

Abstract
Synergistic engagement of the B cell receptor (BCR) and Toll-like receptor 9 (TLR9) in response to DNA-containing antigens underlies the production of many autoantibodies in systemic autoimmune diseases. However, the molecular basis of this synergistic engagement is not known. Given that these receptors are spatially segregated, with the BCR on the cell surface and TLR9 in endocytic vesicles, achieving synergy must involve novel mechanisms. We show that upon antigen binding the BCR initiates signaling at the plasma membrane and continues to signal to activate MAP kinases as it traffics to autophagosome-like compartments. The internalized BCR signals through a phospholipase D-dependent pathway to recruit TLR9-containing endosomes to the autophagosome via the microtubular network. The recruitment of TLR9 to the autophagosomes was necessary for hyper-activation of MAP kinases. This novel mechanism for BCR-induced TLR9 recruitment resulting in B cells hyper-responses may provide new targets for therapeutics for autoimmune diseases.
Introduction
B cell responses are initiated by the binding of multivalent antigens to the highly diversified, clonally distributed B cell receptors (BCRs). Antigen-induced clustering of the BCRs triggers a signaling cascade that involves the activation of at least four major signaling pathways that include phospholipase C (PLC), the Rho family of GTPases, ras and phosphatidylinositol-3-kinase (PI3K) (Campbell, 1999; Kurosaki, 1999). Combinations of these signaling pathways lead to the activation of MAP kinases and transcription of a variety of genes associated with B cell activation (Dal Porto et al., 2004; Schulze-Luehrmann and Ghosh, 2006). In addition, the BCR, under the influence of signaling cascades, efficiently transports antigens to a MHC-class II-containing, multivesicular, intracellular compartment where the antigens are proteolytically cleaved and the resulting peptides are assembled with MHC class II molecules into complexes for recognition by helper T cells (Lanzavecchia, 1990). Recent studies provided evidence that the correct targeting of the BCR to class II-containing intracellular compartments was dependent on the activation of phospholipase D (PLD) (Snyder and Pierce, 2006), a lipase implicated in the intracellular trafficking of a variety of surface receptors (Melendez and Allen, 2002; Jenkins and Frohman, 2005). Antigen binding to the BCR also appears to result in the clustering or fusion of intracellular vesicles resulting in large LAMP-1 positive, MHC-class II-rich compartments into which the BCR traffics, although the molecular basis of this subcellular reorganization is not known (Lankar et al., 2002; Siemasko and Clark, 2001). Lastly, it was shown recently that in antigen presenting cells MHC-class II-containing intracellular compartments continuously fuse with the autophagosomes, providing means for presentation of cytosolic proteins on MHC-class II molecules (Schmid et al., 2007).
It has becoming increasingly clear that signaling through the BCR is regulated or fine-tuned by an array of signaling receptors receiving information from the B cells’ environment (Dal Porto et al., 2004). In particular, receptors of the innate immune systems have been shown to significantly influence the outcome of antigen engagement by the BCR and may, in fact, contribute to the immune dysregulation observed in autoimmune diseases (Bernasconi et al., 2003). Of particular interest are the Toll-like receptors (TLRs), germ line encoded innate immune system receptors that recognize conserved molecular patterns of microorganisms (Akira et al., 2001). Here we focus on the interactions of the BCR with TLR9 that recognizes unmethylated CpG-DNA motifs present in viral and bacterial DNA. As for the BCR, TLR9 initiated signaling ultimately results in the activation of the MAP kinases, p38 and JNK, and NF-κB, although through signaling pathways distinct from those triggered by the BCR (Campbell, 1999; Krieg, 2002; Krieg et al., 1995; Kurosaki, 1999). Indeed, CpG DNA-induced TLR9 signaling has been shown to synergize with antigen-induced BCR signaling in the phosphorylation of p38 and JNK, and NF-κB activation (Yi et al., 2003). Moreover, this synergistic response was also evident when measured by cell proliferation and Ig secretion (Yi et al., 2003). The synergistic engagement of TLR9 and the BCR in response to DNA-containing antigens has been implicated in the activation of autoimmune B cells (Leadbetter et al., 2002; Viglianti et al., 2003). Many of the autoantigens targeted in systemic lupus erythematosus (SLE) contain self DNA, histones, RNA or ribonucleoproteins that are thought to be released from apoptotic cells. Moreover, TLR9 has been shown to play a critical role in regulating DNA-specific autoantibody production in mouse models of lupus by mechanisms that involve the simultaneous engagement of TLR9 and the BCR (Christensen et al., 2005; Christensen et al., 2006). The relevance of TLR9 signaling in B cells to human diseases is underscored by the observation that genetic variations in TLR9 are linked to SLE susceptibility (Tao et al., 2007). Consequently, blocking TLR9 signaling in autoreactive B cells has been suggested as a therapy for SLE (Lenert, 2006). A role for TLRs in autoantibody response does not appear to be limited to the DNA-binding TLR9, but also includes the endosomal RNA-binding TLR, TLR7. RNA-associated autoantigens activate B cells to produce antibodies in a BCR-and TLR7-dependent manner (Lau et al., 2005). Recently, the first direct genetic evidence for a role of TLR7 in autoimmune diseases was provided in mice by the finding that a duplication of the TLR7 gene resulted in RNA-specific autoantibody response (Pisitkun et al., 2006). It was subsequently shown that a simple two fold increase in expression of TLR7 alone was sufficient to accelerate autoimmunity (Deane et al., 2007). Thus, it is becoming essential to understand the molecular basis of the interaction of BCR and TLRs in response to DNA- and RNA- containing antigens to understand the molecular basis of autoantibody responses. At present, little is known about the cellular or molecular mechanisms that underlie TLR9- and TLR7- enhanced signaling of the BCR and given the spatial segregation of the BCR on the plasma membrane and both TLR9 and TLR7 in early endosomal compartments (Ahmad-Nejad et al., 2002), it is not obvious how hyperresponses to DNA- or RNA- containing antigens are achieved.
In this study we explore the effect of ligating the BCR and TLR9 on both the strength and the spatial distribution of the resulting BCR- and TLR9-mediated signaling. We provide evidence that following antigen binding and internalization the BCR recruits TLR9 from multiple small endosomes to large autophagosome-like compartments in a microtubule dependent process, where signaling to p38 phosphorylation occurs. Thus, the hyper-response of B cells to DNA-containing antigens appears to involve a novel mechanism of physical recruitment of TLR9 to the MHC-class II antigen processing compartments where the BCRs deliver antigens.
