus | 53.77 | 43.43 | 66.97 |
| θpetiolaris | 87.07 | 67.55 | 119.52 |
| θancestor | 5.24 | 3.37 | 7.25 |
| T | 4.48 | 4.02 | 4.97 |
| m1 | 0.027 | 0.007 | 0.054 |
| m2 | 0.017 | 0.002 | 0.047 |
| S | 0.26 | 0.17 | 0.38 |
| Demographic quantity | High point | HPD90Lo | HPD90Hi |
| Nef —H. annuus | 2.77×106 | 2.24×106 | 3.45×106 |
| Nef —H. petiolaris | 4.49×106 | 3.48×106 | 6.16×106 |
| Nef —common ancestor | 2.70×105 | 1.74×105 | 3.74×105 |
| Nef —H. annuus initial | 7.09×104 | 4.69×104 | 1.02×105 |
| Nef —H. petiolaris initial | 1.99×105 | 1.68×105 | 2.23×105 |
| Divergence time | 9.23×105 | 8.28×105 | 1.03×106 |
| Nef m: H. petiolaris→ H. annuus | 0.36 | 0.09 | 0.72 |
| Nef m: H. annuus→ H. petiolaris | 0.36 | 0.03 | 1.01 |
Few robust estimates of long-term effective migration rates are available among plant species, and those that are available do not indicate high levels of long-term gene flow among species known to hybridize. Sweigart and Willis (2003) attributed extremely high genetic diversity (average over two loci silent-site θ = 0.077) in Mimulus guttatus at least partially to asymmetric introgression from its sympatric congener M. nasutus. However, they did not provide a quantitative estimate of Nef m, and alternative hypotheses to interspecific gene flow were not formally tested. Stadler et al. (2005, 2008) examined genetic variation at 13 loci in three Lycopersicon species and could not reject an isolation model (Wang et al. 1997) of no gene flow following initial species divergence (although patterns of linkage disequilibrium indicate that at least some genomic regions may have been subject to introgression). Based on 10 loci, Zhang and Ge (2007) found no clear evidence of introgression among several closely related rice species. One exception can be seen in Hawaiian silverswords, in which Lawton-Rauh et al. (2007) document gene flow rates of Nm = 0.14 and Nm = 0.39 between two recently (less than 500,000 years ago) diverged species.
This general lack of quantitative evidence for long-term effective migration between plant species may be due in part to the very recent availability of multilocus sequence data in natural populations of nonmodel organisms, and of the analytical and computational tools needed to analyze these data in an “isolation with migration” framework (Hey 2006). Here we have presented what is likely the most comprehensive analysis to date of historical demography and long-term effective migration between hybridizing plant species, and our results suggest significant introgression. In this regard, comparisons to intraspecific estimates of effective migration can be informative. Morjan and Rieseberg (2004) collected estimates of within-species Nef m for a range of taxa. For plants, their estimates range from 0.02 to 90.4, with a mean of 1.8 and a median of 1.1. Our estimates for introgression from H. petiolaris into H. annuus (Nef m = 0.31) and H. annuus into H. petiolaris (Nef m = 0.60) are larger than 18% and 41% of these intraspecific Nef m estimates, respectively. Morjan and Rieseberg (2004) also broke their data down by mating system and geographic scale. If we focus only on the most relevant comparisons, outcrossing species with Nef m estimated at the species-wide level (as opposed to among local populations), results are comparable; H. petiolaris into H. annuus and H. annuus into H. petiolaris estimates are larger than 15% and 34% of intraspecific estimates, respectively. The data analyzed by Morjan and Rieseberg (2004) generally used distance-based methods and simple island models of population structure equating Fst (or an Fst analog) to Nef m, and the usual caveats of this methodology apply (Whitlock and McCauley 1999)—most notably, Fst may underestimate (neutral) Nef m if the markers used are linked to loci under divergent selection, or it may overestimate Nef m if populations have recently fragmented and measures of Fst reflect past conditions rather than a current equilibrium. Nonetheless, it seems clear that H. annuus and H. petiolaris have undergone widespread introgression throughout the approximately one million years since their initial divergence.
