


Abstract
Bisulfite treatment can be used to ascertain the methylation states of individual cytosines in DNA. Ideally, bisulfite treatment deaminates unmethylated cytosines to uracils, and leaves 5-methylcytosines unchanged. Two types of bisulfite-conversion error occur: inappropriate conversion of 5-methylcytosine to thymine, and failure to convert unmethylated cytosine to uracil. Conventional bisulfite treatment requires hours of exposure to low-molarity, low-temperature bisulfite (‘LowMT’) and, sometimes, thermal denaturation. An alternate, high-molarity, high-temperature (‘HighMT’) protocol has been reported to accelerate conversion and to reduce inappropriate conversion. We used molecular encoding to obtain validated, individual-molecule data on failed- and inappropriate-conversion frequencies for LowMT and HighMT treatments of both single-stranded and hairpin-linked oligonucleotides. After accounting for bisulfite-independent error, we found that: (i) inappropriate-conversion events accrue predominantly on molecules exposed to bisulfite after they have attained complete or near-complete conversion; (ii) the HighMT treatment is preferable because it yields greater homogeneity among sites and among molecules in conversion rates, and thus yields more reliable data; (iii) different durations of bisulfite treatment will yield data appropriate to address different experimental questions; and (iv) conversion errors can be used to assess the validity of methylation data collected without the benefit of molecular encoding.
INTRODUCTION
Analysis of methylation states in genomic DNA has provided insights into biological phenomena as disparate as genomic imprinting (1,2), human disease (3,4) and atypical floral morphologies (5,6). In eukaryotic genomes, DNA methylation usually involves addition of a methyl group to the 5-carbon of cytosine, yielding 5-methylcytosine. Because both cytosine and 5-methylcytosine are complementary to guanine, conventional sequencing does not distinguish between them. Of the various methods typically used to assess DNA methylation states, only one—bisulfite treatment of genomic DNA, followed by PCR amplification, cloning and sequencing of individual PCR amplimers (7)—yields information on the methylation states of individual cytosines on individual DNA molecules. This detailed information is essential to address many questions in epigenetics (8–10).
Bisulfite treatment is expected to deaminate cytosine to uracil (Figure 1a), and to leave 5-methylcytosine unchanged (11,12) (Figure 1b). When bisulfite-treated DNA is amplified by PCR, 5-methylcytosine on the template strand pairs with guanine on the newly synthesized strand; converted cytosine, which is uracil, pairs with adenine. The methylation patterns of individual DNA molecules therefore can be inferred from the sequences of subcloned PCR products (12). A cytosine site is interpreted as having been unmethylated if it is occupied by thymine and as having been methylated if it is occupied by cytosine.
Figure 1.

Responses of methylated and unmethylated cytosines to bisulfite treatment. (a) Unmethylated cytosines treated with bisulfite are either converted to uracil, or fail to be converted and remain as cytosine. (b) the 5-methylcytosines treated with bisulfite either do not undergo conversion, or are inappropriately converted to thymine.
The most reliable analyses of data from bisulfite-treated DNA are those that account for both types of conversion error: failed conversion and inappropriate conversion. The better studied of these errors—failed conversion—is said to occur when an unmethylated cytosine fails to be deaminated, and thus appears in resulting data as if it had been methylated (Figure 1a). Because 5-methylcytosine in somatic cells of mammals occurs exclusively or almost exclusively at CpG cytosines (13), the failed-conversion frequency for bisulfite treatment of mammalian DNA is indicated by the fraction of non-CpG cytosines that appear as cytosines in sequence data. When not explicitly incorporated as a parameter in data analysis, failed conversion can inflate estimates of methylation densities, and can undermine efforts to determine the sequence motif preferences of DNA methyltransferases. The failed-conversion frequency can typically be reduced by increasing the duration of bisulfite treatment (14), by increasing the number of thermal denaturation steps used during conversion (9,15), or both.
A second type of error—inappropriate conversion—is said to occur when a methylated cytosine is deaminated, yielding thymine (Figure 1b) (14,16). Like uracils that result from deamination of cytosines, thymines that arise through inappropriate conversion of 5-methylcytosine will pair with adenine during PCR. As a result, 5-methylcytosines that undergo inappropriate conversion will be misinterpreted as unmethylated. When inappropriate conversion occurs and is ignored in data analysis, it will lead to underestimates of genomic methylation densities. In contrast, when inappropriate conversion occurs and its frequency is known, it can be included as a parameter in the data analysis. Information on failed- and inappropriate-conversion frequencies is therefore essential for inference from detailed DNA methylation patterns.
Two previous studies have explicitly investigated failed- and inappropriate-conversion frequencies under the conventional bisulfite-conversion protocol, which uses 5.5 M bisulfite and 55°C. We term these conditions ‘LowMT’ (low molarity/temperature). Grunau et al. (14) treated enzymatically methylated DNA under LowMT conditions, and reported inappropriate-conversion frequencies that may be as high as 6%. Shiraishi and Hayatsu (16) reported a comparable inappropriate-conversion frequency under similar conditions for conversion of DNA from a densely methylated tumor cell line. Neither analysis was able explicitly to exclude alternate explanations for these events. Bisulfite-independent phenomena, including incomplete methylation in cancer cell lines, incomplete enzymatic methylation of synthetic DNA, and errors during PCR amplification of bisulfite-converted DNA, could, potentially, mimic conversion errors and lead to overestimates of inappropriate-conversion frequencies.
Shiraishi and Hayatsu (16) also introduced an alternative to the LowMT bisulfite-conversion protocol. They reported that applying high-molarity (9 M), high-temperature (70°C) bisulfite treatment of much shorter duration—a protocol that we here term ‘HighMT’ (high molarity/temperature) (16,17)—decreases the time required for conversion, and may reduce inappropriate-conversion frequencies without appreciable increases in failed-conversion frequencies. Despite the potential advantages of the HighMT treatment over the conventional, LowMT protocol, its conversion dynamics remain largely unexplored. A few papers have cited the original work of Shiraishi and Hayatsu (16), but only one study (18) provides data from DNA converted under HighMT conditions. No published study has provided a comprehensive analysis of conversion errors under HighMT conditions.
We investigated conversion error with the goal of understanding how its dynamics can inform the design of bisulfite treatment protocols and the interpretation of resulting data. We treated methylated, synthetic oligonucleotides under the LowMT and HighMT treatments, and compiled failed- and inappropriate-conversion counts for individual molecules sampled after various durations of bisulfite treatment. Unlike genomic DNAs, whose apparent methylation patterns represent the combined consequences of biological variability and experimental error, oligonucleotides have methylation patterns that can be synthetically specified, and experimentally confirmed. Using synthetic oligonucleotides therefore allowed us to eliminate variation due to biological phenomena, and to conduct a more direct investigation of conversion error than would be feasible with genomic DNAs.
We obtained data from single-stranded oligonucleotides, as well as from hairpin-linked oligonucleotides that provide information on both the top and bottom strands of individual DNA molecules. We validated our data by using molecular batchstamps and barcodes (19, Burden et al., manuscript in preparation) to distinguish valid from contaminant and redundant sequences, and by measuring the frequencies of several processes that could mimic inappropriate conversion.
Here, we address five specific questions:
Is there evidence for inappropriate conversion under both the HighMT and LowMT treatments, even in analyses that control for bisulfite-independent error?
Do inappropriate-conversion events occur throughout the conversion process, or do they occur primarily on molecules that are already well-converted?
Is either the LowMT or the HighMT protocol generally preferable?
How can knowledge of frequencies for both inappropriate- and failed-conversion inform the design of treatment protocols?
Under what circumstances are conversion errors useful?
METHODS
Design and assembly of molecularly encoded oligonucleotides
Design and assembly of double-stranded oligonucleotides
To build a DNA substrate with which to measure conversion-error frequencies, we designed and ordered from GeneLink, Hawthorne, NY (http://www.genelink.com/) four single-stranded synthetic oligonucleotides. Two of these, methylated top strand (‘TM’) and methylated bottom strand (‘BM’), contained 5-methylcytosine at all 10 of their CpG sites.
The sequences of the oligonucleotides were based on a region of the human FMR1 promoter.
TM was 139 nucleotides long and had sequence:
5′-Phos CATGTCCACTTGAAGAGAGAGGGXGGGGCXGAGGGGCTGAGCCXGXGGGGGGAGGGAACAGXGTTGAT …
CAXGTGAXGTGGTTTCAGTGTTTACACCXGCAGXGGGCXGCCCAACAAATTCACGAACCGATGGGATATGT 3′.
BM was 125 nucleotides long and had sequence:
5′ ATCGGTTCGTGAATTTGTTGGGXGGCCXGCTGXGGGTGTAAACACTGAAACCAXGTCAXGTGATC …
AAXGCTGTTCCCTCCCCCXGXGGGCTCAGCCCCTXGGCCCXGCCCTCTCTCTTCAAGTGG 3′.
In the sequences above, ‘X’ indicates a methylated CpG cytosine.
The other two oligonucleotides, (‘TU’), unmethylated top strand, and (‘BU’), unmethylated bottom strand, were identical in sequence to TM and BM, except that they contained cytosines at the sites that were occupied by 5-methylcytosine in TM and BM.