Results
DNA-containing antigens stimulate hyper MAP Kinase and NF-κB responses in B cells
To understand the molecular basis of B cells hyper responses to DNA-containing antigens, we ascertained the effect of ligating the BCR and TLR9 either independently or together on the phosphorylation of p38 and the degradation of I-κB. To do so, mouse splenic B cells were treated with either: F(ab')2 anti-mouse IgM (anti-IgM) alone to crosslink the BCR; CpG DNA alone to stimulate TLR9; anti-IgM and CpG DNA together to simultaneously engage the BCR and TLR9 or with an anti-IgM-CpG DNA conjugate that could potentially coligate the BCR and TLR9. Consistent with earlier published findings (Yi et al., 2003), B cells activated with both anti-IgM and CpG DNA showed a larger increase in p38 phosphorylation and I-κB degradation as compared to B cells treated with either anti-IgM or CpG DNA alone (Fig. S1). This effect was slightly more pronounced when B cells were treated with an anti-IgM-CpG DNA conjugate. The enhanced responses to the combination of CpG DNA and anti-IgM and to the anti-IgM-CpG conjugate were dependent on the TLR9 and were not observed in B cells from TLR9-deficient mice (Fig. S1). TLR9 signaling to CpG is blocked by chloroquine and bafilomycin as is the enhanced signaling observed to anti-IgM plus CpG and to anti-IgM-CpG DNA (Fig. S1). We also determined the effect of BCR and TLR9 coligation on the expression of phospho-p38 in cells by confocal microscopy, as described in the methods section. A larger number of B cells showed p38 phosphorylation when activated by both anti-IgM and CpG DNA or with the anti-IgM-CpG DNA conjugate as compared to either anti-IgM or CpG DNA alone (Fig. 1A). We also observed that the B cells stimulated with either anti-IgM and CpG DNA or with the anti-IgM-CpG DNA conjugate responded more rapidly than cells stimulated with CpG DNA and anti-IgM alone. Neither TLR9-induced p38 phosphorylation nor TLR9 enhancement of with BCR signaling were observed in B cells from MyD88-deficient mice (Fig. 1A). Similar results were obtained analyzing B cells for JNK phosphorylation (data not shown). Taken together these results show that a larger number of B cells reach a threshold for activation and do so more quickly when stimulated with a mixture of CpG DNA and antigen or with a CpG DNA-containing antigen. We also determined the effect of TLR9 and BCR signaling on later functional responses, measuring the cell surface expression of CD40, CD54 and α6-integrin 48 hr after stimulation. The co-stimulatory molecule CD40 and α6 integrin were maximally upregulated in the presence of the anti-IgM-CpG conjugate (Fig. 1C). However, anti-IgM induced CD54 expression was selectively downregulated in the presence of CpG either alone or as part of the anti-IgM-CpG conjugate (Fig. 1C). Taken together these results indicate that DNA-containing antigens affect B cell responses at several levels and suggest that the modulating effects of TLR9 on downstream BCR signaling events are complex. For the remaining analysis we focus on the early signaling events including p38 activation.
Figure 1. BCR and TLR9 costimulation reduces the threshold for p38 phosphorylation and increases CD40 and α6-integrin expression.

(A). The percent of mouse splenic B cells staining for p-p38- Alexa 488 with time is shown for B cells from wild type mice following treatment with: CpG DNA alone (open squares); anti-IgM alone (closed squares); anti-IgM plus CpG DNA (closed circles) and the anti-IgM-CpG DNA conjugate (open circles). Approximately 250 cells were examined by confocal microscopy for each condition in 3 independent experiments as described in Methods. Error bars represent standard deviation.
(B). Same as in (A) except B cells from MyD88-deficient mice were used.
(C). Mouse splenic B cells were either untreated or incubated with: 10µg/ml anti-IgM alone; 3µM stimulatory CpG DNA alone; both 10µg/ml anti-IgM and 3µM CpG DNA or 10µg/ml anti-IgM-CpG DNA conjugate for 48 h. Cells were stained for B220 and CD40 or α6 integrin or CD54 using appropriately fluorescent antibodies specific for each. Shown are the CD40, α6 integrin and CD54 staining in the B220 positive population.
TLR9 is recruited to autophagosome-like compartments upon BCR crosslinking
To learn where synergistic signaling occurred, the subcellular distribution of TLR9 in resting and activated splenic B cells was determined by confocal microscopy. In resting B cells TLR9 was present in small intracellular vesicles (Fig. 2A, B) that colocalized with early endosome markers, antigen 1, (EEA1) and transferrin receptor (TfR) (Fig. 3). In CpG DNA stimulated B cells, TLR9 remained in endosomes (Fig. 2A,B). Remarkably, crosslinking the BCR with anti-IgM alone or in combination with CpG DNA or with the anti-IgM-CpG DNA conjugate resulted in the relocalization of TLR9 into a single large intracellular structure (Fig. 2A,B). TLR9 staining was specific and was not detected in B cells from TLR9-deficient mice under any condition (Fig. 4C and S2). The relocalization of TLR9 induced by both anti-IgM and anti-IgM-CpG DNA occurred in a time-dependent fashion that was initiated within 5 min and complete by 30 min, (Fig. S3) consistent with the observed time course of p38 phosphorylation (Fig. 1A). Similar results were obtained in HEL-specific transgenic B cells when activated with HEL either alone or in combination with CpG indicating that the recruitment of TLR9 occurs in response to a bonafide antigen and is not restricted to signals transduced by crosslinking the BCR by anti-Ig (Fig. S4). We also observed that the recruitment of TLR9 was absolutely dependent on BCR crosslinking and was not observed in B cells stimulated with CpG alone for any length of time up to 8h (Fig. 4A). However, anti-IgM treatment of B cells stimulated for 8 h with CpG resulted in the recruitment of TLR9 (data not shown) suggesting that relocalization of TLR9 in B cells is completely governed by the antigen engagement of the BCR. Moreover, TLR9 recruitment was not dependent on TLR9 signaling and was observed following BCR crosslinking in B cells from MyD88-deficient mice (Fig 4B).
Figure 2. Subcellular location of TLR9 changes upon BCR crosslinking.
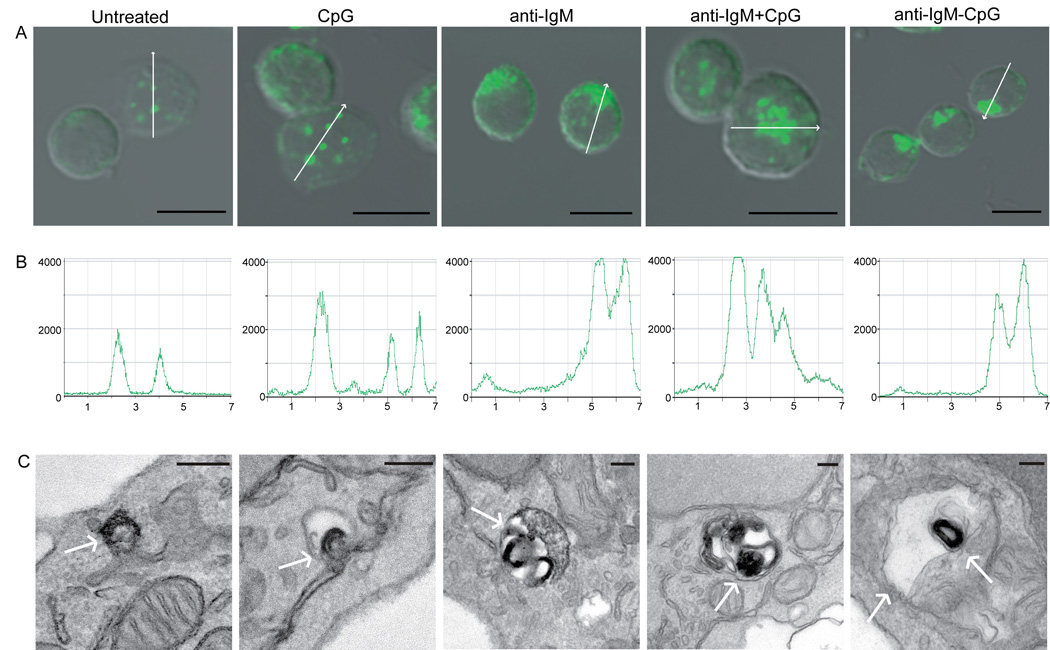
(A). Mouse splenic B cells were either untreated or stimulated with: 3µM CpG DNA alone; 10µg/ml anti-IgM alone; 10µg/ml anti-IgM plus 3µM CpG or 10µg/ml anti-IgM-CpG conjugate for 60 min. The merged confocal fluorescent Alexa 488 and differential interference contrast (DIC) images are shown. Over 500 cells were analyzed for each condition in 20 independent experiments and representative images are shown. Scale bars represent 5µm.
(B). The intensity analyses of Alexa 488- anti-TLR9 in representative cells in the direction shown by the white arrow in A.
(C). The immunoelectron microscopy images of cells stained with HRP-anti-TLR9 for cells treated as in panel A. The HRP reaction product DAB, visible as a dark stain, represents the location of TLR9 as shown by white arrow. Scale bars represent 100nm.
Figure 3. Colocalization of TLR9 to early endosome markers in resting cells.