As discussed above, multilocus studies often show variation among genes or genomic regions in rates of introgression (Hey 2006). In the most extreme examples, some loci show fixed differences with little or no evidence of introgression, whereas others show very high levels of introgression with little or no interspecific differentiation (e.g., Geraldes et al. 2006). These patterns may be due to genetic factors that contribute to reproductive isolation and species differences (Wu 2001); or they may be due to chromosomal factors such as rearrangements or proximity to centromeres, which may have direct negative fitness consequences in hybrids (in the case of rearrangements) or may extend the negative effects of genic factors through recombination suppression (Noor et al. 2001; Rieseberg 2001). In our case, although all 18 loci show evidence of gene flow at some point in the history of these two species, there is also some variation among loci. Although more than half of our loci show no fixed differences between H. annuus and H. petiolaris (see Table 4), the most significant outlier, (3724), shows six fixed differences and no shared polymorphisms between the two species. This locus is also by far the least variable of the 18 analyzed here. It bears little similarity to any other genes of known function—the closest Arabidopsis BLAST hit is to an unknown protein with very low similarity (see online Supplementary Table S2). We are currently gathering mapping data for all 18 loci, and hope to examine possible causes of among-locus variation in more detail in the near future.
Although confidence intervals for effective migration rates from H. annuus into H. petiolaris and vice versa are largely overlapping and not significantly different from each other, MLEs differ by a factor of two. It is not immediately clear why introgression from H. annuus into H. petiolaris might be higher than in the other direction. There are clear asymmetries in F1 hybrid frequencies in hybrid zones, with H. annuus much more likely to be the maternal parent, but there does not appear to be a bias toward either parental species in backcrosses (Rieseberg et al. 1998). Evidence of asymmetrical introgression has also been found between H. annuus and another annual sunflower species, H. debilis (Rieseberg et al. 1991b).
Our average estimates of silent site sequence diversity are 0.025 and 0.033 for H. annuus and H. petiolaris, respectively, with the highest estimates within each species being 0.045 and 0.063, respectively. These estimates are well above the average of 0.0152 for land plants reported by Lynch (2006), and the only individual species’ values included in Lynch’s (2006) analysis that are higher than H. petiolaris’ 18-locus average are based on single (or in one case two) highly variable loci. Helianthus petiolaris’ average is also higher than all vertebrate values reported by Lynch, and higher than all but four invertebrate values based on more than one to two loci. Our molecular clock calibration of 1 × 10−8 substitutions/site/year, estimated from of EST sequence comparisons and fossil calibrations in flowering plants (M. S. Barker and L. H. Rieseberg, unpubl. ms.) is close to Lynch’s (1997) estimate of nuclear synonymous substitution rate in land plants of 7.31 × 10−9 substitutions/site/year. However, our mutation rate estimate is for all sites, and sequence diversity/divergence at silent sites only is approximately 60% higher than overall diversity/divergence (data not shown), leading to a corresponding silent site mutation rate estimate of 1.6 × 10−8 substitutions/site/year, roughly twice as high as Lynch’s (1997) estimate. If we apply the Lynch (1997) estimate to our dataset, halving the mutation rate estimate will result in a doubling of our estimates of current and ancestral effective population sizes as well as divergence time (estimates of Nef m will be unaffected because the mutation rate cancels out when multiplying model parameters of population size and gene flow). A two million year divergence time is not consistent with rough estimates of divergence time based on cpDNA (Rieseberg et al. 1991a), which are largely in agreement with our current estimate of one million year. Assuming our estimate is an accurate one, sunflowers and related taxa may have a fairly high average nuclear mutation rate compared to other flowering plants. However, it is worth pointing out that our estimates of 1 × 10−8 and 1.6 × 10−8 substitutions/site/year for all sites and silent sites, respectively, are roughly in line with some recent estimates in flowering plants (Koch et al. 2000; Kay et al. 2006), and considerably slower than a recent estimate for noncoding sequence near the tb1 maize domestication gene (Clark et al. 2005). Given these recent estimates, our clock calibration does not seem unreasonable.
Helianthus annuus and H. petiolaris represent the deepest split within the North American annual sunflowers (Rieseberg 1991). Schilling et al. (1998) suggested that the genus Helianthus originated in the southeastern United States. Although North American annual sunflowers are nested within the perennial members of the genus, they appear to occupy a fairly basal position, and it may be the case that the annual sunflowers originated further to the east than the center of their current distribution. Expansion into their much larger ranges in the central and western United States would explain the dramatic population size increases documented here. Effective population sizes may also be increased due to hybridization/introgression with other species, especially for H. annuus (Rieseberg et al. 1990; Kim and Rieseberg 1999; Carney et al. 2000; Whitney et al. 2006). In addition, the method implemented in IM assumes no population structure within each species, a condition likely to be violated in almost every natural example. Population structure within species should increase effective population size estimates, although the sensitivity of estimates to this assumption is not clear, and in some cases species with likely violations still have reasonable estimates of population sizes (Hey 2005).