To assemble double-stranded molecules from one version of the top strand oligonucleotide (TM or TU) and one version of the bottom strand oligonucleotide (BM or BU), 1.65 μl of 500 μM solutions of each strand were combined in 30 μl of Buffer EB (Qiagen, Valencia, CA), heated to 85°C for 3 min, and then cooled to room temperature. The annealed, double-stranded oligonucleotide was designed to basepair at 125 nt, with the top strand overhanging the bottom strand by 5 nt at the 5′-end, and by 9 nt at the 3′ end.
Molecular encoding with hairpin linkers
Hairpin linkers were covalently linked to the TM:BM oligonucleotide using the protocol previously described (19). Hairpin linkers contained a batchstamp that was unique to each experiment, thus enabling us to detect contamination, and a random barcode, thus enabling us to detect sequence redundancy (Figure 2).
Figure 2.

A hairpin-linked TM:BM oligonucleotide. The 5-methylcytosines are present at each of 10 CpG sites on both top, TM, and bottom, BM, strands of TM:BM, and are indicated by ‘Me’. Each hairpin linker contains a batchstamp common to all molecules processed in a given experiment, and a randomly generated barcode (19). Hairpin-linked molecules have primer binding sites on the top and bottom strands of the oligonucleotide, permitting subsequent PCR amplification.
A second configuration of hairpin oligonucleotides was used for an experiment to evaluate the frequency of failed-conversion with bisulfite solutions used soon after opening compared with those used 22 months after opening, and to evaluate the possible impact of complementary strands on the frequency of conversion error. For this experiment, TU was annealed to a complementary oligonucleotide that was ordered as a truncated version of BU. This shorter bottom strand contained the last 53 nt of the BU sequence depicted above. Annealing of truncated BU to TU thus yielded a complex that contained 53 complementary bases, with a 5-nt overhang at the 5′ end and an 81-nt overhang at the 3′-end. Hairpin ligation of this complex produced a molecularly encoded molecule that had appreciable numbers of unmethylated and methylated cytosines in both single- and double-stranded regions.
Molecular encoding with end-codes
Oligonucleotide molecules to be used in experiments with single-stranded DNA were batchstamped and barcoded through covalent attachment of an ‘end-code’ (Burden et al., manuscript in preparation; Figure 3). Each end-code oligonucleotide contained a defined batchstamp specific to that experiment, a randomly generated barcode, and a 5-nt overhang complementary to the 5′-overhang of the top strand of the double-stranded oligonucleotide. End-coders were combined with annealed TM:BU oligonucleotides at room temperature in a 1:16.7 molar ratio, and treated with T4 ligase, as per the manufacturer's; instructions (NEB, Ipswich, MA). After 1 hour, the ligase was heat-inactivated at 65°C for 20 min. The ligation step is expected to bind the end-coder to the top strand of the annealed oligonucleotide, using the 5′-phosphate of TM. End-coders differ from hairpin linkers in two ways: (i) they bear a reverse-primer binding site and (ii) they lack a 5′-phosphate, thus ensuring that they attach covalently only to the top strand of TM:BU. The end-coded top strand contains both primer binding sites, and thus can be used to detect top-strand conversion errors in the protocol we describe here.
Figure 3.

An end-coded TM oligonucleotide ligated while base-paired with BU. The 5-methylcytosines are present at 10 CpG sites on the top strand, TM, and are indicated with ‘Me’. The end-coder (Burden et al., manuscript in preparation) contains a batchstamp common to all molecules processed in a given experiment, and a randomly generated barcode. The end-coder is attached to the top, TM, but not the bottom, BU, strand of TM:BU. Thus, the bottom strand will separate from the top strand under denaturating conditions. Oligonucleotides labeled with end-coder have a forward primer binding site on TM, and a reverse-primer binding site of 21 nt, indicated here in purple, on the end-coder itself (after Figure 1 of Burden et al., manuscript in preparation).
Bisulfite-independent treatment of molecularly encoded oligonucleotides
We used oligonucleotide molecules encoded with end-codes to measure frequencies of errors that arise through bisulfite-independent phenomena. We examined sequences from molecules amplified either with no prior chemical treatment, or after exposure to 0.3 N sodium hydroxide (NaOH) for 50 min at 42°C. This NaOH treatment was designed to mimic the total NaOH exposure that molecules encounter as part of the bisulfite conversion protocol (20 min for pre-bisulfite denaturation and 30 min for post-bisulfite desulfonation).
Bisulfite treatment of molecularly encoded oligonucleotides
Treatment with HighMT Bisulfite
To prepare the HighMT bisulfite solution (16), 2.08 g of NaHSO3 (sodium bisulfite) and 0.67 g 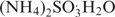 (ammonium sulfite monohydrate) were dissolved in 5 ml of 45% NH4HSO3 in water (ammonium bisulfite; Spectrum Chemical Manufacturing Corporation, Gardena, CA), to a final volume of 6.0 ml, and pH of 5.4 and held at 70°C for 10 min.
(ammonium sulfite monohydrate) were dissolved in 5 ml of 45% NH4HSO3 in water (ammonium bisulfite; Spectrum Chemical Manufacturing Corporation, Gardena, CA), to a final volume of 6.0 ml, and pH of 5.4 and held at 70°C for 10 min.
In preparation for HighMT treatment, molecularly encoded oligonucleotides (20 μl) were first denatured in 0.3 N NaOH at 42°C for 20 min, added to the HighMT bisulfite solution (180 μl), and held at 70°C for durations ranging from 5 to 200 min.
Ammonium bisulfite solutions change in color upon exposure to air. They have previously been reported to undergo spontaneous decomposition (20) during storage, and, in particular, during prolonged exposure to oxygen. The freshness of ammonium bisulfite may also impact conversion kinetics. In one experiment, the cytosine-conversion rate differed significantly—but by less than a factor of 2 (data not shown)—between DNA treated with HighMT solution prepared using a freshly opened bottle, and DNA treated with HighMT solution that contained ammonium bisulfite from a bottle that was first opened 22 months earlier but was of the same lot number. We recommend that ammonium bisulfite solutions be sampled under a hood, that bottles of ammonium bisulfite stocks be replaced often, and that existing stocks be protected from air.
Treatment with LowMT Bisulfite
For the LowMT treatment, molecularly encoded oligonucleotides were first denatured in 0.3 N NaOH at 42° for 20 min. To prepare the LowMT bisulfite solution, 4.05 g of sodium bisulfite were dissolved in 8 ml of deionized water (dH2O). In a separate tube, 50 mg hydroquinone were dissolved in 25 ml of dH2O. Next, 500 μl of the hydroquinone solution were combined with 230 μl of 10 N NaOH, and an additional 970 μl of dH2O. For LowMT treatment of single-stranded molecules, the 20 μl DNA–NaOH solution was combined with 180 μl of the sodium bisulfite solution, and held at 55°C for 4–20 h, with no thermal denaturation step. Conventional treatment of hairpin-linked DNA used the LowMT conditions, as was described previously (15) and included 10 thermal denaturation steps.
Post-bisulfite cleanup, PCR amplification and sequencing
Bisulfite-treated molecules were desulfonated and purified as described previously (15). Hairpin-linked molecules were amplified using initiating primer 5′ … TATCCCATCAATTCATAAATTT … 3′ and reverse primer 5′ … GGTTTGTGAATTTGTTGGG … 3′. Molecules encoded with end-codes were amplified using initiating primer 5′ … TATCCCATCAATTCATAAATTT … 3′ and a reverse primer corresponding to the end-coder used in each experiment (Figure 3). We used HotStarTaq Master Mix (Qiagen) for all amplification reactions. PCR products were then subcloned using the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA), and inserts from individual colonies were amplified using T7 and M13R primers. Colony PCR products were cycle-sequenced using either the T7 primer or the M13R primer, and Big-Dye Terminator v3.1 (ABI, Foster City, CA), then sequenced on an ABI3100, using a 36 cm POP6 array (Comparative Genomics Center, University of Washington, Seattle, WA). We used information from molecular batchstamps and barcodes on the end-coders to exclude contaminant and redundant sequences from each data set prior to analysis (19, Burden et al., manuscript in preparation).
Data analysis
Calculation of point estimates and 95% intervals on bisulfite-independent error frequencies and failed-conversion frequencies
For those experiments that yielded non-zero event counts in all categories, we calculated point estimates as the number of events divided by the number of opportunities, and calculated 95% confidence intervals under the binomial distribution.
For protocols that yielded event counts of 0 for one or more categories, we calculated point estimates on counts adjusted using the pseudocounts method originally proposed by LaPlace (21). Under the pseudocounts method, each observed count is treated as if it were greater by 1, and the denominator is calculated as the true sampled number, plus the total number of data groups. This approach makes it possible to avoid calculating a point estimate of 0% in cases where it is not plausible that the true frequency is 0. For instance, even if we observed 1000 successful conversions and zero failures, this sample size would be far too small to conclude that the true failed-conversion frequency was 0. A more plausible explanation would be that we had sampled too few cytosines to measure the small but non-zero frequency. For this example, LaPlace's; pseudocounts method would yield a point estimate of (0 + 1)/(1000 + 2), or 0.1%.
