

Abstract
The β-subunits of voltage-gated potassium (Kv) channels are members of aldo-keto reductase (AKR) superfamily. These proteins regulate inactivation and membrane localization of Kv1 and Kv4 channels. The Kvβ proteins bind to pyridine nucleotides with high affinity; however, their catalytic properties remain unclear. Here we report that recombinant rat Kvβ2 catalyzes the reduction of a wide range of aldehydes and ketones. The rate of catalysis was slower (0.06 to 0.2 min−1) than that of other AKRs, but displayed the expected hyperbolic dependence on substrate concentration, with no evidence of allosteric cooperativity. Catalysis was prevented by site-directed substitution of Tyr-90 with phenylalanine, indicating that the acid-base catalytic residue, identified in other AKRs, has a conserved function in Kvβ2. The protein catalyzed the reduction of a broad range of carbonyls including aromatic carbonyls, electrophilic aldehydes and prostaglandins, phospholipid and sugar aldehydes. Little or no activity was detected with carbonyl steroids. Initial velocity profiles were consistent with an ordered bi-bi rapid-equilibrium mechanism in which NADPH binding precedes carbonyl binding. Significant primary kinetic isotope effects (2.0 – 3.1) were observed under single and multiple turnover conditions, indicating that the bond-breaking chemical step is rate-limiting. Structure-activity relationships with a series of para-substituted benzaldehydes indicated that the electronic interactions predominate during substrate binding and that no significant charge develops during the transition state. These data strengthen the view that Kvβ proteins are catalytically-active AKRs that impart redox-sensitivity to Kv channels.
Most eukaryotic cells express a diverse range of voltage-gated potassium (Kv) channels (1). In excitable cells, such as nerves and muscles, Kv channels regulate the generation and the duration of action potential, firing patterns, pacemaking and neurotransmitter release (2-4). In non-excitable cells, these channels participate in volume regulation, hormonal secretion as well as proliferation and apoptosis (5-8). Native mammalian Kv channels are large macromolecular complexes composed of 4 pore-forming transmembrane proteins (Kv α subunits) that regulate K+ efflux. The cytosolic domains of the Kv 1 and 4 α subunits associate with auxiliary β-subunits that regulate channel assembly and function. In mammals, 3 distinct Kvβ genes have been identified. These genes encode proteins with a highly conserved C-terminus and a variable N-terminal domain. The conserved C-terminus of Kvβ proteins folds into an (α/β)8 or triosephosphate isomerase (TIM) barrel motif, which is the most common structural scaffolding among enzymes involved in metabolism and biosynthesis (9). The TIM barrel also forms an efficient fold for high affinity binding of flavin and pyridine nucleotides that participate in catalyzing oxidation-reduction reactions (9). The TIM barrel structure of Kvβ proteins bears a strong structural resemblance and sequence similarity to proteins of the aldo-keto reductase (AKR) superfamily and the catalytic features of the AKR active site are conserved in the C-terminal domain of Kvβ proteins (10-12). On the basis of this homology, the Kvβ proteins have been classified as a distinct family (AKR6) within the AKR superfamily of proteins (13).
Our previous studies show that like other AKRs, the Kvβ proteins bind to pyridine nucleotides with high affinity (14;15). We have found that binding of reduced nucleotides supports N-terminus mediated inactivation of Kvα currents by Kvβ, whereas oxidized nucleotides remove inactivation of K+ currents generated by Kv α-β subunits (16). These observations support the notion that the β-subunits impart metabolic sensitivity to Kv currents. Such coupling might allow the Kv channel to sense changes in cell metabolism and oxygenation (17). Nevertheless, it remains unclear whether binding of pyridine nucleotide to Kvβ proteins by itself is required for the regulation of Kv activity or if catalysis is necessary to change the redox state of the nucleotide bound to Kvβ. Clearly, further studies on the catalytic activity and substrate-specificity of Kvβ are required to understand how it regulates Kv function.
Several lines of evidence suggest that Kvβ proteins are catalytically active and display AKR properties. The AKR active site is conserved in Kvβ proteins and the catalytic residues are positioned for efficient carbonyl binding. Moreover, substitution of the active site residues of the protein markedly affects the inactivating properties of Kvβ (15;18;19). Indeed, weak catalytic activity of Kvβ2 with aromatic aldehydes has been reported before (18). Nevertheless, the catalytic and kinetic mechanisms of the protein remains poorly understood and its substrate specificity is unknown. Specifically, it is not clear whether the protein displays cooperativity; which steps limit the overall catalytic rate; and which structural features of the substrate facilitate binding to the active site. The current study was therefore designed to identify the reaction sequence, determine the kinetic mechanism of Kvβ catalysis and to examine the substrate-specificity of the protein. Our results show that the Kvβ follows a rapid equilibrium bi-bi ordered reaction mechanism. These properties may be important aspects of Kvβ function in the regulation of Kv channel.