Population structure in the ancestral species may artificially inflate divergence time estimates. However, if the ancestor to annual sunflowers had a limited range in the southeastern United States, dramatic structure seems less likely. Microsatellite-based estimates of differentiation on the regional and species level in both H. annuus and H. petiolaris bear this out. Estimates of Nef m among southwestern and western populations of each species are approximately one (Schwarzbach and Rieseberg 2002; Welch and Rieseberg 2002; Gross et al. 2003); and an AMOVA of central and western populations of H. annuus and H. petiolaris indicates that only 3–4% of genetic variation is distributed among populations within species (Yatabe et al. 2007). In addition, we divided H. annuus and H. petiolaris sampling populations into three regions for each species based on geographical proximity and ran AMOVAs for each locus analyzed here. An average of 5.4% of sequence variation is distributed among regions within species, compared to 29.6% distributed between species and 65% distributed within regions, indicating that deep structure within each species is not likely to be a problem here. Finally, our divergence estimate is in line with previous estimates for this group (Rieseberg et al. 1991a).
Inbreeding effective population sizes for H. annuus and H. petiolaris are 1.8 million and 2.4 million, respectively. These numbers, although quite large, are certainly well below the total census population sizes of each species, in which individual populations of many thousands of individuals are not uncommon. Both species also possess life-history traits associated with larger inbreeding effective population sizes, including an outcrossing mating system and extensive seed banks (Nunney 2002; Vitalis et al. 2004). It should be mentioned that our sampling regime reflects our interest in hybrid speciation in Helianthus in the southwestern United States as part of a larger study; so although a significant portion of the range of each species is represented here, we have not included samples from the central United States. However, as discussed above, geographic structure within each species range is rather limited. As a result, we do not expect our results to be strongly affected by sampling regime.
The method implemented in IM also allows us to estimate the posterior probability density of the numbers of migration events and mean times of migration events for each locus in both directions (Won and Hey 2005). All of our loci show some evidence of migration, and posterior probability densities for mean times of migration for all 18 loci are shown in Figure 3. Considering the loci overall there is significant probability density throughout the entire history of these two species, but an increase in probability density begins roughly 300,000 years before present. This corresponds with the arrival of bison in North America 200,000–300,000 years before present (Shapiro et al. 2004). Bison are thought to have historically been a primary dispersal agent for sunflowers, as seeds became tangled in their matted hair and were transported long distances during the regular movements of the bison (Asch 1993). In addition, bison are known to have created a variety of natural disturbances, such as wallows and trails (Barsness 1985), in which both sunflower species are commonly found (Asch 1993). The arrival of bison provides an intriguing possible explanation for the dramatic population expansions of both H. annuus and H. petiolaris, as well as their increasing rates of hybridization and introgression.
Figure 3.