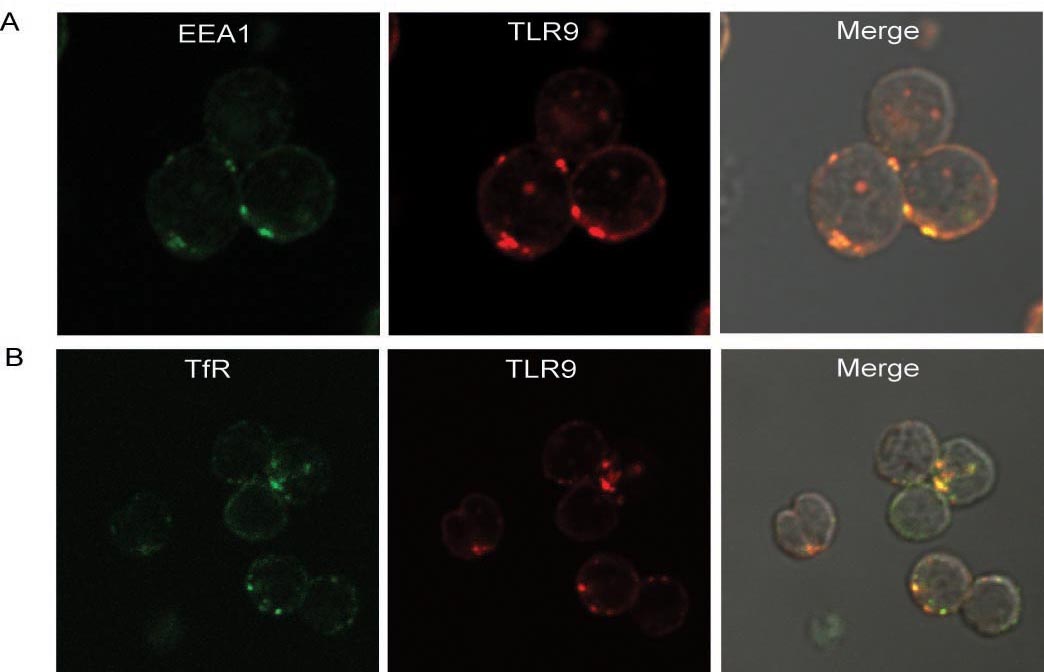
(A) Mouse splenic B cells were stained with antibodies specific for EEA1 labeled with Alexa 488 or for TLR9 labeled with Alexa 647. Shown are the confocal images of Alexa 488 and Alexa 647 merged with DIC images. (B) Mouse splenic B cells were stained with antibodies specific for transferrin receptor (TfR) labeled with Alexa 555 or for TLR9 labeled with Alexa 647. Shown are the confocal images of Alexa 555 and Alexa 488 merged with DIC images. Approximately 200 cells were analyzed from 3 independent experiments and a representative image is given.
Figure 4. TLR9 recruitment is dependent on BCR signaling but independent of TLR9 signaling.
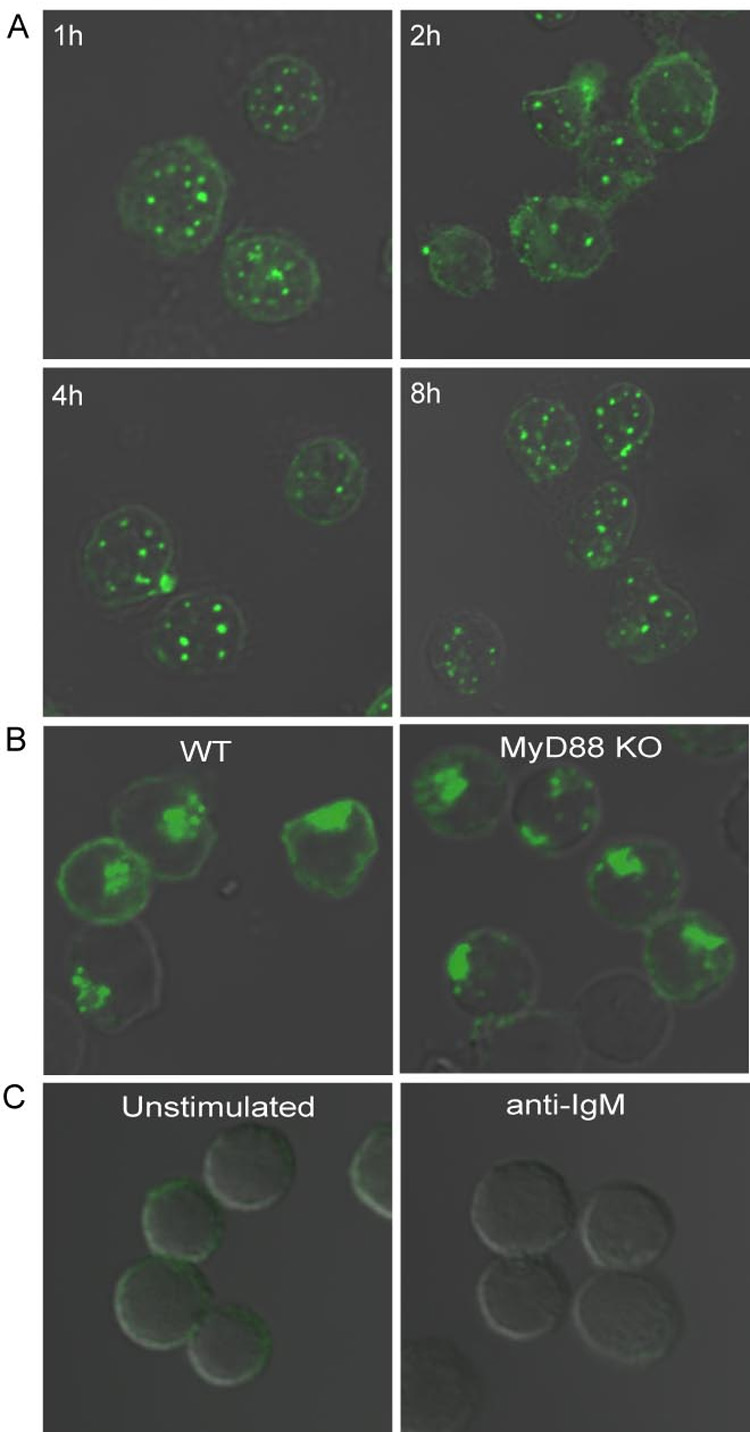
(A). Mouse splenic B cells were incubated with 3µM CpG for 1 to 8 hr and stained for TLR9 as described. Shown in the panel are DIC images merged with confocal images of Alexa 488.
(B). Wild-type and MyD88- deficient B cells were incubated with 10µg/ml anti-IgM for 60 min, and stained for TLR9 as described. Shown are the DIC images merged with confocal images of Alexa 488.
(C). TLR9- deficient B cells were either untreated or incubated with 10µg/ml anti-IgM for 60 min, and stained for TLR9 as described. Shown are the DIC images merged with confocal images of Alexa 488.
Approximately 200 cells were analyzed for each condition in 5 independent experiments and representative images are provided.
To further characterize the compartments to which TLR9 was recruited, B cells were analyzed by electron microscopy following staining with HRP-labeled TLR9- specific antibodies developed with DAB. In resting B cells and in CpG DNA-stimulated B cells, TLR9 was present in small endosomal vesicles approximately 100 nm in size (Fig. 2C). In contrast, in B cells stimulated through the BCR, the vast majority of TLR9 was present in double membrane enclosed organelles resembling autophagosomes ranging from 300 to 500 nm in size (Fig. 2C). Autophagosomes have been reported to be constitutively formed in human B cells lines (Schmid et al., 2007), however, in primary B cells autophagosomes are not well characterized. In many cell types autophagosomes function in the degradation and elimination of damaged organelles and proteins (Levine, 2005; Levine and Klionsky, 2004). To determine if the BCR-induced recruitment of TLR9 to autophagosome is a mechanism to degrade TLR9, B cells were stimulated with anti-IgM for up to 24 hr and stained for TLR9. Even after 24 hr of stimulation through the BCR, TLR9 remained in the autophagosome-like structures (Fig. S5). Moreover, quantification of the intracellular pool of TLR9 by immunoblotting showed that TLR9 was not degraded over time following BCR crosslinking and actually increased somewhat in activated B cells (Fig. S5).