To establish 95% credible intervals on pseudocounts-adjusted observations, we took 10 000 random draws from a β-distribution, with α = number of failed-conversions and β = number of successful conversions, and then used the ‘quantile’ function in the freely available statistical analysis package, R, to find the region that contained 95% of these random draws. In Tables 1–5, the abbreviation ‘CI’ is used to indicate 95% confidence intervals for point estimates calculated directly, and to indicate 95% credible intervals for point estimates calculated using pseudocounts.
Table 1.
Error counts in sequences derived from untreated oligonucleotides
| Observed nucleotide count (%; 95%CI) | Expected nucleotide (n) |
||||
|---|---|---|---|---|---|
| A (1232) | T (1064) | C (1120) | G (2632) | 5 mC (560) | |
| A | 1230 (99.8%; 99.1, 99.9%) | 3 (0.28%; 0.06, 0.78%) | 0 (0.09%; 0.002, 0.32%) | 0 (0.04%; 0.008, 0.14%) | 0 (0.2%; 0.005, 0.63%) |
| T | 0 (0.08%; 0.002, 0.3%) | 1057 (99.3%; 98.7, 99.7%) | 10 (1%; 0.45, 1.7%) | 0 (0.03%; 0.001, 0.14%) | 0 (0.17%; 0.04, 0.64%) |
| C | 0 | 3 | 1108 | 2 | 560 |
| (0.08%; 0.002, 0.29%) | (0.28%; 0.055, 0.82%) | (98.7; 98, 99.4%) | (0.11%; 0.02, 0.27%) | (99.4%; 98.9, 99.9%) | |
| G | 2 | 1 | 2 | 2630 | 0 |
| (0.24%; 0.04, 0.58%) | (0.09%; 0.002, 0.52%) | (0.1%; 0.05, 0.66%) | (0.2%; 0.004, 0.65%) | (0.2%; 0.1, 0.7%) | |
Error counts were recorded from 56 single-stranded oligonucleotide molecules, excluding the 5′-overhang and primer-binding regions. For each molecule, we collected information from 22 adenines, 19 thymines, 20 unmethylated non-CpG cytosines, 47 guanines and 10 methylated CpG cytosines. For columns in which one or more values were 0, percentages were calculated using the ‘pseudocounts’ method originally introduced by LaPlace (21). Under this method, each value is treated as if it were greater by 1, and the denominator is calculated as the true value plus the total number of groups. Values shown in parentheses below event counts for each nucleotide indicate mean error rates and 95% CI on point estimates computed directly, or 95% credible intervals on point estimates computed using the pseudocounts method (see Methods section).
Table 2.
Error counts in sequences derived from NaOH-treated oligonucleotides
| Observed nucleotide count (%; 95%CI) | Expected nucleotide (n) |
||||
|---|---|---|---|---|---|
| A (1320) | T (1140) | C (1200) | G (2820) | Possible 5 mC (600) | |
| A | 1314 | 0 | 1 | 4 | 0 |
| (99.2%; 98.7–99.6%) | (0.08%; 0.002, 0.31%) | (0.08%; 0.02–0.46%) | (0.18%; 0.06–0.36%) | (0.16%; 0.004, 0.61%) | |
| T | 0 | 1137 | 11 | 0 | 1 |
| (0.07%; 0.018, 0.27) | (99.8%; 99.8–99.9%) | (0.92%; 0.5–1.7%) | (0.03%; 0, 0.13%) | (0.33%; 0.04, 0.92%) | |
| C | 3 | 2 | 1182 | 7 | 599 |
| (0.3%; 0.08–0.7%) | (0.26%; 0.05–0.65%) | (98.5%; 97.6–99.1%) | (0.28%; 0.12–0.5%) | 99.8%; 99.4–99.9% | |
| G | 3 | 1 | 6 | 2809 | 0 |
| (0.3%; 0.08–0.7%) | (0.2%; 0.02–0.48%) | (0.5%; 0.18–1.1%) | (99.6%; 99.3–99.8%) | (0.16%; 0.004, 0.61%) | |
Error counts were recorded from 60 single-stranded oligonucleotide molecules, excluding the 5′ overhang and primer-binding regions. For each molecule, we collected information from 22 adenines, 19 thymines, 20 unmethylated non-CpG cytosines, 47 guanines and 10 methylated, CpG cytosines. See Methods section and the legend of Table 1 for information on calculation of point estimates and their CIs.
Table 3.
Error frequencies for single-stranded molecules under HighMT treatment
| Protocol | Seq. | % Failed conversions; Counts (95% credible interval) | % Inappropriate conversions; Counts (95% credible interval) |
|---|---|---|---|
| HighMT 5 min | 29 | 63.3 | 0.17 |
| 349 (59.3, 67.3) | 1 (0, 1.2) | ||
| HighMT 15 min | 39 | 27.4 | 0.089 |
| 203 (24.2, 31) | 1 (0, 0.94) | ||
| HighMT 30 min | 29 | 7.4 | 1.2 |
| 41 (5.4, 9.8) | 4 (0, 3.1) | ||
| HighMT 80 min | 22 | 0.23 | 0.29 |
| 0 (0, 1.3) | 1 (0,1.6) | ||
| HighMT 200 min | 27 | 0.19 | 6.1 |
| 0 (0, 1) | 17 (3.7, 9.9) |
Results for each single-stranded molecule were tallied from 10 methylated, CpG cytosines and 19 unmethylated, non- CpG cytosines. The point estimate for each error frequency is given in the first line of each box; the second line shows the raw event count and the 95% credible interval on the frequency estimate (see Methods section), calculated using the β-distribution, with distribution parameter α as the number of conversion errors, distribution parameter β as the number of properly converted unmethylated cytosines in the case of the failed-conversion frequency, or the number of properly non-converted 5-methylcytosines in the case of the inappropriate-conversion frequency. Failed-conversion frequencies and their credible intervals were calculated directly, except for observations of zero, for which we used the pseudocounts correction. Inappropriate-conversion frequencies were calculated as the observed percent minus the bisulfite-independent error frequency.
Table 4.
Error frequencies for single-stranded molecules under LowMT bisulfite treatment
| Protocol | Seq. | % Failed conversions; Counts (95% credible interval) | % Inappropriate conversions; Counts (95% credible interval) |
|---|---|---|---|
| LowMT 4 h | 21 | 37.3 | 0.31 |
| 149 (32.7, 42.2) | 1 (0, 2.5) | ||
| LowMT 8 h | 52 | 36.5 | 1.2 |
| 361 (33.5, 39.6) | 7 (0, 2.7) | ||
| LowMT 20 h | 58 | 6.5 | 3.1 |
| 72 (5.1, 8.0) | 19 (1.7, 4.8) |
Results for each single-stranded molecule were tallied from 10 methylated, CpG cytosines and 19 unmethylated, non-CpG cytosines. The point estimate for each error frequency is given in the first line of each box; the second line shows the raw event count, and the 95% credible interval on the frequency estimate, calculated using the β-distribution. Failed-conversion frequencies and their credible intervals were calculated directly. Inappropriate-conversion frequencies were calculated as the observed percent minus the bisulfite-independent error frequency.
Table 5.
Error frequencies for hairpin-linked molecules under HighMT bisulfite treatment
| Protocol | Seq. | % Failed conversions; Counts (95% credible interval) | % Inappropriate conversions; Counts (95% credible interval) |
|---|---|---|---|
| HighMT 40 min | 41 | 8.4 | 1.4 |
| 187 (7.3, 9.6) | 13 (0.13, 2.7) | ||
| HighMT 60 min | 12 | 3.2 | 0.67 |
| 21 (2.0, 4.7) | 2 (0, 2.4) | ||
| HighMT 90 min | 41 | 0.86 | 1.7 |
| 19 (0.51, 1.3) | 15 (0.29, 3) |
Results for each double-stranded molecule were tallied from 20 methylated, CpG cytosines and 54 unmethylated, non-CpG cytosines. The point estimate for each error frequency is given in the first line of each box; the second line shows the event count, and the 95% CI on this frequency estimate. Failed-conversion frequencies and their CI were calculated directly. Inappropriate-conversion frequencies were calculated as the observed percent minus the bisulfite-independent error frequency, as described in Methods section.
Calculation of point estimates and 95% CI on inappropriate-conversion frequencies
Apparent inappropriate-conversion events can result both from bisulfite-independent phenomena, and from bona fide conversion errors. For each conversion protocol, we calculated the point-estimate of the inappropriate-conversion frequency as the apparent inappropriate-conversion frequency minus the bisulfite-independent error frequency.
We compiled data on the total error, and the bisulfite-independent error, and used R to calculate the 95% binomial confidence interval on each inappropriate-conversion frequency estimate.
Investigating the time course of conversion for cytosine and 5-methylcytosine
We used linear regression to investigate the time course of conversion for methylated and unmethylated cytosines.
To examine the time course of conversion for cytosine, we inferred the best-fit line for the log10-transformed failed-conversion counts observed for single-stranded oligonucleotides treated under the HighMT protocol, and for hairpin-linked oligonucleotides and plasmid DNA, as reported by Shiraishi and Hayatsu (16). The contribution of each mean to the regression was weighted by the total number of cytosines examined.
To examine the time course of inappropriate conversion for 5-methylcytosine, we first inferred the best-fit line for the number of 5-methylcytosines surviving in single-stranded DNA subjected to 0 min, 5 min, 15 min, 30 min, 80 min and 200 min of treatment with HighMT bisulfite. To investigate whether inappropriate-conversion events occurred over the entire range of treatment durations, we then excluded data from the 200-min sampling point from the analysis, and asked whether there was evidence of a significant inverse relationship between treatment duration and number of surviving 5-methylcytosines.