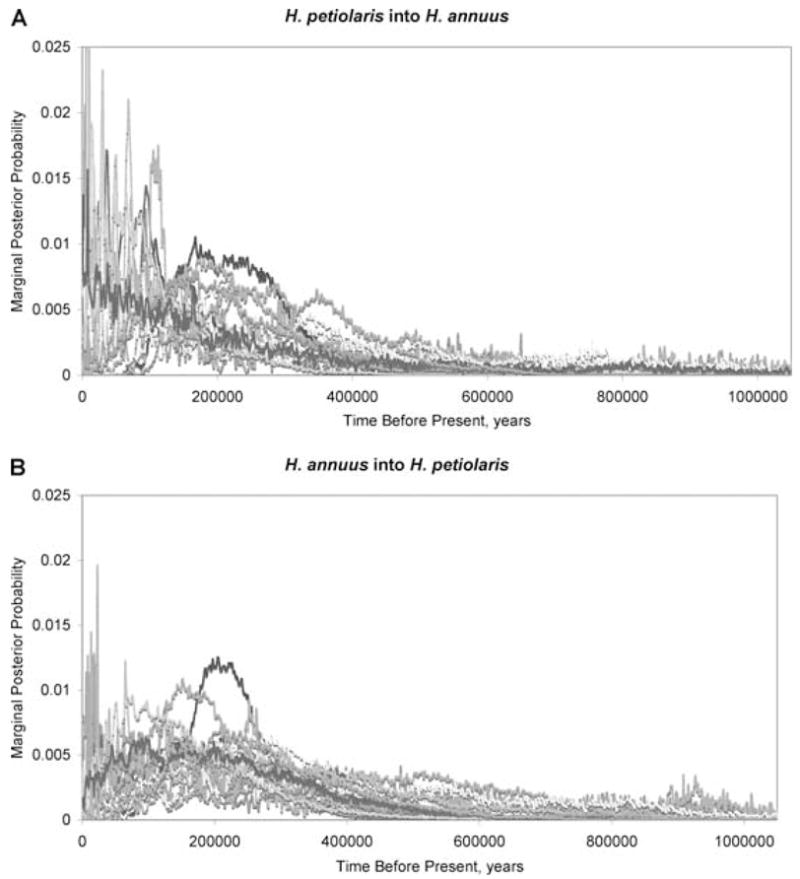
Marginal posterior probability densities for the mean time of all inferred migration events by locus for (A) H. petiolaris into H. annuus and (B) H. annuus into H. petiolaris.
IM assumes no intralocus recombination, although it is not completely clear how violations of this assumption will affect the estimation of each parameter in IM. To examine this, we performed IM analyses using the same program settings but including the entire length of sequence for each locus. Due to computational limitations, a single haplotype was randomly selected from each individual. Results are shown in Table 6. The most striking change is that estimates of current effective population sizes are much larger—58% larger for H. annuus and 85% larger for H. petiolaris. This pattern may be expected if variants actually created by recombination are inferred to be the result of repeated mutation. Differences among other estimates were not statistically significant, although ancestral effective population size and divergence time both decreased somewhat, and the MLE of gene flow from H. annuus into H. petiolaris decreased by 40%. Bull et al. (2006) found similar results (increased current effective population sizes, no significant changes in other parameters) when they compared a full dataset of four loci, three of which show evidence of recombination, to reduced datasets with recombinant haplotypes or sequence blocks removed. Further research into the effect of recombination on parameter estimation in IM and programs with similar methodologies, as well as comparisons between these methods and summary-based methods that incorporate recombination (e.g., Becquet and Przeworski 2007), will be very valuable.
Recently, a number of related models have been proposed in which chromosomal rearrangements can reduce gene flow and potentially contribute to speciation through the suppression of recombination (Noor et al. 2001; Rieseberg 2001; Navarro and Barton 2003). Empirical evidence for these models has been strongest in Drosophila (Hey and Nielsen 2004; Machado et al. 2007a; Noor et al. 2007), although evidence has also been found in sunflowers (Rieseberg et al. 1999), Anopheles mosquitos (Stump et al. 2007), and other systems. In their 14-locus analysis, Hey and Nielsen (2004) found much of the among-locus variation in rates of gene flow between D. pseudoobscura and D. persimilis could be explained by proximity to inversions that distinguish the two species. Sunflowers show extremely rapid rates of chromosomal evolution, and H. annuus and H. petiolaris differ by a minimum of 11 chromosomal rearrangements (Burke et al. 2004). However, Yatabe et al. (2007) found no correlation between location of microsatellite markers on collinear versus rearranged chromosomes and genetic distance between H. annuus and H. petiolaris, as might be expected if chromosomal rearrangements reduce recombination rates, thereby increasing the sizes of genomic regions prevented from introgressing. They suggested that the lack of a correlation between genetic distance and collinear versus rearranged chromosomes may be due to a very small genomic unit of isolation between the two species, possibly as a result of extensive hybridization and concomitant opportunities for recombination (Yatabe et al. 2007). Of the 18 loci used here, five map to collinear loci, 11 map to rearranged loci, and two have not been mapped. Although we obviously do not have the genomic coverage to address this question in detail with our sequence data, it is worth pointing out that we also find no relationship between sequence divergence and chromosome type (Table 7). In fact, the three least divergent loci all map to rearranged chromosomes. Overall gross divergence at loci on rearranged chromosomes is slightly lower than at loci on collinear chromosomes, although net divergence is slightly higher due to slightly lower variation within species.
Table 7.