We next determined whether the process of autophagy plays a role in the recruitment of TLR9 to autophagosome-like compartments. B cells were stimulated in the presence of class III PI-3- kinase inhibitors, 3-methyladenine (3-MA) or wortmannin that inhibit autophagy (Blommaart et al., 1997; Seglen and Gordon, 1982). In the presence of 3-MA or wortmannin, TLR9 remained in the small endosomes and did not localized to the autophagosome-like compartments (Fig. S6). We did not observe any effect of 3-MA or wortmannin on the BCR internalization at the concentrations that blocked TLR9 recruitment (Fig S6 and data not shown). Thus, blocking autophagy blocked BCR recruitment of TLR9. The BCR induced recruitment of TLR9 to the autophagosome-like compartments appeared selective and did not involve all endosomes. In anti-IgM stimulated B cells EM images showed many endosomal vesicles approximately 50–100 nm in size that were not recruited to the autophagosome-like compartment (Fig.S7). In addition, by confocal microscopy many TfR-containing endosomes did not colocalize with TLR9 in anti-IgM stimulated B cells (Fig. S7).
MAP Kinase phosphorylation occurs at distinct subcellular sites following TLR9 versus BCR crosslinking
To determine the intracellular sites from which the BCR and TLR9 signaled to MAP kinase activation, B cells were stimulated with anti-IgM and CpG either alone or together or with the anti-IgM-CpG DNA conjugate. Cells were stained for either phosphorylated p38 (p-p38) (Fig. 5) or phosphorylated JNK (p-JNK) (Fig. S8) and analyzed by confocal microscopy. No p-p38 was detected in untreated B cells or B cells treated with nonstimulatory control CpG DNA (Fig. 5). In cells stimulated with anti-IgM, p-p38 appeared primarily in a single large intracellular structure, resembling autophagosome-like compartments to which TLR9 was recruited following BCR crosslinking (Fig. 5). Similarly, in cells treated with a combination of anti-IgM and CpG DNA or with the anti-IgM-CpG DNA conjugate, the p-p38 appeared in similar autophagosome-like compartments (Fig. 5). In contrast, B cells stimulated with CpG DNA alone showed punctate p-p38 staining (Fig. 5) consistent with the endosomal location of TLR9 (Fig. 2). Similar results were obtained for p-JNK (Fig. S8). Similar results were also obtained for HEL-specific transgenic B cells (Fig. S4). The p-p38 colocalized with the BCR in a time-dependent fashion beginning at 5 min and continuing through 45 min (Fig. S9) consistent with the observed time course of p38 phosphorylation (Fig. 1). In B cells from TLR9- or MyD88-deficient mice CpG DNA did not induce p38 phosphorylation, however, anti-IgM triggered p38 phosphorylation in autophagosome-like compartments remained intact (data not shown). Taken together these results indicate that the observed enhanced signaling of B cells in response to anti-IgM and CpG DNA or anti-IgM-CpG DNA conjugate occurs from autophagosome-like compartments to which TLR9 is recruited.
Figure 5. Phosphorylation of p38 occurs in distinct subcellular compartments following BCR and TLR9 stimulation.
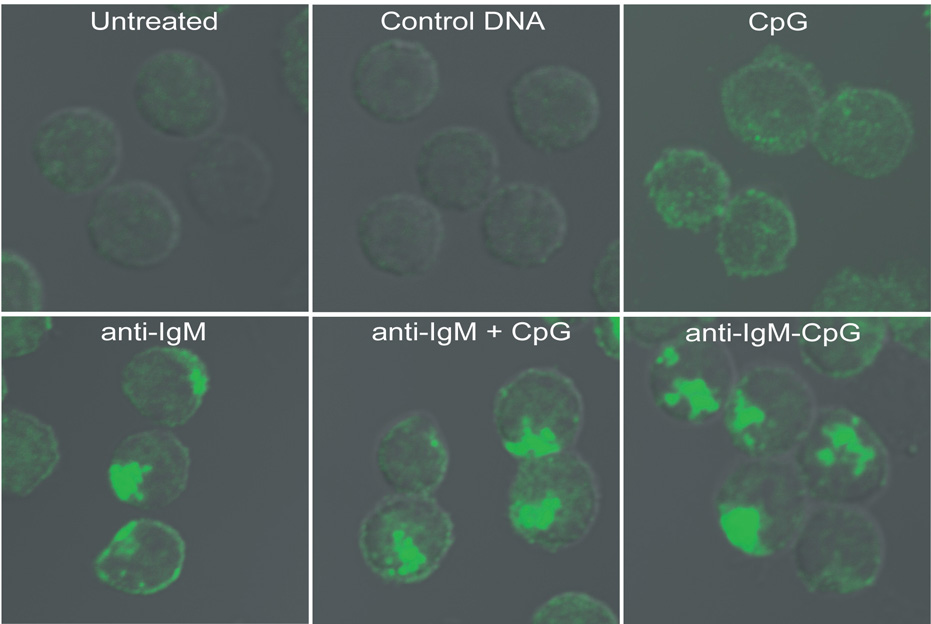
Mouse splenic B cells were either untreated or incubated with: 3µM control GpC DNA; 3µM stimulatory CpG DNA alone; 10µg/ml anti-IgM alone; both 10µg/ml anti-IgM and 3µM CpG DNA; or 10µg/ml anti-IgM-CpG DNA conjugate for 60 min. In each case the cells were fixed, permeabilized and stained for p-p38 using rabbit antibodies specific for p-p38, followed by Alexa 488-labeled goat antibodies specific for rabbit Ig. Shown are the DIC images merged with confocal images of Alexa 488. Approximately 250 cells were analyzed for each condition in 6 independent experiments and representative images are provided.
Internalized BCR colocalizes with TLR9 in LAMP-1-positive compartments and signals for p38 phosphorylation
To directly determine if the BCR and TLR9 colocalize following BCR crosslinking, B cells were imaged following incubation at 37°C for 60 min with Cy5-conjugated anti-IgM to label and crosslink the surface BCR and with Cy3-conjugated CpG DNA to activate and track TLR9. We observed colocalization of the Cy3-CpG and Cy5-anti-IgM in large intracellular compartments in the majority of B cells (Fig. 6A). We also observed co-localization of the Cy3-CpG and labelled BCR in HEL-specific transgenic B cells in response to HEL (Fig. S10). B cells labelled with Cy5-anti-IgM were fixed and permeabilized 60 min following treatment and three-color fluorescent microscopy was used to determine the subcellular location of the Cy5-anti-IgM, TLR9 and p-p38 or the Cy5-anti-IgM, TLR9 and LAMP-1. The internalized BCR colocalized both with TLR9 and p-p38 (Fig. 6B). Moreover, internalized BCR and TLR9 also colocalized with markers for MHC-class II antigen-processing compartments in B cells, LAMP-1 (Fig. 6C) and invariant chain, Ii (data not shown). The fluorescent intensities of the images in Figs. 6A, B were quantified and the overlap is given (Fig. S11). Taken together these results indicate that following crosslinking the BCR is internalized into LAMP-1- and Ii- positive autophagosome-like compartments to which TLR9 is recruited and where activation of MAP kinases occurs.
Figure 6. Internalized BCR colocalizes with TLR9 in LAMP-1 positive compartments.