Investigating potential variation in inappropriate-conversion frequencies across treatment protocols and across cytosine sites
We used the Fisher's; Exact Test for several analyses, including our comparison of the frequencies of conversion error under different treatment protocols, and our investigation of potential site–site variation in these frequencies. For those data sets where sample sizes were large, we used simulated P-values to make computation tractable.
Fisher's; Exact Test takes whole-number arguments, so it was not feasible to use inappropriate-conversion measurements that had been adjusted to account for contributions from bisulfite-independent error. Because we have no evidence that bisulfite-independent error frequencies vary among sites, we chose to perform the Fisher's; Exact Test on unadjusted, raw counts of apparent inappropriate-conversion events.
RESULTS AND DISCUSSION
Validated sequence data confirm that apparent inappropriate-conversion events can arise under bisulfite treatment of both single-stranded and hairpin-linked DNA
We measured the frequency of apparent inappropriate conversion events on our methylated, synthetic oligonucleotide after various bisulfite-treatment protocols. Our results from LowMT and HighMT treatment of these molecules confirm prior reports that apparent inappropriate-conversion events can arise at appreciable frequencies during bisulfite treatment of single-stranded DNA (14,16), and indicate that such events can also arise on hairpin-linked molecules. Under various bisulfite conversion protocols described in subsequent sections, we estimated inappropriate-conversion frequencies to range from 0.09% to 6.1%.
Before proceeding to an analysis of these inappropriate-conversion frequencies, we asked whether some fraction of apparent-conversion events arose on our oligonucleotides through bisulfite-independent phenomena. Bisulfite-independent error was not considered explicitly in prior investigations of inappropriate-conversion frequencies (14,16).
Bisulfite-independent errors do occur, but at <1% of 5-methylcytosines
Errors introduced by impurities in 5-methylcytosine used for oligonucleotide synthesis
If cytosines or thymines were either systematically or randomly incorporated at 5-methylcytosine positions during oligonucleotide synthesis, they would mimic the products of inappropriate conversion, yielding overestimates of the inappropriate-conversion frequency.
A systematic error in the incorporation of 5-methylcytosine into our oligonucleotide would cause all or nearly all molecules examined to contain apparent inappropriate-conversion events at the same CpG. In the 146 single-stranded sequences examined after various durations of HighMT treatment, no single site was observed to have undergone apparent inappropriate conversion more than four times. We conclude that our oligonucleotide contains no systematic errors at 5-methylcytosine positions.
We asked whether random misincorporation of either cytosine or thymine occurred at some 5-methylcytosine positions. We learned from a high-performance liquid chromatogram (HPLC) supplied by the oligonucleotide manufacturer, GeneLink (www.genelink.com), that the purity of the phosphoramidite precursor of 5-methylcytosine used to construct our synthetic oligonucleotides was at least 99.8%. This purity level implies that mistaken incorporation of cytosine could have occurred at no >0.2% of 5-methylcytosine positions.
To further address the possibility that thymines were incorporated at random at some 5-methylcytosine positions during oligonucleotide synthesis, we amplified, subcloned and sequenced oligonucleotides with no prior chemical treatment. We found that, as expected, no thymine was present at any the 560 5-methylcytosine positions examined in these 56 molecules (Table 1). We conclude, using the binomial distribution, that misincorporation of thymine occurred at <0.54% of 5-methylcytosine sites.
Errors during sequencing
DNA sequencing is a standard component of protocols designed to collect DNA methylation patterns. To investigate how often errors were introduced during the sequencing and base-calling processes, we performed repeat sequencing on a subset of our clones. The T7 and M13R primers amplify molecules subcloned with the Topo-TA Cloning Kit (Qiagen); either primer can be used to cycle-sequence the resulting colony amplimers. Using PCR products from each of 71 subclones, we sequenced once with M13R and once with T7. We examined the concordance of sequences collected from single clones using these two different primers.
Sequences recovered using the T7 primer confirmed 24 of the 25 apparent inappropriate-conversion events, and 168 of the 173 apparent failed-conversion events that we observed in sequences recovered using the M13R primer. Thus, the vast majority (97%; C I= 94–98.9%) of conversion errors observed in sequences recovered using M13R were confirmed by resequencing with T7. We conclude that the vast majority of apparent conversion errors arose by a process other than sequencing error.
Apparent conversion errors produced by PCR
PCR amplification is used twice during standard bisulfite-sequencing protocols: the first PCR reaction amplifies genomic or oligonucleotide DNA immediately after bisulfite conversion; the second prepares individual subcloned PCR products for sequencing. Thus, all molecules examined contain information on the upper bound of the PCR error frequency. Our above-described finding that all 560 of the 5-methylcytosine sites examined in untreated DNA had base-paired with guanine during PCR establishes a similar upper bound on the PCR error frequency at those sites (Table 1). We conclude that PCR error could yield apparent inappropriate-conversion events at no >0.54% of 5-methylcytosines.
While examining sequences derived from untreated oligonucleotides, we noticed unexpectedly high error frequecies for non-CpG cytosine sites, which were expected to be occupied by unmethylated cytosines. C→T errors were observed at 0.89% (10 of 1120; 0.42–1.5%) and 0.96% (11 of 1140; 0.48–1.6%) of cytosine positions in sequences from untreated and NaOH-treated oligonucleotides, respectively. C→T error frequencies for pooled data from these two data sets (21 of 2320; 0.91%; CI = 0.56–1.3%) differ significantly from the C→T error frequency (21 of 8664; 0.24%; 0.15–0.35% observed for non-CpG cytosines in PCR products from the FMR1 locus in genomic DNA (P = 3.11 × 10−5, data not shown). We have no information that explains why the C→T error frequency is high for unmethylated cytosines in our oligonucleotides compared to those in genomic DNA. Whatever its cause, an error frequency of this magnitude will have a relatively minor impact on our estimates of the failed-conversion frequency.
Errors introduced by incomplete deprotection of 5-methylcytosine after olignucleotide synthesis
We investigated whether apparent inappropriate-conversion events could have arisen through the sodium hydroxide (NaOH) treatment that is used to denature DNA prior to bisulfite treatment and/or during the NaOH treatment used to desulfonate DNA after bisulfite treatment.
Our concern about the potential impact of NaOH on the apparent inappropriate-conversion frequency arose from details of oligonucleotide synthesis. During the synthesis of oligonucleotides, isobutyrl-methylcytosines, rather than 5-methylcytosines, are incorporated at 5-methylcytosine positions. Newly synthesized oligonucleotides are subjected to high-temperature ammonium hydroxide, with the goal of replacing the isobutyrl-amine group with an amino group, yielding 5-methylcytosine (22). Any isobutyrl-methylcytosines that failed to be deprotected in ammonium hydroxide would remain as isobutyrl-methylcytosines, rather than the intended 5-methylcytosines. Exposure of any remaining isobutyrl-methylcytosines to the NaOH before or after bisulfite treatment would likely replace the isobutyrl-amine group of isobutyrlmethylcytosine with oxygen, rather than the amino group expected under NH4OH treatment, yielding thymine rather than 5-methylcytosine, and mimicking inappropriate conversion.
To investigate the potential impact of failed deprotection on our error-rate estimates, we treated single-stranded oligonucleotides with 0.33 N NaOH for 30 min at 42°C, and sequenced subclones. Of the 600 5-methylcytosine positions examined, 599 appeared to contain cytosine, and one appeared to contain thymine (Table 2). Thus, we can conclude that errors arising during NaOH treatment of the oligonucleotide could yield apparent inappropriate-conversion events at not more than 0.78% of 5-methylcytosine positions.
Total contribution of bisulfite-independent phenomena to the frequency of apparent inappropriate-conversion events
The results described above indicate that no single bisulfite-independent phenomenon made a large contribution to the apparent inappropriate-conversion frequency. However, they do not exclude the possibility that small contributions from several different bisulfite-independent phenomena sum to make a substantial contribution. To address this possibility, we calculated an upper bound on the overall frequency of bisulfite-independent errors that would mimic inappropriate conversion.
The value of this upperbound is available from sequences collected from NaOH-treated oligonucleotides. Those molecules contain any errors that arose not only during NaOH-treatment, but also during oligonucleotide synthesis, and during subsequent PCR amplification. Our finding that the NaOH-treated molecules were observed to have 5-methylcytosine at 599 of the 600 expected positions thus excludes a combined synthesis-, deprotection- and PCR-error frequency of >0.78%.
We applied the above estimate of the bisulfite-independent error frequency in our analysis of data from each bisulfite treatment protocol, using the approach described in ‘Methods’ section. This approach allows us essentially to ‘correct for’ bisulfite-independent error, and thus to estimate the inappropriate-conversion frequency for each treatment duration.
Investigating bisulfite-conversion error by analysis of individual molecules
Published analyses by our group (15,24) and others for instance (23) have typically excluded individual molecules that have large numbers of conversion failures, with the goal of reducing the number of unmethylated cytosines misinterpreted to be methylated. Here, we consider the possibility that all molecules, including these typically excluded ones, can provide useful information relevant to the dynamics of bisulfite-conversion error. Therefore, we examined both population-mean and single-molecule error frequencies for single-stranded and hairpin-linked molecules treated under the LowMT and HighMT conditions.