Comparison of polymorphism and divergence data for loci on collinear versus rearranged chromosomes.
| H. annuus | H. petiolaris | |||||
|---|---|---|---|---|---|---|
| π (silent) | θ (silent) | π (silent) | θ (silent) | Gross divergence | Net divergence | |
| Collinear, avg. (n= 5) | 0.02851 | 0.03870 | 0.03578 | 0.05088 | 2.736 | 0.779 |
| Rearranged, avg. (n= 11) | 0.02625 | 0.03363 | 0.03522 | 0.04514 | 2.690 | 0.877 |
Supplementary Material
Acknowledgments
We are very grateful to B. Blackman, A. Schwarzbach, and M. Welch for sharing their collections, to Z. Lai for assistance in marker development, and to B. Gross and N. Kane for helpful discussions. We are also grateful to the Indiana University High Performance Systems group for the use of their high-performance computing systems for IM analyses. This work was supported by a National Institutes of Health Ruth L. Kirschstein Postdoctoral Fellowship (5F32GM072409-02) to JLS and grants from the National Science Foundation (DEB-0314654 and DBI0421630) the National Institutes of Health (GM059065) to LHR.
LITERATURE CITED
- Arnold ML. Natural hybridization as an evolutionary process. Annu Rev Ecol Syst. 1992;23:237–261. [Google Scholar]
- Asch DL. Common sunflower (Helianthus annuus L.): The pathway toward domestication. Proceedings of the 58th Annual Meeting of the Society for American Archaeology; St. Louis, MO. 1993. pp. 1–15. [Google Scholar]
- Barsness L. Head, hides, and horns: the compleat buffalo book. Texas Christian Univ. Press; Fort Worth, TX: 1985. [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annu Rev Ecol Syst. 1985;16:113–148. [Google Scholar]
- Becquet C, Przeworski M. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 2007;17:1505–1519. doi: 10.1101/gr.6409707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerkle CA, Rieseberg LH. Low intraspecific variation for genomic isolation between hybridizing sunflower species. Evolution. 2001;55:684–691. doi: 10.1554/0014-3820(2001)055[0684:livfgi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Bull V, Beltran M, Jiggins CD, McMillan WO, Bermingham E, Mallet J. Polyphyly and gene flow between non-sibling Heliconius species. BMC Biol. 2006;4:11. doi: 10.1186/1741-7007-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Lai Z, Salmaso M, Nakazato T, Tang SX, Heesacker A, Knapp SJ, Rieseberg LH. Comparative mapping and rapid karyotypic evolution in the genus Helianthus. Genetics. 2004;167:449–457. doi: 10.1534/genetics.167.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney SE, Gardner KA, Rieseberg LH. Evolutionary changes over the fifty-year history of a hybrid population of sunflowers (Helianthus) Evolution. 2000;54:462–474. doi: 10.1111/j.0014-3820.2000.tb00049.x. [DOI] [PubMed] [Google Scholar]
- Clark RM, Tavare S, Doebley J. Estimating a nucleotide substitution rate for maize from polymorphism at a major domestication locus. Mol Biol Evol. 2005;22:2304–2312. doi: 10.1093/molbev/msi228. [DOI] [PubMed] [Google Scholar]
- Counterman BA, Noor MAF. Multilocus test for introgression between the cactophilic species Drosophila mojavensis and Drosophila arizonae. Am Nat. 2006;168:682–696. doi: 10.1086/508632. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sinauer Associates; Sunderland, MA: 2004. [Google Scholar]
- Crow KD, Munehara H, Kanamoto Z, Balanov A, Antonenko D, Bernardi G. Maintenance of species boundaries despite rampant hybridization between three species of reef fishes (Hexagrammidae): implications for the role of selection. Biol J Linn Soc. 2007;91:135–147. [Google Scholar]
- de Casas RR, Cano E, Balaguer L, Perez-Corona E, Manrique E, Garcia-Verdugo C, Vargas P. Taxonomic identity of Quercus coccifera L. in the Iberian Peninsula is maintained in spite of widespread hybridisation, as revealed by morphological, ISSR and ITS sequence data. Flora. 2007;202:488–499. [Google Scholar]
- Dorado O, Rieseberg LH, Arias DM. Chloroplast DNA introgression in southern California sunflowers. Evolution. 1992;46:566–572. doi: 10.1111/j.1558-5646.1992.tb02061.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies from molecular sequences—inference and reliability. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes A, Ferrand N, Nachman MW. Contrasting patterns of introgression at X-linked loci across the hybrid zone between subspecies of the European rabbit (Oryctolagus cuniculus) Genetics. 2006;173:919–933. doi: 10.1534/genetics.105.054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer CJ. Practical Markov Chain Monte Carlo. Stat Sci. 1992;7:473–483. [Google Scholar]