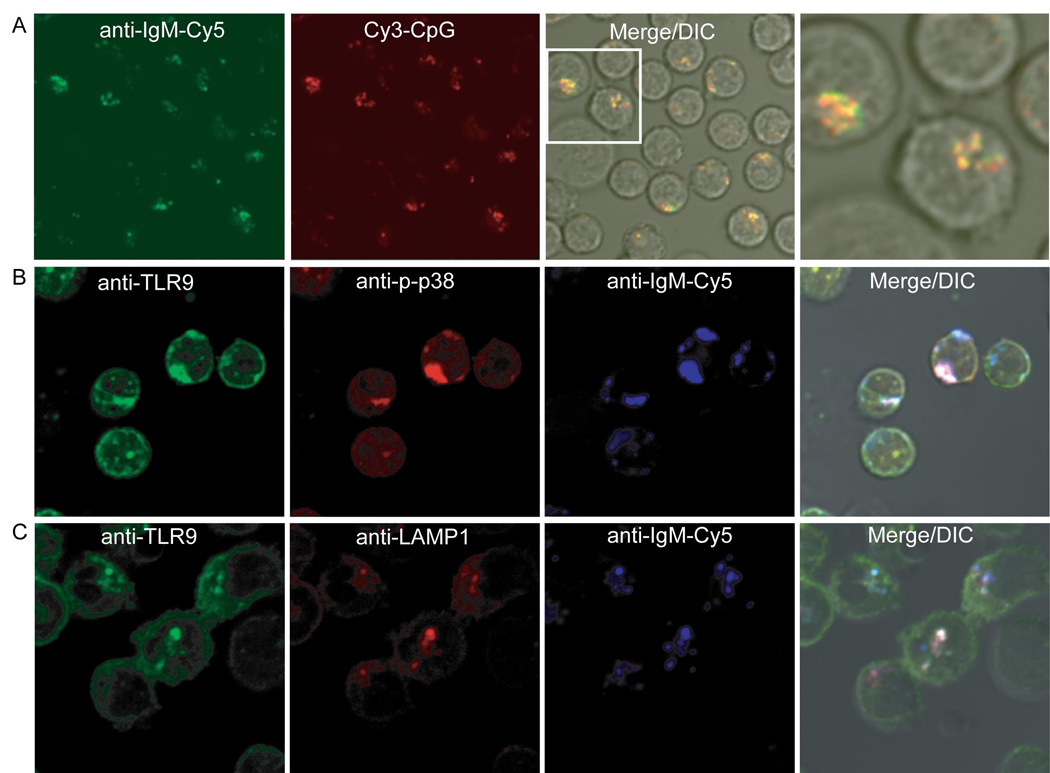
(A). Mouse splenic B cells were stimulated with 3µM Cy3-labeled CpG and 10µg/ml Cy5-labeled anti-IgM for 60 min and analyzed by fluorescent confocal microscopy. Shown are merged DIC and confocal images of Cy3-CpG and Cy5-anti-IgM.
(B). Mouse splenic B cells were incubated with 10µg/ml Cy5-anti-IgM for 60 min, fixed, permeabilized and stained as described for TLR9 and p-p38. Shown are the merged DIC and confocal images of Cy5-anti-IgM, Alexa 488 for TLR9 and Alexa 565 for anti-p-p38.
(C). Same as in (B) except cells were stained for TLR9 and LAMP1 and shown are the merged DIC and confocal images of Cy5-anti-IgM, Alexa 488 for TLR9 and Alexa 565 for LAMP-1.
Approximately 200 cells were analyzed for each condition in 4 independent experiments and representative images are provided.
Recruitment of TLR9 to autophagosome-like compartments requires BCR internalization and signaling through PLD
The requirements for BCR-mediated recruitment of TLR9 were further delineated. We first examined the need for BCR internalization for the recruitment of TLR9 to autophagosome-like compartments. Antigen binding to the BCR initiates a PP2-sensitive, Src-family kinase-dependent signaling cascade at the plasma membrane that results in the internalization of the BCR via a clathrin-dependent mechanism and its trafficking to the MHC class II-containing compartments (Clark et al., 2003; Stoddart et al., 2002). Treatment of B cells with PP2 blocked the BCR-induced TLR9 recruitment to the autophagosome-like compartments (Fig. S12). Moreover, treatment of B cells with monodansylcadaverine (MDC) that blocks BCR internalization but not early signaling as measured by induction of protein phosphorylation (Fig. S13) also blocked the BCR-induced TLR9 recruitment to the autophagosome-like compartments (Fig. S12). These results indicate that the physical internalization of BCRs into autophagosome-like compartments is key to the recruitment of TLR9. Concerning the nature of the BCR signal required for TLR9 recruitment, recruitment was completely blocked in the presence of calcium chelator, EGTA (data not shown). However, treatment of B cells with ionomycin and PMA to mimic a part of downstream BCR signaling was insufficient for TLR9 recruitment (Fig. S12) indicating a signaling for recruitment is more complex.
It has been shown that proper trafficking of the BCR to MHC-class II-containing antigen processing compartments relies on the activity of phospholipase D (PLD) (Snyder and Pierce, 2006), an enzyme that plays a role in endocytosis, vesicular trafficking and fusion (Jenkins and Frohman, 2005; Snyder and Pierce, 2006). When PLD activity is blocked BCR are internalized following antigen binding but fail to traffic properly to the class II compartments (Snyder and Pierce, 2006). PLD is activated following antigen binding to the BCR resulting in hydrolysis of phosphatidylcholine into phosphatidic acid (PA) and free choline (Snyder and Pierce, 2006). To ascertain whether BCR-mediated TLR9 recruitment to autophagosome-like compartments requires PLD activity, cells were treated anti-IgM in the presence of n-butanol, a PLD inhibitor or with t-butanol as a control and the subcellular location of TLR9 was determined. Treatment of B cells with 0.3% (v/v) n-butanol completely blocked BCR- mediated recruitment of TLR9 to autophagosome-like compartments (Fig. 7A). The effect appeared specific in that n-butanol did not affect induction of protein phosphorylation or BCR internalization (Fig. S12) induced by BCR crosslinking. In controls, t-butanol had no effect on TLR9 recruitment. PA can also be generated from diacyl glycerol through the action of DAG kinases (DGK) or can be converted into DAG by the phosphatidic acid phosphohydrolases (PAPs) (Jenkins and Frohman, 2005). To ascertain if only PLD-produced PA is required for TLR9 recruitment or if DAG-converted PA also plays a role, cells were stimulated in the presence of the DGK inhibitor R59949. The DGK inhibitor did not block the BCR-mediated TLR9 recruitment suggesting the PLD-produced PA is required (Fig 7A). On the other hand, treatment of cells with CpG alone in the presence of the PAP inhibitor propranolol, resulted in the recruitment of TLR9 to autophagosome-like compartments (Fig 7A). Taken together these results indicate that BCR crosslinking activates PLD that together with PA regulates the recruitment of TLR9 from endosome to autophagosome-like compartments.
Figure 7. BCR induced TLR9 recruitment to autophagosome-like compartments requires PLD activity and the microtubular network.