Single-stranded DNA treated under the HighMT protocol
We measured conversion-error frequencies for end-coded, single-stranded molecules subjected to HighMT bisulfite for treatment durations ranging from 5 to 200 min. We anticipated, based on the work of Shiraishi and Hayatsu (16), that 200 min of HighMT bisulfite treatment would be in excess of the time required to achieve complete or near-complete conversion of unmethylated cytosines. We hypothesized that inappropriate-conversion events accrue during prolonged treatment of molecules that have already achieved complete or near-complete conversion, and examined individual molecules to test this possibility.
Our results confirm the finding of Shirashi and Hayatsu (16) that the conversion of unmethylated cytosines in single-stranded DNA occurs very rapidly under HighMT treatment. We found that 92.6% (100−7.4) conversion of unmethylated cytosines was achieved after only 30 min of treatment (Table 3). At 80 and 200 min, conversion of unmethylated cytosines was complete (980 of 980 total unmethylated cytosines; 100%, CI = 99.7–100%) (Figure 4d and e). This indicated that the final 120 min of exposure were unnecessary.
Figure 4.

Single-stranded oligonucleotides exposed to HighMT bisulfite. Distributions of failed-conversion counts for molecules treated in HighMT bisulfite for 5 min (a), 15 min (b), 30 min (c), 80 min (d) and 200 min (e). We use n to indicate the number of molecules examined; conversion failures are summarized by the mean number of failed-conversion events per molecule, and the SD on this mean. Sunflower plots of individual-molecule failed- and inappropriate-conversion counts for molecules treated with HighMT bisulfite for 5 min (f), 15 min (g), 30 min (h), 80 min (i) and 200 min (j). A single point indicates one molecule, a line indicates two and a z-petaled ‘flower’ indicates z-molecules.
We reasoned that if inappropriate-conversion events occur predominantly on molecules that have already achieved complete conversion, then the inappropriate-conversion frequency from the 200 min treatment should be disproportionately greater than the frequencies observed under shorter treatments. For treatments up to and including 80 min, inferred inappropriate-conversion frequencies ranged from 0.09% to 1.2% (Table 3), with no clear relationship between failed- and inappropriate-conversion counts for individual molecules (Figure 4f–i). The total number of apparent inappropriate-conversion events in samples taken at up to 80 min was small (7 of 1190; 0.42%, CI = 0–0.82%, Table 3). By 200 min, however, the inappropriate-conversion frequency had reached 6.1% (17 of 270; CI = 3.7–9.9%), and differed significantly from the frequency observed for the earlier samples (P = 2.2 × 10−8). We conclude that bona fide inappropriate-conversion events occur rarely—if at all—on our single-stranded oligonucleotide when treated with HighMT bisulfite for up to and including 80 min.
These data are consistent with our hypothesis that the inappropriate-conversion events detected in the 200 min sample occurred primarily during the last 120 minutes of bisulfite treatment on molecules that had already achieved complete conversion.
Single-stranded DNA treated under the LowMT protocol
We investigated conversion-error frequencies for single-stranded oligonucleotides subjected to LowMT conditions for 4 h, 8 h and 20 h. The mean failed-conversion frequency for the population was 37.3% after 4 h of treatment, and had declined to 6.5% of unmethylated cytosines by 20 h (Table 4). The 4-h and 8-h treatments both produced molecules with extremely broad distributions of failure counts, with SDs of 6.21 and 5.45, respectively (Figure 5). These were in sharp contrast to the small SDs (range: 1–2.5; Figure 4) observed for single-stranded molecules under the HighMT treatment. In both the 4-h and 8-h samples, a small number of molecules had already achieved complete conversion. Nevertheless, in both samples, nearly 60% of molecules remained unconverted at nearly half of their unmethylated cytosines (Figure 5a and b). By 20 h of treatment, the distribution of failure counts had shifted markedly (Figure 5c). Nearly 80% of molecules had achieved complete conversion, though a small subpopulation of molecules still had up to 10 unmethylated cytosines that had not yet been converted. Like single-stranded molecules sampled after short durations of HighMT treatment, single-stranded molecules sampled after short durations of LowMT treatment showed no clear relationship between the failed- and inappropriate-conversion counts on individual molecules (Figure 5d and e).
Figure 5.

Single-stranded oligonucleotides exposed to LowMT bisulfite. Distributions of failed-conversion counts for bisulfite treatments of 4 h (a), 8 h (b) and 20 h (c). Sunflower plots of individual molecule failed- and inappropriate-conversion counts for treatments of 4 h (d), 8 h (e) and 20 h (f). We use n to indicate the number of molecules examined; conversion failures are summarized by the mean number of failed-conversion events per molecule and the SD on this mean. A single point indicates one molecule, a line indicates two and a z-petaled ‘flower’ indicates z-molecules.
In the previous section describing single-stranded DNA treated under HighMT conditions, we identified 80 min as the duration by which most molecules have achieved complete conversion, and beyond which inappropriate-conversion events accrue. We therefore sought to identify a LowMT treatment duration that would achieve a similar combination of low frequencies for both failed and inappropriate conversion on single-stranded DNA.
For the 4-h treatment, the inferred inappropriate-conversion frequency was 0.31% (1 event out of 210 opportunities; CI = 0–2.6%), indicating that few inappropriate-conversion events occurred during the first 4-h of treatment. For the 8-h sample, the inferred inappropriate-conversion frequency was 1.2% (7 events out of 520 opportunities; CI = 0–2.7%), suggesting that inappropriate conversion had begun to occur. By 20 h, the inferred inappropriate-conversion frequency had climbed to 3.1% (19 events out of 580 opportunities; CI = 1.7–4.8%; Table 4). In this sample, molecules with no conversion failures had an inappropriate-conversion frequency of 3.6% (17 of 440; CI = 1–5.2%); molecules with one-or-more conversion failures had an inappropriate-conversion frequency of 1.2% (2 of 140; CI = 0.016–3.6%). This difference was small and did not lead to statistical significance (P = 0.27), but hinted that the zero-failure molecules had begun to experience inappropriate conversion.
To further explore this possibility, we pooled data from the three LowMT treatment durations. In view of our hypothesis that individual molecules, not populations, accrue inappropriate-conversion events after attaining high levels of conversion of their unmethylated cytosines, and in light of our finding that single-stranded molecules were broadly distributed in their times to complete conversion under the LowMT treatment, we reasoned that well-converted molecules from all three time samples might have accrued inappropriate-conversion events. Combining data from the 4-h, 8-h and 20-h treatments, we compared inappropriate-conversion event counts on zero-failure molecules to counts for one-or-more failure molecules. Inappropriate-conversion counts differed significantly between molecules with zero-failures (20 of 610 opportunities; 3.1%, CI = 1.8–4.6%) and those molecules with one-or-more failures (7 of 700; 0.83%, CI = 0.23–1.6%, P = 0.006; Figure 5).
We conclude that both LowMT and HighMT treatments, when prolonged beyond the time required for complete or nearly complete conversion, can lead to unnecessarily large numbers of inappropriate conversion events on single-stranded molecules. Moreover, under the LowMT treatment, single-stranded molecules that attain complete conversion early during bisulfite treatment can begin to accrue inappropriate-conversion events before conversion is complete for the majority of molecules. This phenomenon will lead to unnecessarily large inappropriate-conversion frequencies.
Hairpin-linked DNA treated under HighMT conditions
We hairpin-linked synthetic oligonucleotides, (19) and sampled molecules after 40 min, 60 min and 90 min of HighMT bisulfite exposure.
Like single-stranded molecules, populations of hairpin-linked molecules under HighMT conditions exhibited declining failed-converison counts with increasing treatment duration. Molecules were well-converted after 40 min of treatment (2027 out of 2214; 91.6%, CI = 90.3–92.6%), and almost completely converted after 90 min of treatment (99.14%; 2195 of 2214; CI = 98.9–99.5%, Table 5). The distributions of per-molecule failure counts for hairpin-linked molecules under HighMT treatment had SDs ranging from 0.78 to 2.6 (Figure 6a–c). These SDs are similar to those we observed for single-stranded DNA under HighMT treatment, but are substantially smaller than those observed for single-stranded DNA under LowMT treatment. This result suggested that the distribution of failure counts within a given sample might be determined primarily by the bisulfite treatment conditions—that is, HighMT versus LowMT—rather than by the strandedness of the DNA.
Figure 6.

Hairpin-linked oligonucleotides exposed to HighMT bisulfite. Distributions of failed-conversion counts for bisulfite treatments of 40 min (a), 60 min (b) and 90 min (c). We use n to indicate the number of molecules examined; conversion failures are summarized by the mean number of failed-conversion events per molecule and the SD on this mean. Sunflower plots of individual-molecule failed- and inappropriate-conversion counts for bisulfite treatments of 40 min (d), 60 min (e) and 90 min (f). A single point indicates one molecule, a line indicates two and a z-petaled ‘flower’ indicates z-molecules.