- Gross BL, Schwarzbach AE, Rieseberg LH. Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae) Am J Bot. 2003;90:1708–1719. doi: 10.3732/ajb.90.12.1708. [DOI] [PubMed] [Google Scholar]
- Gross BL, Kane NC, Lexer C, Ludwig F, Rosenthal DM, Donovan LA, Rieseberg LH. Reconstructing the origin of Helianthus deserticola: survival and selection on the desert floor. Am Nat. 2004;164:145–156. doi: 10.1086/422223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano TA. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Heiser CB. Hybridization in the annual sunflowers: Helianthus annuus x H. argophyllus. Am Nat. 1951a;85:65–72. [Google Scholar]
- Heiser CB. Hybridization in the annual sunflowers: Helianthus annuus x H. debilis var. cucumerifolius. Evolution. 1951b;5:42–51. [Google Scholar]
- Hey J. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 2005;3:965–975. doi: 10.1371/journal.pbio.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. Recent advances in assessing gene flow between diverging populations and species. Curr Opin Genet Dev. 2006;16:592–596. doi: 10.1016/j.gde.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Wakeley J. A coalescent estimator of the population recombination rate. Genetics. 1997;145:833–846. doi: 10.1093/genetics/145.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Won YJ, Sivasundar A, Nielsen R, Markert JA. Using nuclear haplotypes with microsatellites to study gene flow between recently separated Cichlid species. Mol Ecol. 2004;13:909–919. doi: 10.1046/j.1365-294x.2003.02031.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. Academic Press; New York: 1969. pp. 21–132. [Google Scholar]
- Kay KM, Whittall JB, Hodges SA. A survey of nuclear ribosomal internal transcribed spacer substitution rates across angiosperms: an approximate molecular clock with life history effects. BMC Evol Biol. 2006;6:9. doi: 10.1186/1471-2148-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Rieseberg LH. Genetic architecture of species differences in annual sunflowers: implications for adaptive trait introgression. Genetics. 1999;153:965–977. doi: 10.1093/genetics/153.2.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol Biol Evol. 2000;17:1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- Lai Z, Livingstone K, Zou Y, Church SA, Knapp SJ, Andrews J, Rieseberg LH. Identification and mapping of SNPs from ESTs in sunflower. Theor Appl Genet. 2005a;111:1532–1544. doi: 10.1007/s00122-005-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Nakazato T, Salmaso M, Burke JM, Tang SX, Knapp SJ, Rieseberg LH. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics. 2005b;171:291–303. doi: 10.1534/genetics.105.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton-Rauh A, Robichaux RH, Purugganan MD. Diversity and divergence patterns in regulatory genes suggest differential gene flow in recently derived species of the Hawaiian silversword alliance adaptive radiation (Asteraceae) Mol Ecol. 2007;16:3995–4013. doi: 10.1111/j.1365-294X.2007.03445.x. [DOI] [PubMed] [Google Scholar]
- Liu AZ, Burke JM. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics. 2006;173:321–330. doi: 10.1534/genetics.105.051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart A, Lachaise D, Coyne JA. Multilocus analysis of introgression between two sympatric sister species of Drosophila: Drosophila yakuba and D. santomea. Genetics. 2005;171:197–210. doi: 10.1534/genetics.104.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol Biol Evol. 1997;14:914–925. doi: 10.1093/oxfordjournals.molbev.a025834. [DOI] [PubMed] [Google Scholar]
- Lynch M. The origins of eukaryotic gene structure. Mol Biol Evol. 2006;23:450–468. doi: 10.1093/molbev/msj050. [DOI] [PubMed] [Google Scholar]
- Machado CA, Kliman RM, Markert JA, Hey J. Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Mol Biol Evol. 2002;19:472–488. doi: 10.1093/oxfordjournals.molbev.a004103. [DOI] [PubMed] [Google Scholar]