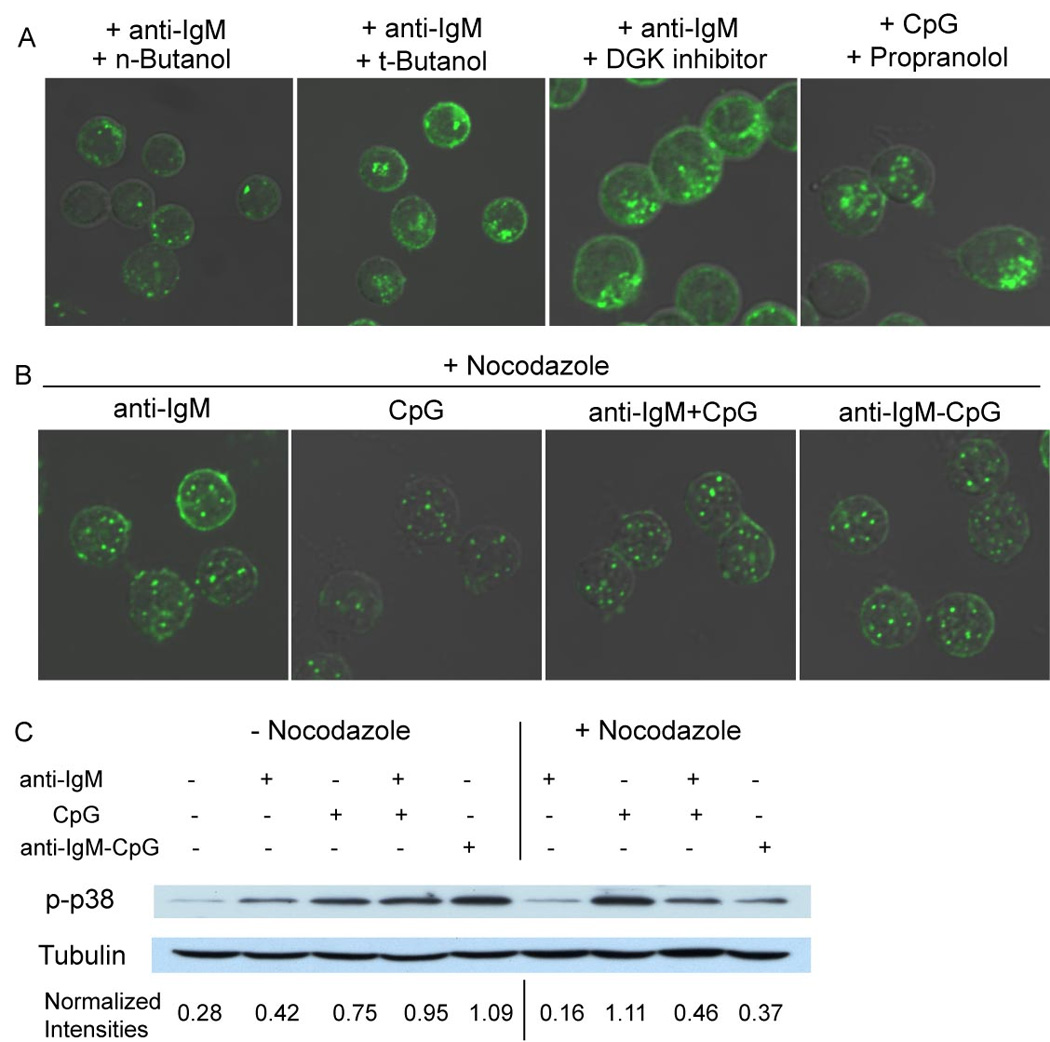
(A). Mouse splenic B cells were incubated with: 10µg/ml anti-IgM in the presence of 0.3% (v/v) n-Butanol; 0.3% (v/v) t-Butanol; or 10µM DGK inhibitor R59949. Alternatively, cells were incubated with 3µM CpG in the presence of 100µM PAP inhibitor Propranolol for 60 min. Cells were stained for TLR9 as described. Merged confocal and DIC images of Alexa 488 are shown.
(B). Cells were incubated with 10µg/ml anti-IgM; 3µM CpG DNA alone; both 10µg/ml anti-IgM and 3µM CpG DNA or 10µg/ml anti-IgM-CpG DNA conjugate in the presence of nocodazole (100µg/ml) for 60 min. Cells were stained for TLR9 as described. Shown are confocal and DIC images of Alexa 488.
Approximately 200 cells were analyzed for each condition in 5 independent experiments and representative images are provided.
(C). Mouse splenic B cells were either untreated or incubated with: 10µg/ml anti-IgM alone; 3µM stimulatory CpG DNA alone; both 10µg/ml anti-IgM and 3µM CpG DNA or 10µg/ml anti-IgM-CpG DNA conjugate either in the absence or presence of nocodazole for 60 min. Cytoplasmic extracts were prepared and analyzed by SDS-PAGE and immunoblotting, probing with p-p38-specific antibodies. Blots were stripped and reprobed with tubulin-specific antibodies. The intensity of the p-p38 and tubulin bands were quantified using ImageJ software and expressed as a ratio of p-p38 to tubulin for each condition.
TLR9 moves from endosome to autophagosome-like compartments with the microtubular network
We next explored the cellular mechanism by which TLR9-containing endosomes were moved to the autophagosome-like compartments. A role of microtubular networks has been well established in the movement of secretory granules in cytotoxic T lymphocytes and peptide-loaded MHC class II-containing endosomal tubules in dendritic cells (Boes et al., 2002; Stinchcombe et al., 2006). It was also shown that Syk phosphorylates α-tubulin upon BCR crosslinking (Peters et al., 1996), however the function of phosphorylated α-tubulin is not known. We observed that treatment of B cells with the microtubule-disrupting agent, nocodazole, blocked the recruitment of TLR9 to autophagosome-like compartments upon BCR crosslinking (Fig. 7B), however BCR-mediated signaling and BCR internalization were not affected (Fig. S13). To determine if the BCR-mediated recruitment of TLR9 to autophagosome-like compartments is required for the observed modulated signaling, phosphorylation of p38 was determined in the presence of nocodazole. Nocodazole treatment blocked by 50 to 75 percent the increase in p38 phosphorylation triggered by treatment with anti-IgM plus CpG or the anti-IgM-CpG conjugate indicating that TLR9 recruitment was necessary for enhanced signaling (Fig. 7C). Nocodazole treatment, however, also reduced anti-IgM induced p38 phosphorylation while increasing somewhat the CpG-induced p38 phosphorylation (Fig. 7C). Taken together these results indicated that BCR-induced recruitment of TLR9 to the autophagosome-like compartments is important for enhanced signaling to p38 in response to DNA-containing antigens. However, disabling the microtubular network also influenced the quality of downstream signaling pathways emanating from both the BCR and TLR9.
Discussion
Multiple studies have suggested the importance of synergistic engagement of TLR9 and the BCR in hyper B cells responses in autoimmunity (Leadbetter et al., 2002; Viglianti et al., 2003). A growing number of studies indicate that TLRs play important roles in defining B cell responses to both T-dependent and T-independent antigens and that both DNA- and RNA-containing antigen complexes stimulate autoantibody production in a BCR and TLR9-or TLR7-dependent manner (Leadbetter et al., 2002; Viglianti et al., 2003). However, the mechanism by which these spatially segregated receptors interact in response to DNA- and RNA- containing antigens has not been previously addressed. Earlier reports suggested that the BCR might function as a shuttle for the delivery of DNA and RNA to the TLR9- and TLR7-containing compartments, however, no evidence was provided for such a mechanism (Leadbetter et al., 2002; Viglianti et al., 2003).
The results presented here provide evidence for a novel pathway by which B cells regulate responses to DNA-containing antigens, namely, by governing the subcellular location of TLR9. In our study we show that in resting B cells TLR9 is localized in punctate, small endosomal compartments and signals in these compartments to MAP kinase activation following stimulation by CpG alone. However, the location of TLR9 is dramatically altered in cells in which the BCR is crosslinked by antigen. Following BCR crosslinking the BCR initiates signaling at the plasma membrane and then traffics to LAMP-1-positive autophagosome-like compartments where the BCR signals to MAP kinase activation. TLR9 is recruited to these compartments where it colocalizes with the BCR. The recruitment of TLR9 to the autophagosome is required for enhanced signaling as the hyperactivation of MAP kinases to the anti-IgM and CpG DNA was not observed when TLR9 recruitment was blocked by the tubulin-depolymerizing agent, nocodazole.