Together, the 40-, 60- and 90-min samples contained a total of 30 inappropriate-conversion events out of the 1871 readable 5-methylcytosines examined, yielding an inferred inappropriate-conversion frequency of 1.4% (CI = 0.91–2.1%; Table 5), indicating that inappropriate conversion can occur on hairpin-linked molecules treated under HighMT conditions.
Hairpin-linked DNA treated under the published hairpin-bisulfite PCR protocol
We asked whether inappropriate conversion can also occur during treatment of hairpin-linked DNA treated under our published hairpin-bisulfite PCR protocol [(15), modified in (19)], which uses LowMT conditions, and includes 10 thermal denaturation steps. We examined 71 encoded, hairpin-linked oligonucleotides subjected to this treatment.
All of the oligonucleotides treated using our published protocol had nearly complete conversion of their unmethylated cytosines (3829 of 3834; 99.87%; CI = 99.7–99.9%). The distributions of per-molecule failure counts were extremely narrow: only five molecules had any failed conversions, and no molecule had more than one (Figure 7). Of the 1368 5-methylcytosine sites that could be read unambiguously from eletropherograms for these molecules, 50 appeared as thymine, yielding an inferred inappropriate-conversion frequency of 3.5% (CI = 2.7–4.9%).
Figure 7.

Hairpin-linked DNA treated under the published hairpin-bisulfite PCR protocol. (a) Distribution of failed-conversion counts. We use n to indicate the number of molecules examined; conversion failures are summarized by the mean number of failed-conversion events per molecule and the SD on this mean. (b) Sunflower plot of individual-molecule failed- and inappropriate-conversion counts. A single point indicates one molecule, a line indicates two and a z-petaled ‘flower’ indicates z-molecules.
Most of the molecules collected under the published hairpin-bisulfite PCR protocol were completely converted (Figure 7). Thus, it seemed possible that many or all of these molecules had been treated beyond the point necessary to achieve complete or near-complete conversion, and that most or all of the inappropriate-conversion events occurred on molecules that had already achieved complete conversion. The small number of molecules with one-or-more failures prohibited a statistical examination of this possibility.
Indeed, the inappropriate-conversion frequency was higher for the molecules with no conversions failures (48 of 1268 5-methylcytosine positions examined), than for the molecules that had one conversion failure (2 of 100 examined), but this result did not reach statistical significance (P = 0.58).
Site-site variation in conversion-error frequencies
One advantage of investigating conversion errors on validated, individual molecules is the opportunity to measure site-specific error frequencies. Harrison et al. (25) noted that the conversion rate for a given cytosine can depend on its sequence context. Here, we examined our data from validated single-stranded oligonucleotides to investigate whether or not failed-conversion frequencies differed among the sites on our single-stranded molecules and whether this effect differed between the LowMT and HighMT treatments.
Site–site variation in failed-conversion frequencies
The LowMT treatment yielded significant heterogeneity in site-specific failed-conversion counts for the 4 h (P = 0.0045), 8 h (P = 0.00045) and 20 h (P = 0.007) time points (Figure 8a–c). These results confirm the finding of Harrison et al. (25) that individual sites can differ in their conversion rates when exposed to conditions similar to the LowMT conditions reported here.
Figure 8.

Single-stranded DNA treated under LowMT conditions. Cytosine site-specific failed-conversion counts for LowMT-treated, single-stranded DNA examined after 4 h (a), 8 h (b) and 20 h (c). Fraction of unmethylated cytosines surviving at two relatively rapidly converted sites (1 and 2) and two relatively slowly converted sites (7 and 9) after treatment for 4 to 20 h (d). The ‘N’ indicates the total number of molecules examined.
Two of the more rapidly converted sites, 1 and 2, had attained nearly complete conversion by 4 h of treatment (Figure 8d). In contrast, two of the more slowly converted sites, 7 and 9, remained poorly converted at 8 h. Even by 20 h, one of these—site 9—had yet to be converted in 10 of 58 (16%) of sequences (Figure 8c and d). Although it seems likely that prolonged treatment would reduce the frequency of failed-conversions at these more slowly converted sites, such prolonged treatment would likely also further increase the inappropriate-conversion frequency, which, by 20 h, had already reached 3.1% (Table 4). Multiple thermal-denaturation steps would likely reduce the site–site heterogeneity observed after LowMT treatment for 20 h. We have not, however, systematically explored this possibility.
The HighMT treatment, as compared to the LowMT treatment, yielded remarkably little variation among sites in the time to complete conversion (Figure 9). Although sites differed somewhat in their frequencies of conversion when examined early in the treatment, by 30 min, sampled molecules contained no evidence of site-site variation in failed-conversion counts (P = 0.15). The time course of conversion observed was similar for sites 1 and 2, which were converted fairly rapidly, and sites 7 and 9, which were comparatively slow in their conversion (Figure 9d).
Figure 9.

Single-stranded DNA treated under HighMT conditions. Cytosine site-specific failed-conversion counts for HighMT-treated, single-stranded DNA examined after 5 min (a), 15 min (b) and 30 min (c). Fraction of unmethylated cytosines surviving at two relatively rapidly converted sites (1 and 2), and two relatively slowly converted sites (7 and 9) after treatment for 5–200 min (d). We use n to indicate the total number of sequences examined.
We conclude that the HighMT treatment, compared to the LowMT treatment, yields substantially less site–site heterogeneity in the time to complete conversion of unmethylated cytosines.
Site–site variation in inappropriate-conversion frequencies
We summed site-specific inappropriate-conversion counts across all treatment durations and used the Fisher's; Exact Test to investigate whether the frequency of inappropriate-conversion differed across 5-methylcytosine sites. We found no evidence of significant site–site variation under either the HighMT treatment (P = 0.995; Figure 10a), or the LowMT treatment (P = 0.86; Figure 10b).
Figure 10.

Inappropriate-conversion counts at 5-methylcytosine sites in single-stranded DNA. End-coded single-stranded oligonucleotides were treated with bisulfite under HighMT (a), and LowMT (b) conditions. The inappropriate-conversion count for each site is the sum of events across all treatment durations for which data are reported in Tables 3 and 4, respectively.
Our power to detect differences among sites was much lower for inappropriate conversion than for failed conversion because these events were less common, and because each sequence contained fewer sites informative about inappropriate conversion (10 for inappropriate conversion versus 19 for failed conversion). Even so, site–site variation in inappropriate-conversion frequencies appeared to be small. Thus, under protocols that yield low inappropriate-conversion frequencies, the impact of site–site variation on data analysis is likely to be negligible.
Conversion dynamics and the design of bisulfite-treatment protocols
Choosing bisulfite-conversion conditions
Shiraishi and Hayatsu (16) recommended HighMT treatment as a way to accelerate the bisulfite-conversion process, and to reduce inappropriate-conversion frequencies, while still achieving good conversion of unmethylated cytosines. We have confirmed with validated data collected from synthetic oligonucleotides that the HighMT treatment can indeed yield samples with low mean frequencies of both failed and inappropriate conversion.
Our investigation of errors on individual molecules and at individual sites revealed an additional and unanticipated advantage of the HighMT compared to the LowMT treatment: the HighMT method produces markedly less variation among molecules and among sites in the time to complete conversion. This difference between the LowMT and HighMT treatments is highly relevant in light of our finding that inappropriate-conversion events accumulate predominantly on molecules that have already attained complete or near-complete conversion. Under the LowMT method, prolonged treatment is necessary to attain high levels of conversion for all sites and all molecules. Such prolonged treatment has the disadvantage of subjecting molecules in the well-converted subpopulation to increased numbers of inappropriate-conversion events.
Modulating bisulfite-conversion error frequencies to address a specific biological question
Our results from both single-stranded and hairpin-linked DNA suggest that HighMT treatment yields low—but non-zero—frequencies of conversion error. By examining the relationship between failed- and inappropriate-conversion frequencies, it should be possible to optimize treatment duration to suit a specific biological question.
For experiments where even a very small number of methylated cytosines would provide important biological information, it will be desirable to minimize the misclassification of unmethylated cytosines as methylated. For instance, it remains uncertain whether or not there is a low level of non-CpG methylation in mammalian somatic or embryonic stem cells (26) and whether or not CpG methylation occasionally occurs in the promoters of genes on the active X chromosome of human females. To collect data useful to address these issues, it is desirable to choose a protocol and treatment duration with a low failure frequency, and to tolerate the concomitantly high inappropriate-conversion frequency. Our analysis of failed- and inappropriate-conversion frequencies suggests that HighMT treatment at durations that yield >99.9% conversion of unmethylated cytosines will meet this goal (Figure 11).
Figure 11.

Inferred time courses of conversion of cytosine, and inappropriate conversion of 5-methylcytosine, in single-stranded DNA under HighMT bisulfite conditions. For each treatment duration examined, we indicate the fractions of surviving 5-methylcytosines (squares) and cytosines (diamonds), as given in Table 3. Error bars show 95% intervals for these point estimates (see Methods section). Vertical dashed lines indicate the treatment durations inferred as sufficient to achieve 99%, 99.5% and 99.9% conversion of cytosines, as extrapolated from the best-fit curve. To determine the relationship of the inappropriate-conversion frequency to treatment duration, we inferred the best fit line from all six time points, including the 0 time point. This line indicated a significant relationship between treatment duration and inappropriate-conversion frequency (P = 0.0076). As described in the text, this significant relationship was driven entirely by data from the 200 min sample. When we excluded this 200 min sample from the analysis (uppermost line, truncated at 80 min), there was no evidence of a significant relationship.