- Machado CA, Haselkorn TS, Noor MAF. Evaluation of the genomic extent of effects of fixed inversion differences on intraspecific variation and interspecific gene flow in Drosophila pseudoobscura and D. persimilis. Genetics. 2007a;175:1289–1306. doi: 10.1534/genetics.106.064758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado CA, Matzkin LM, Reed LK, Markow TA. Multilocus nuclear sequences reveal intra- and interspecific relationships among chromosomally polymorphic species of cactophilic Drosophila. Mol Ecol. 2007b;16:3009–3024. doi: 10.1111/j.1365-294X.2007.03325.x. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybrid speciation. Nature. 2007;446:279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- Mayr E. Animal species and evolution. Belknap Press of Harvard Univ. Press; Cambridge, MA: 1963. [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the ADH locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Meyerhans A, Vartanian JP, Wainhobson S. DNA recombination during PCR. Nucleic Acids Res. 1990;18:1687–1691. doi: 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morjan CL, Rieseberg LH. How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Mol Ecol. 2004;13:1341–1356. doi: 10.1111/j.1365-294X.2004.02164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Barton NH. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution. 2003;57:447–459. doi: 10.1111/j.0014-3820.2003.tb01537.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MAF, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci USA. 2001;98:12084–12088. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MAF, Garfield DA, Schaeffer SW, Machado CA. Divergence between the Drosophila pseudoobscura and D. persimilis genome sequences in relation to chromosomal inversions. Genetics. 2007;177:1417–1428. doi: 10.1534/genetics.107.070672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunney L. The effective size of annual plant populations: the interaction of a seed bank with fluctuating population size in maintaining genetic variation. Am Nat. 2002;160:195–204. doi: 10.1086/341017. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins SE, Stranger BE, Mitchell-Olds T, Aguade M. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics. 2004;166:373–388. doi: 10.1534/genetics.166.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. Homoploid reticulate evolution in Helianthus (Asteraceae)—evidence from ribosomal genes. Am J Bot. 1991;78:1218–1237. [Google Scholar]
- Rieseberg LH. Chromosomal rearrangements and speciation. Trends Ecol Evol. 2001;16:351–358. doi: 10.1016/s0169-5347(01)02187-5. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Hybrid speciation in wild sunflowers. Ann Mo Bot Gard. 2006;93:34–48. [Google Scholar]
- Rieseberg LH, Wendel JF. Introgression and its consequences in plants. In: Harrison RG, editor. Hybrid Zones and the Evolutionary Process. Oxford Univ. Press; New York: 1993. pp. 70–109. [Google Scholar]
- Rieseberg LH, Baird SJE, Desrochers AM. Patterns of mating in wild sunflower hybrid zones. Evolution. 1998;52:713–726. doi: 10.1111/j.1558-5646.1998.tb03696.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Beckstrom-Sternberg S, Doan K. Helianthus annuus ssp texanus has chloroplast DNA and nuclear ribosomal RNA genes of Helianthus debilis ssp cucumerifolius. Proc Natl Acad Sci USA. 1990;87:593–597. doi: 10.1073/pnas.87.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Beckstrom-Sternberg SM, Liston A, Arias DM. Phylogenetic and systematic inferences from chloroplast DNA and isozyme variation in Helianthus Sect Helianthus (Asteraceae) Syst Bot. 1991a;16:50–76. [Google Scholar]
- Rieseberg LH, Choi HC, Ham D. Differential cytoplasmic versus nuclear introgression in Helianthus. J Hered. 1991b;82:489–493. [Google Scholar]
- Rieseberg LH, Desrochers AM, Youn SJ. Interspecific pollen competition as a reproductive barrier between sympatric species of Helianthus (Asteraceae) Am J Bot. 1995;82:515–519. [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics. 1999;152:713–727. doi: 10.1093/genetics/152.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal DM, Rieseberg LH, Donovan LA. Re-creating ancient hybrid species’ complex phenotypes from early-generation synthetic hybrids: three examples using wild sunflowers. Am Nat. 2005;166:26–41. doi: 10.1086/430527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Schilling EE, Paner JL. A revised classification of subtribe Helianthinae (Asteraceae : Heliantheae). I. Basal lineages. Bot J Linn Soc. 2002;140:65–76. [Google Scholar]