A number of earlier studies indicated that TLR9 is located in late endosomes or lysosomes where it signals upon CpG DNA stimulation, consistent with the endosomal location of TLR9 in primary splenic B cells presented here (Ahmad-Nejad et al., 2002; Tabeta et al., 2006). However, a more detailed analysis provided evidence that TLR9 was concentrated in the endoplasmic reticulum (ER) of dendritic cells, macrophages and a B cell line and that upon stimulation with CpG DNA, TLR9 redistributed from the ER to CpG-DNA containing endosomes (Latz et al., 2004; Leifer et al., 2004). The mechanism underlying this redistribution of TLR9 is not known, however, a relationship between the ER and endosomes in TLR9 signaling was also suggested by the finding that a mutation in the gene encoding the 12-membrane spanning protein, UNC93B, a highly conserved resident ER protein, resulted in a complete deficiency in signaling via TLRs 3, 7 and 9 but did not alter their sub-plasmalemmal vesicular compartmentalization (Tabeta et al., 2006). The UNC93B mutation also resulted in deficiencies in antigen cross presentation, a process that involves both autophagy and endocytic trafficking. Moreover, recently it has been shown that TLR3, 7 and 9 physically interact with UNC93B, and this interaction is mediated through the transmembrane domain of at least TLR3 and 9 (Brinkmann et al., 2007). Taken together these findings suggest a link between the ER and endosomal TLR9 signaling that will be of interest to explore.
The results provided here indicate that the antigen bound BCR and TLR9 colocalize and signal from autophagosome-like compartments. Recently evidence was provided that autophagosomes play an important role in intracellular TLR signaling. In pDCs TLR7 gains access to single strand RNA viruses and viral replication intermediates through autophagy (Lee et al., 2007). In B cells autophagosomes have been less well characterized, however, recent results showed that a relationship exists between the class II-loading compartments and autophagosomes that results in the delivery of cytosolic antigens to class II-loading compartments (Schmid et al., 2007). It remains to be determined if similar mechanisms are at play for the relocalization of TLR9 to class II-loading compartments where BCR traffics DNA-containing antigens. It will also be of interest to determine if BCR-induced relocalization of TLR9 to autophagosome-like compartments is limited to TLR9 or is also observed with other intracellular TLRs. We provide evidence here that the recruitment of TLR9 was selective to the extend that TfR-positive endosomes were not recruited along with TLR9 positive endosomes. However, the regulation of the location of the TLR3 and 7 is of interest. Indeed, TLR3 that recognizes dsRNA is constitutively present in multivesicular bodies of a B cell line (Matsumoto et al., 2003).
The studies presented here support a model for B cell hyperresponses to DNA-containing antigen in which the antigen-engaged, internalized BCR signals to recruit TLR9 to autophagosome-like compartments to allow TLR9 to survey the antigen for its DNA ligand. If the ligand is present in the BCR-bound antigen, TLR9 is engaged and activated. How does this process result in autoantibody production? The enhanced signaling from autophagosomes through the BCR and TLR9 may contribute to autoantibody production by reducing the threshold for B cell activation. Alternatively, the subcellular location of TLR9 could influence its specificity for ODNs as suggested by the observation that a chimeric TLR9 receptor that localized to the cell surface rather than endosomes responded to both self and nonself DNA (Barton et al., 2006). In addition, in plasmacytoid dendritic cells the subcellular location to which CpG is delivered dictates the outcome of MyD88-IRF7 signaling (Honda et al., 2005). Additional studies indicated that crosslinking the BCR that we now know recruits TLR9 to autophagosomes resulted in the B cells’ ability to respond to a broader array of CpG-containing ODNs, including methylated DNA as occur in self DNA (Goeckeritz et al., 1999). Thus, it is possible that TLR9 recruitment to the autophagosomes by the BCR may have a significant effect on the ability of the BCR to discriminate self and nonself DNA-containing antigens.
Another important aspect of the recruitment of TLR9 vesicles to BCR-containing autophagosome-like compartments is the possible need to regulate recruitment. Presumably, the TLR9 is induced to locate to the class II loading compartments rather than being constitutively present in that compartment to allow regulation of the TLR amplification of BCR signaling. It will be of interest to determine if BCR coreceptors such as the inhibitory FcγRIIB that has emerged as a key factor in autoimmunity (Schmidt and Gessner, 2005), regulates TLR recruitment by the BCR. In addition, since multivesicular bodies in B cells are the subcellular sites in which the MHC class II molecules are assembled with peptides derived from the BCR-bound antigen, recruitment of TLR9 into the class-II loading compartments along with the antigen might influence the ability of the BCR to present DNA-containing antigens to helper T cells.
Lastly, an important aspect of the studies presented here is the evidence that the BCR continues to signal after its internalization to class II loading compartments and moreover, that the intracellular signaling is required for TLR9 recruitment. A number of cell surface receptors have been shown to continue to signal as they are internalized and that intracellular signaling was essential for the function of the receptor (Grimes et al., 1996; Vieira et al., 1996; Wang et al., 2002). To date, the intracellular signaling of the immune receptors has not been investigated in detail and a few studies suggested that signaling was extinguished once the receptor was internalized. Here we provide evidence that the BCR-initiated phosphorylation of p38 continues as the BCR is internalized into the cells, first into small endosomal vesicles then into class-II loading compartments. Thus, the spatial segregation of the BCR’s activation of p38 may play an important role in facilitating interaction with TLR9. It is also possible that the intracellular location of late BCR signaling may play additional roles in regulating B cell activation.
METHODS
Reagents, Cells and Mice
Single-stranded phosphorothioate CpG-DNA (5’-TCCATgACgTTCCTgACgTT-3’), control GpC-DNA (5’-TCCATgAgCTTCCTgAgCTT-3’) and 5’-amino modified oligos were purchased from Operon Biotechnologies, Huntsville, AL. Cy3-labeled CpG DNA was purchased from Tri-Link Biotechnologies, San Diego, CA. PP2, bafilomycin, propranolol and R59949 were purchased from Calbiochem, San Diego, CA. Monodansylcadaverine and chloroquine were purchased from Sigma-Aldrich, St. Louis, MO. F(ab')2 goat antibodies specific for mouse anti-IgM (anti-IgM), and HRP-conjugated goat antibodies specific for mouse IgG and rabbit IgG were purchased from Jackson Immuno Research Laboratories, West Grove, PA. Antibodies specific for p-JNK, were purchased from Cell Signaling Technology, Danvers, MA. Phospho-p38-specific antibodies were purchased from R&D systems Inc., Minneapolis, MN. I-κB- and LAMP1- specific antibodies were from Santa Cruz Biotechnologies Inc., Santa Cruz, CA. Alexa 488 labeled goat antibodies specific for mouse Ig and rabbit Ig, and Alexa 565-labeled antibodies specific for rat Ig and rabbit Ig and Zenon antibody-labeling kit were purchased from Invitrogen Corp., Carlsbad, CA. The TLR9-specific mAb, clone-26C593.2, was obtained from Imgenex Corp., San Diego, CA. The TLR9-specific mAb, clone-26C593.2, was conjugated to HRP using a Zenon antibody labeling kit. Another TLR9-specific mAb, clone 5G5 from Hycult Biotechnology was also used and similar results were obtained (data not shown). Breeding pairs for TLR9- and MyD88- deficient mice were provided by Dr. Shizuo Akira, Osaka University, Osaka, Japan. Mice were bred and maintained in our animal facility. C57/BL6 mice were purchased from Jackson laboratory, Bar Harbor, ME. HEL transgenic mice MD4 were purchased from Taconic farms Inc. B cells were purified from spleens of mice as previously described (Chaturvedi et al., 2002).
Synthesis of anti-IgM-CpG conjugate
Anti-IgM was incubated with a 20-fold molar excess of succinimidyl-6-hydrazinonicotinate acetone hydrazone, SANH, Solulink Biotechnologies, San Diego, CA, at 25°C for 3 h to modify the amino side chains of lysine residues by incorporating hydrazine groups. Residual SANH was removed by chromatography on a G-25 column (Amersham Pharmacia Biotech, Piscataway, NJ). 5’-amino modified CpG DNA or control DNA was incubated with succinimidyl-4-formylbenzoate, SFB, Solulink Biotechnologies, San Diego, CA, at 25°C for 3h to incorporate a benzaldehyde moiety. Residual SFB was removed by chromatography on a G-25 column. The resulting 5’-modified DNA was mixed with the modified anti-IgM at a 5:1 molar ratio and incubated overnight at 25°C. The anti-IgM-CpG conjugate was purified by size exclusion chromatography. The anti-IgM–CpG conjugate samples were determined to be lipopolysaccharide free by Limulus amebocyte lysate assay (BioWhittaker, Walkersville, MD). Immunoblotting was performed as previously described (Snyder and Pierce, 2006).