For experiments where even a very small number of unmethylated cytosines would provide important biological information, it will generally be desirable to minimize the number of inappropriate-conversion events. Some cell lines have been reported to have nearly complete DNA methylation at some loci (16). These findings are in contrast to data from human somatic cells, which do not indicate comparably high densities in methylated regions (15). To assess whether complete or near-complete methylation really does occurs in some biological cases, or whether it is instead an artifact of conversion error, it is necessary to choose conditions that yield low inappropriate-conversion frequencies. For this goal, too, HighMT conditions seem preferable. Specifically, our data indicate that for our single-stranded oligonucleotide treated under HighMT conditions, durations of ∼50–80 min would yield successful conversion frequencies between 98% and 99.9%, with few or no inappropriate-conversion events. From Shiraishi and Hayatsu's; (16) work with plasmid DNA, we predict that a HighMT treatment duration of 20 to 30 min would yield a comparable pair of failed- and inappropriate-conversion frequencies. Below, we discuss some parameters that may contribute to variation among DNAs of interest in the treatment duration required to achieve a given level of conversion.
HighMT treatment is surprisingly robust to differences in sequence concentration, complexity and secondary structure of the target DNA
Previous findings indicate that bisulfite conversion is most efficient for unpaired cytosines (11). Protocols for conversion of single-copy loci in genomic DNA typically achieve this unpairing with a NaOH denaturation step prior to bisulfite treatment. Under the LowMT protocol, this initial NaOH treatment seems insufficient to yield high levels of conversion for all molecules; at least one thermal-denaturation step is often included in pursuit of this goal. We are not aware of a detailed study of how the importance of thermal denaturation varies among target loci. It seems likely that DNA concentration and sequence complexity, GC content, and the presence or absence of a hairpin linker are key parameters. These parameters will influence the rate at which complementary strands renature after NaOH treatment, and, in particular, the extent to which cytosines remain unpaired upon introduction of bisulfite, and thus available for efficient conversion.
When genomic DNA is subjected to NaOH treatment, complementary strands are denatured, and diffuse away from one another. It is unlikely that complementary strands of single-copy loci in mammalian genomes will meet and renature during the subsequent bisulfite treatment. For repetitive elements, the renaturation rate is markedly higher (27) than for single-copy loci, so it is possible that they at least partially renature during bisulfite treatment, potentially slowing the conversion process (14). It seems likely that the one or more thermal denaturation steps often used with conventional bisulfite treatment [as applied in (9)], may promote conversion by successively denaturing DNA, until further renaturation is disfavored by the reductions in complementarity that accrue through conversion of unmethylated cytosines to uracils.
Because thermal-denaturation steps are absent from the HighMT protocol, we were especially surprised by two of our findings under this treatment. First, the rate of conversion under HighMT conditions differed by a factor of only 2 between our highly concentrated, single-stranded oligonucleotide and the less-concentrated and larger, more complex plasmid examined by Shiraishi and Hayatsu (16) (Figure 12). If renaturation were impeding the conversion process in proportion to the sequence complexity and the concentration of the target DNA, the initial conversion rates for these two DNAs would differ by several orders of magnitude, rather than by a factor of only 2. Second, the rate of conversion under HighMT conditions was greater by only a factor of 1.5 for our single-stranded olignucleotide, compared to our hairpin-linked oligonucleotide. This is surprising in that the hairpin-linked molecule is expected to ‘snap back’ to its double-stranded state immediately upon removal from denaturing conditions, thus impairing conversion until there is substantial loss of complementarity. These two results suggest that HighMT conditions lead to partial or complete DNA denaturation, and/or that bisulfite conversion under HighMT conditions can occur on paired cytosines. Our data do not allow us to distinguish between these two possibilities.
Figure 12.

Comparison of conversion rates for unmethylated cytosines in plasmid DNA, singlestranded oligonucleotides and hairpin-linked oligonucleotides. Failed-conversion rate data are compared here for end-coded, single-stranded oligonucleotides (Table 3), hairpin-linked oligonucleotides (Table 5), and for plasmid DNA [from Figure 2 of Shiraishi and Hayatsu (16)]. Data were log-transformed and best-fit lines estimated as described in Methods section. Solid lines indicate treatment durations bounded by recorded data; dashed lines indicate extrapolations from recorded data.
Initial results indicate that eukaryotic genomic DNAs, like the prokaryotic DNA described by Shiraishi and Hayatsu (16) and the synthetic DNA described here, undergo rapid conversion under HighMT conditions. Shiraishi and Hayatsu (16) applied HighMT treatment to human genomic DNA, and achieved >99% conversion by 40 min. We have confirmed this finding, and extended it to the FMR1 promoter region in hairpin-linked genomic DNAs that were encoded with batch-stamps and barcodes. We examined the conversion dynamics of DNA purified from a lymphoblastoid cell line established using cells from a male affected with fragile X syndrome (GM03200A; Coriell). Non-CpG cytosines in the examined region of the FMR1 locus in this genomic DNA had a time course of conversion under HighMT treatment that was similar to that reported above for hairpin-linked oligonucleotides (Figure 12), with 99.5% conversion achieved by 80 min of treatment.
Proteinase K digestion was a step in the isolation procedure for the genomic DNAs reported on here and in Shirashi and Hayatsu (16). Such proteinase digestion has been recommended by Warnecke et al. (28) as a means to reduce conversion-rate variation that may arise if proteins remain bound to purified DNA. We have not investigated, and Shiraishi and Hayatsu (16) did not report on, whether conversion rates under HighMT conditions differ among genomic DNAs isolated under different protocols. These are important variables to investigate in future work applying HighMT treatment to genomic DNAs. Regardless of the results of these investigations, it is clear that genomic DNA subjected to HighMT treatment can undergo rapid conversion in both single-stranded and hairpin-linked configurations.
The ability of HighMT conditions rapidly to convert genomic and synthetic DNAs that are both hairpin-linked and GC rich suggests that most DNAs may be amenable to rapid conversion under this protocol. The durations of HighMT treatment required for conversion of a broad range of target loci at various DNA concentrations likely span a narrow range—possibly <2 hours (Figure 12).
Conversion error as information
Methylation patterns, particularly those on densely methylated molecules, often vary substantially within populations of cells. PCR amplification sometimes fails to capture this epigenetic diversity, yielding data sets composed mostly or exclusively of multiple sequences that are derived from a single template molecule. Using molecular-encoding techniques, we have identified the two phenomena that are principally responsible for the lack of pattern diversity in these data sets. One of these, template redundancy, occurs when the products of a given PCR amplification are dominated by the amplimers of a single genomic template from the intended experiment. The other, PCR contamination, occurs when an amplification reaction is contaminated, and sometimes even dominated, by copies of one or a few amplimers from a previous reaction (19). We have applied molecular-encoding techniques to single-stranded DNA, as described here (Methods section), and more fully elsewhere (Burden et al., manuscript in preparation).
For methylation data gathered in the absence of molecular encoding, it has typically been difficult to distinguish valid data sets from those compromised by PCR redundancy and contamination. Conversion errors can sometimes help to fill this void insofar as they independently mark individual genomic template molecules, and thus can alert experimenters to likely contamination and redundancy. This insight was recently applied to methylation patterns collected from the leptin promoter in sperm DNA from mouse (24).
The expected methylation features of the DNA of interest determine whether failed- or inappropriate-conversion events will be informative about possible contamination and redundancy. In experiments with mammalian somatic DNA, failed-conversion events will be of greater utility. In these DNAs, methylation is expected to occur mostly or exclusively at CpG cytosines. Thus, any non-CpG cytosines that appear as cytosines in resulting sequence data almost certainly result from failed-conversion, rather than from bona fide methylation. When failed-conversion events occur, they happen prior to PCR amplification on individual genomic template molecules. Consequently, when the same pattern of failed-conversion events appears on two or more molecules, PCR redundancy—rather than biological identity—is most likely the cause.
We illustrate this point with six sequences collected through PCR amplification of end-coded oligonucleotides. Like mammalian DNA, these oligonucleotides have methyl groups at cytosines within CpG dinucleotides, but not on other cytosines. As we describe in previous sections, these molecules were first encoded with a molecular batchstamp, and a set of random barcodes, and then subjected to bisulfite conversion (Figure 13). The three sequences in Figure 13a all contain the same, intended batch-stamp, indicating that they are not contaminants. However, these three sequences bear an identical barcode, indicating that they are almost certainly copies of a single template molecule. Their identical patterns of failed-conversion events are consistent with the information from their molecular codes, and indicate that these sequences are clones of one another, and should be considered as one molecule—not three—in the analysis of these data.
Figure 13.

Conversion errors can distinguish between clonal and unique sequences. (a) Three clonal sequences from end-coded, single-stranded oligonucleotides (Burden et al., manuscript in preparation). All three sequences bear the expected GTATGTGT batchstamp, indicating that they are from the intended experiment. However, they have the same barcode, TTAGATA, indicating that they are clones. Their shared pattern of failed-conversion events confirms their common origin from a single template molecule. (b) Three unique sequences from end-coded, single-stranded oligonucleotides. Each sequence bears the GTATGTGT batchstamp (bold-faced) used in this experiment, and a unique barcode. These sequences also differ in their patterns of failed-conversion events (dark gray boxes), confirming that they proceeded through the conversion and amplification processes independently.