- Schilling EE, Linder CR, Noyes RD, Rieseberg LH. Phylogenetic relationships in Helianthus (Asteraceae) based on nuclear ribosomal DNA internal transcribed spacer region sequence data. Syst Bot. 1998;23:177–187. [Google Scholar]
- Schwarzbach AE, Rieseberg LH. Likely multiple origins of a diploid hybrid sunflower species. Mol Ecol. 2002;11:1703–1715. doi: 10.1046/j.1365-294x.2002.01557.x. [DOI] [PubMed] [Google Scholar]
- Shapiro B, Drummond AJ, Rambaut A, Wilson MC, Matheus PE, Sher AV, Pybus OG, Gilbert MTP, Barnes I, Binladen J, et al. Rise and fall of the Beringian steppe bison. Science. 2004;306:1561–1565. doi: 10.1126/science.1101074. [DOI] [PubMed] [Google Scholar]
- Stadler T, Roselius K, Stephan W. Genealogical footprints of speciation processes in wild tomatoes: demography and evidence for historical gene flow. Evolution. 2005;59:1268–1279. [PubMed] [Google Scholar]
- Stadler T, Arunyawat U, Stephan W. Population genetics of speciation in two closely related wild tomatoes (Solanum section lycopersicon) Genetics. 2008;178:339–350. doi: 10.1534/genetics.107.081810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump AD, Pombi M, Goeddel L, Ribeiro JMC, Wilder JA, Torre AD, Besansky NJ. Genetic exchange in 2La inversion heterokaryotypes of Anopheles gambiae. Insect Mol Biol. 2007;16:703–709. doi: 10.1111/j.1365-2583.2007.00764.x. [DOI] [PubMed] [Google Scholar]
- Sweigart AL, Willis JH. Patterns of nucleotide diversity in two species of Mimulus are affected by mating system and asymmetric introgression. Evolution. 2003;57:2490–2506. doi: 10.1111/j.0014-3820.2003.tb01494.x. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods) Sinauer; Sunderland, MA: 1999. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR. Population genetics and microevolutionary theory. Wiley-Liss; Hoboken, NJ: 2006. [Google Scholar]
- Turner TL, Hahn MW. Locus- and population-specific selection and differentiation between incipient species of Anopheles gambiae. Mol Biol Evol. 2007;24:2132–2138. doi: 10.1093/molbev/msm143. [DOI] [PubMed] [Google Scholar]
- Vitalis R, Glemin S, Olivieri I. When genes go to sleep: the population genetic consequences of seed dormancy and monocarpic perenniality. Am Nat. 2004;163:295–311. doi: 10.1086/381041. [DOI] [PubMed] [Google Scholar]
- Wakeley J. Distinguishing migration from isolation using the variance of pairwise differences. Theor Popul Biol. 1996;49:369–386. doi: 10.1006/tpbi.1996.0018. [DOI] [PubMed] [Google Scholar]
- Wakeley J, Hey J. Estimating ancestral population parameters. Genetics. 1997;145:847–855. doi: 10.1093/genetics/145.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RL, Wakeley J, Hey J. Gene flow and natural selection in the origin of Drosophila pseudoobscura and close relatives. Genetics. 1997;147:1091–1106. doi: 10.1093/genetics/147.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Welch ME, Rieseberg LH. Patterns of genetic variation suggest a single, ancient origin for the diploid hybrid species Helianthus paradoxus. Evolution. 2002;56:2126–2137. doi: 10.1111/j.0014-3820.2002.tb00138.x. [DOI] [PubMed] [Google Scholar]
- Whitlock MC, McCauley DE. Indirect measures of gene flow and migration: Fst not equal 1/(4Nm+1) Heredity. 1999;82:117–125. doi: 10.1038/sj.hdy.6884960. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. Am Nat. 2006;167:794–807. doi: 10.1086/504606. [DOI] [PubMed] [Google Scholar]
- Won YJ, Hey J. Divergence population genetics of chimpanzees. Mol Biol Evol. 2005;22:297–307. doi: 10.1093/molbev/msi017. [DOI] [PubMed] [Google Scholar]
- Wu CI. The genic view of the process of speciation. J Evol Biol. 2001;14:851–865. [Google Scholar]
- Yatabe Y, Kane NC, Scotti-Saintagne C, Rieseberg LH. Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, Helianthus annuus and H. petiolaris. Genetics. 2007;175:1883–1893. doi: 10.1534/genetics.106.064469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LB, Ge S. Multilocus analysis of nucleotide variation and speciation in Oryza officinalis and its close relatives. Mol Biol Evol. 2007;24:769–783. doi: 10.1093/molbev/msl204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.