Confocal Microscopy
Purified splenic B cells attached to poly-lysine coated chamber slides (Bio-Coat), were treated with the indicated stimuli at 37°C for the indicated times, fixed with 3.7% paraformaldehyde for 20 min at 25°C, permeabilized with 0.2% TX-100 in PBS-BSA, blocked with 50 µg/ml of purified normal goat IgG for 20 min at 25°C and incubated with antibodies specific for phosphorylated p38 or TLR9 for 2 h at 25°C. The Alexa flour conjugated goat antibodies specific for rabbit Ig or goat antibodies for mouse Ig were added in the dark for 45 min at 25°C. The coverslips were mounted in Prolong Antifade (Invitrogen) and examined by fluorescence microscopy using Zeiss 510 Meta confocal microscope (Carl Zeiss). Approximately 250 cells were analyzed for each experimental condition. Cells were scored positive by an investigator who was blinded to the experimental protocol when the fluorescence appeared to be several fold above background.
To follow the uptake of anti-IgM and CpG, mouse B cells were suspended in RPMI plus 0.5% BSA and allowed to attach to poly-L-lysine-coated coverslip chambers (LAB-TEK, Nalge Nunc international, Naperville IL). Cells were incubated with 3uM Cy3-labeled CpG DNA and Cy5-labeled anti-IgM in RPMI plus 2% serum at 37°C for various times and visualized by time-lapse fluorescence microscopy using a Zeiss 510 Meta confocal microscope equipped with a heated stage, a heated air circulation system and an objective heater (Carl Zeiss). A Zeiss Plan-Apochromat 63X oil-immersion objective was used for image acquisition.
Electron Microscopy
Mouse splenic B cells were activated in suspension culture as indicated at 37 °C for 60 min, rinsed with PBS and fixed in periodate-lysine-paraformaldehyde fixative containing 0.35% glutaraldehyde for 1 h at 25°C. Cells were washed and permeabilized with PBS containing 0.02% saponin for 5 min at 25°C and incubated with HRP labeled TLR9 specific mAb (clone-26C593.2) in PBS containing 0.02% saponin for 2 h. Cells were washed in PBS, fixed in 0.1 M sodium phosphate containing 1.5% glutaraldehyde and 5% sucrose (pH 7.4) for 1 h at 25°C, washed with 50 mM Tris-HCl (pH 7.4) plus 7.5% sucrose and developed with metal enhanced DAB substrate (Pierce, Rockford, IL). Cells were rinsed with 50 mM Tris-HCl, (pH 7.4) plus 7.5% sucrose, and pre-fixed in 4% paraformaldehyde plus 2.5% glutaraldehyde in 100 mM sodium phosphate buffer (pH 7.4). Cells were post-fixed in 0.8% potassium ferrocyanide with the 1% osmium tetroxide and washed in phosphate buffer and water. Cells were dehydrated in a graded ethanol series, and embedded in Spurr's resin. Thin sections were cut with an MT-7000 ultramicrotome (RMC, Tucson, AZ). Samples were examined on a Hitachi H7500 TEM at 80 kV and images were captured with a CCD camera (Advanced Microscopy Technologies, Danvers, MA)
Supplementary Material
Acknowledgements
We thank S. Akira, Osaka University, Japan for TLR9-and MyD88-deficient mice and K.Takabayashi and E.Raz, UCSD, San Diego, USA for suggestions in anti-IgM-CpG conjugate synthesis. This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
Footnotes
Competing Financial Interests
Authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32:1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101:4500–4504. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. The Journal of cell biology. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KS. Signal transduction from the B cell antigen-receptor. Curr Opin Immunol. 1999;11:256–264. doi: 10.1016/s0952-7915(99)80042-9. [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Siddiqui Z, Bayiroglu F, Rao KV. A GPI-linked isoform of the IgD receptor regulates resting B cell activation. Nat Immunol. 2002;3:951–957. doi: 10.1038/ni839. [DOI] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Clark MR, Massenburg D, Zhang M, Siemasko K. Molecular mechanisms of B cell antigen receptor trafficking. Ann N Y Acad Sci. 2003;987:26–37. doi: 10.1111/j.1749-6632.2003.tb06030.x. [DOI] [PubMed] [Google Scholar]
- Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeritz BE, Flora M, Witherspoon K, Vos Q, Lees A, Dennis GJ, Pisetsky DS, Klinman DM, Snapper CM, Mond JJ. Multivalent cross-linking of membrane Ig sensitizes murine B cells to a broader spectrum of CpG-containing oligodeoxynucleotide motifs, including their methylated counterparts, for stimulation of proliferation and Ig secretion. Int Immunol. 1999;11:1693–1700. doi: 10.1093/intimm/11.10.1693. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- Jenkins GM, Frohman MA. Phospholipase D: a lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Kurosaki T. Genetic analysis of B cell antigen receptor signaling. Annu Rev Immunol. 1999;17:555–592. doi: 10.1146/annurev.immunol.17.1.555. [DOI] [PubMed] [Google Scholar]
- Lankar D, Vincent-Schneider H, Briken V, Yokozeki T, Raposo G, Bonnerot C. Dynamics of major histocompatibility complex class II compartments during B cell receptor-mediated cell activation. J Exp Med. 2002;195:461–472. doi: 10.1084/jem.20011543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol. 1990;8:773–793. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173:1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenert PS. Targeting Toll-like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res Ther. 2006;8:203. doi: 10.1186/ar1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- Peters JD, Furlong MT, Asai DJ, Harrison ML, Geahlen RL. Syk, activated by cross-linking the B-cell antigen receptor, localizes to the cytosol where it interacts with and phosphorylates alpha-tubulin on tyrosine. J Biol Chem. 1996;271:4755–4762. doi: 10.1074/jbc.271.9.4755. [DOI] [PubMed] [Google Scholar]
- Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor kappa B. Immunity. 2006;25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemasko K, Clark MR. The control and facilitation of MHC class II antigen processing by the BCR. Curr Opin Immunol. 2001;13:32–36. doi: 10.1016/s0952-7915(00)00178-3. [DOI] [PubMed] [Google Scholar]
- Snyder MD, Pierce SK. A mutation in Epstein-Barr virus LMP2A reveals a role for phospholipase D in B-Cell antigen receptor trafficking. Traffic. 2006;7:993–1006. doi: 10.1111/j.1600-0854.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- Stoddart A, Dykstra ML, Brown BK, Song W, Pierce SK, Brodsky FM. Lipid rafts unite signaling cascades with clathrin to regulate BCR internalization. Immunity. 2002;17:451–462. doi: 10.1016/s1074-7613(02)00416-8. [DOI] [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- Tao K, Fujii M, Tsukumo S, Maekawa Y, Kishihara K, Kimoto Y, Horiuchi T, Hisaeda H, Akira S, Kagami S, Yasutomo K. Genetic variations of Toll-like receptor 9 predispose to systemic lupus erythematosus in Japanese population. Ann Rheum Dis. 2007;66:905–909. doi: 10.1136/ard.2006.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pennock S, Chen X, Wang Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol. 2002;22:7279–7290. doi: 10.1128/MCB.22.20.7279-7290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi AK, Yoon JG, Krieg AM. Convergence of CpG DNA- and BCR-mediated signals at the c-Jun N-terminal kinase and NF-kappaB activation pathways: regulation by mitogen-activated protein kinases. Int Immunol. 2003;15:577–591. doi: 10.1093/intimm/dxg058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.