In contrast, the three sequences in Figure 13b are unique, valid sequences. Each of them bears the expected batch-stamp, indicating that each is from the intended experiment. Each sequence also has a barcode different from the barcode on the other two sequences, indicating that each arose from a different template molecule. These sequences also differ in their patterns of unconverted non-CpG cytosines, suggesting that each is the product of a template molecule that proceeded independently through the bisulfite-conversion process. Thus information available from the conversion errors on these molecules confirms the information available from their molecular codes.
Conversion error as a surrogate for molecular encoding is, of course, useful only when errors occur at detectable frequencies. Whether or not this condition is met during a given bisulfite conversion depends on the error frequencies themselves, and on the number of opportunities for error. In some circumstances, it may be desirable to choose conversion conditions that give rise to error frequencies that are sufficient to authenticate resulting sequence data.
CONCLUDING REMARKS
Shiraishi and Hayatsu (16) introduced the HighMT bisulfite-conversion protocol to reduce the time required to collect reliable data on DNA methylation. We evaluated conversion rates for LowMT and HighMT treatment of synthetic oligonucleotides, whose defined methylation patterns enabled us to examine conversion errors without confounding by the biological variation that is typical of genomic DNA (24). Our examination of these bisulfite-treated molecules, validated with molecular encoding, revealed an additional and unexpected benefit of this new protocol: a marked reduction in heterogeneity among sites and among molecules in the time to complete conversion of unmethylated cytosines.
Our findings indicate that:
Inappropriate-conversion events can occur under both HighMT and LowMT treatments;
5-methylcytosines are largely refractory to inappropriate conversion early in bisulfite treatment, and become vulnerable principally when molecules are exposed to bisulfite beyond the time when they achieve complete or near-complete conversion of their unmethylated cytosines. This finding highlights the importance of modulating bisulfite-conversion protocols so that treatment duration does not exceed the time to complete or near-complete conversion for the majority of molecules;
The HighMT protocol is preferable to the LowMT protocol because it decreases the treatment duration required to achieve a given level of conversion and because it yields greater homogeneity across both molecules and sites in the time course of conversion. This greater homogeneity reduces the risk that subpopulations of molecules, and individual sites, will attain complete conversion early during the treatment, and then accrue inappropriate-conversion events before conversion is complete for other molecules and sites. One potential explanation for this greater homogeneity is that the HighMT treatment fosters partial or complete denaturation of DNA, thereby reducing differences among regions in the time spent in the single-stranded state that is susceptible to conversion;
For some analyses of bisulfite-treated DNA, the value of complete conversion of unmethylated cytosines is great, and may warrant attendant increases in the frequency of inappropriate conversion. For other applications, low frequencies of inappropriate conversion will be essential to reliable data. Shiraishi and Hayatsu's; (16) HighMT protocol provides a valuable opportunity for fine-scale modulation of error frequencies to achieve the balance of failed and inappropriate-conversion events that is best-suited to address a specific biological question; and
Even the HighMT protocol will rarely yield data sets that contain no conversion errors at all. Conversion errors introduce apparent diversity among DNA methylation patterns, so recovery of large numbers of sequences with identical methylation patterns strongly suggests the occurrence of PCR contamination and/or redundancy. Conversion errors thus can be used to assess the validity of data sets collected without the benefit of molecular encoding.
We recommend that the HighMT protocol be used to improve both the efficiency and the reliability of bisulfite treatments used to collect epigenetic data.
FUNDING
National Institutes of Health (HD002274, GM077464 to C.D.L.); T32 HG00035 Training Grant (to the University of Washington). Funding for open access charge: NIH (HD002274 and GM077464).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Audrey Fu, Stanley Gartler, Bruce Godfrey, Scott Hansen and Matthew Stephens for their many helpful suggestions; the developers of the bisulfite-treatment protocols evaluated here, especially Marianne Frommer, Susan Clark and Hikoya Hayatsu, for their encouragement and input over many years; and Ali Javed of GeneLink for providing detailed information on oligonucleotide synthesis and the purity of isobutyrl-methylcytosine stocks.
REFERENCES
- 1.Stöger R, Kubicka P, Liu CG, Kafri T, Razin A, Cedar H, Barlow DP. Maternal-specific methylation of the imprinted mouse Igf2r locus identies the expressed locus as carrying the imprinting signal. Cell. 1993;73:61–71. doi: 10.1016/0092-8674(93)90160-r. [DOI] [PubMed] [Google Scholar]
- 2.Swain JL, Stewart TA, Leder P. Parental legacy determines methylation and expression of an autosomal transgene: a molecular mechanism for parental imprinting. Cell. 1987;50:719–727. doi: 10.1016/0092-8674(87)90330-8. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Vogelstein B. Alterations in DNA methylation in human colon neoplasia. Semin. Surg. Oncol. 1987;3:149–151. doi: 10.1002/ssu.2980030304. [DOI] [PubMed] [Google Scholar]
- 4.Laird CD, Jaffe E, Karpen G, Lamb M, Nelson R. Fragile sites in human chromosomes as regions of late-replicating DNA. Trends Genet. 1987;3:274–281. [Google Scholar]
- 5.Jacobsen SE, Meyerowitz EM. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science. 1997;277:1100–1103. doi: 10.1126/science.277.5329.1100. [DOI] [PubMed] [Google Scholar]
- 6.Cubas P, Vincent C, Coen E. An epigenetic mutation responsible for natural variation in oral symmetry. Nature. 1999;401:157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- 7.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat. Genet. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- 9.Stöger R, Kajimura TM, Brown WT, Laird CD. Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum. Mol. Genet. 1997;6:1791–1801. doi: 10.1093/hmg/6.11.1791. [DOI] [PubMed] [Google Scholar]
- 10.Genereux DP, Miner BE, Bergstrom CT, Laird CD. A population-epigenetic model to infer site-specic methylation rates from double-stranded DNA methylation patterns. Proc. Natl Acad. Sci. USA. 2005;102:5802–5807. doi: 10.1073/pnas.0502036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 14.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:e65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laird CD, Pleasant ND, Clark AD, Sneeden JL, Hassan KMA, Manley NC, VaryJr JC, Morgan T, Hansen RS, Stöger R. Hairpin-bisulte PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc. Natl Acad. Sci. USA. 2004;101:204–209. doi: 10.1073/pnas.2536758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiraishi M, Hayatsu H. High-speed conversion of cytosine to uracil in bisulfite genomic sequencing analysis of DNA methylation. DNA Res. 2004;11:409–415. doi: 10.1093/dnares/11.6.409. [DOI] [PubMed] [Google Scholar]
- 17.Hayatsu H, Tsuji K, Negishi K. Does urea promote the bisulfite-mediated deamination of cytosine in DNA? Investigation aiming at speeding-up the procedure for DNA methylation analysis. Nucleic Acids Symp. Ser. 2006;50:69–70. doi: 10.1093/nass/nrl034. [DOI] [PubMed] [Google Scholar]
- 18.Egger G, Jeong S, Escobar SG, Cortez CC, Li TH, Saito Y, Yoo CB, Jones PA, Liang G. Identication of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl Acad. Sci. USA. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miner BE, Stöger RJ, Burden AF, Laird CD, Hansen RS. Molecular barcodes detect redundancy and contamination in hairpin-bisulte PCR. Nucleic Acids Res. 2004;32:e135. doi: 10.1093/nar/gnh132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chertkov BA, Pankratova VN, Dobromyslova NS. Spontaneous decomposition of concentrated ammonium sulfite-bisulfite solutions. Sov. Chem. Ind. 1973;49:383–387. [Google Scholar]
- 21.LaPlace PS. A Philosophical Essay on Probabilities. (1951 edn) Dover, New York: 1814. (trans. by Truscott,F.W. and Emory,F.L., 1951) [Google Scholar]
- 22.Schulhof JC, Molko D, Teoule R. The final deprotection step in oligonucleotide synthesis is reduced to a mild and rapid ammonia treatment by using labile base-protecting groups. Nucleic Acids Res. 1987;15:397–416. doi: 10.1093/nar/15.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishiyama R, Qi L, Lacey M, Ehrlich M. Both hypomethylation and hypermethylation in a 0.2-kb region of a DNA repeat in cancer. Mol. Cancer Res. 2005;3:617–626. doi: 10.1158/1541-7786.MCR-05-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stöger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics. 2006;1:155–162. doi: 10.4161/epi.1.4.3400. [DOI] [PubMed] [Google Scholar]
- 25.Harrison J, Stirzaker C, Clark SJ. Cytosines adjacent to methylated CpG sites can be partially resistant to conversion in genomic bisulfite sequencing leading to methylation artifacts. Ann. Biochem. 1998;264:129–132. doi: 10.1006/abio.1998.2833. [DOI] [PubMed] [Google Scholar]
- 26.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl Acad. Sci. USA. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Britten RJ, Kohne DE. Repeated sequences in DNA. Science. 1968;161:529–540. doi: 10.1126/science.161.3841.529. [DOI] [PubMed] [Google Scholar]
- 28.Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identication and resolution of artifacts in bisulte sequencing. Methods. 2002;27:101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]