Experimental Procedures
Mutagenesis and expression of Kvβ2
The cDNA encoding rat Kvβ2 (rKvβ2) was provided by Dr. Min Li (Johns Hopkins University). The construct for bacterial expression of rKvβ2 [NP_059000] with His-tag at its N-terminus in pET28a vector was generated as described previously (14). Site-directed mutagenesis for Y90F substitution was performed directly in pET28a vector using QuikChange XL site-directed mutagenesis kit (Stratagene). The sequence of the PCR primers was: forward 5′-CGATACGGCGGAGGTCTTCGCAGCTGGCAAGGCTG-3′, and reverse 5′-CAGCCTTGCCAGCTGCGAAGACCTCCGCCGTATCG-3′. The complete sequence of the mutated Kvβ2 insert was confirmed by DNA sequencing. Wild-type and mutant Kvβ2 were expressed in the BL-21(DE3) strain of E.coli and purified to homogeneity as described before.
Protein purification
The AKR C-terminal domain of Kvβ2 protein (amino acids 39-367) was expressed with a His tag at its N-terminus in the BL-21 strain of E.coli. The bacterial pellet was lyzed in ice-cold binding buffer containing (in mM): NaCl, 150; Tris, 20; imidazole, 5; pH 7.9. The supernatant was incubated with Ni-affinity agarose beads (Qiagen) for 2-3 h at 4°C. The Ni-column was washed with binding buffer to 10 times the bed volume and the protein was eluted by imidazole in binding buffer. A small volume from each eluted fraction (2 μl) was separated by SDS-PAGE. The purified proteins were stored at 4°C in 0.2 M potassium phosphate buffer, pH 7.4 and used within 4 - 6 weeks. The concentration of the protein was measured using Bradford's assay (20)
NADPH binding studies
Binding of NADPH to WT or Y90F:Kvβ2 was measured by fluorescence titration as described earlier (14). Briefly, the Kvβ2 protein (15 μg) was equilibrated in 2 ml of 0.2 M potassium phosphate buffer at room temperature for 10 min and changes in fluorescence were measured using an excitation wavelength of 280 nm and emission at 340 nm. Aliquots of NADPH were added and changes in emission were recorded. Fluorescence changes obtained by titrating tryptophan with NADPH were used to correct for inner-filter effect (14). Dissociation constants were calculated from the following equation:
| (1) |
Activity measurements in multiple and single turnover mode
Activity assays were performed in multiple or single turnover mode (18;21). Multiple turnover were measured in quartz microcuvettes with final reaction volume of 250 μl containing Kvβ2 (15-20 μM) and NADPH (150 μM) incubated in 0.2 M phosphate, pH 7.4 at 37°C. The reaction was started by adding the substrate. The loss of NADPH was monitored by following the decrease in absorbance at 340 nm for 15 min using a spectrophotometer (Cary Varian 50). Single turnover activity was measured by monitoring changes in A360 nm to follow oxidation of NADPH bound to Kvβ2. The Kvβ2 protein was pre-equilibrated at 37°C and the enzymatic reaction was initiated by adding the substrate (0.2 – 2 mM). The reaction was monitored continuously by measuring changes in A360 for 30-60 min at 37°C to follow the oxidation of NADPH bound to Kvβ2.
Synthesis of deuterated NADPH
NADPH and NADPD were synthesized by using alcohol dehydrogenase from Thermoanaerobium brockii (Sigma Chemical Co) as described earlier (22). For synthesis, alcohol dehydrogenase 2U (1.21 mg) was incubated with 2 mM NADP+ in 50 mM Tris-HCl (pH7.8) and 300 mM 2-propanol or 2-propanol-d8 (Sigma Chemical Co) for 1 h at 40°C. The progress of the reaction was monitored by measuring changes in absorbance at 340 nm. After completion, the reaction mixture was loaded on an anion exchange column pre-equilibrated with 20 mM Tris-HCl pH 9.0. The column was washed with 25-30 ml of 20 mM Tris-HCl, pH 9.0 and the reduced cofactor NADPH/D was eluted by using 250 mM NaCl, 50 mM Tris, pH 7.8. The absorbance of the eluted fractions at 260 and 340 nm was recorded. Fractions with A260/A340 ratio of > 2.4 were pooled and the pH of the mixture was adjusted to 9.0 with NaOH. Synthesized pyridine nucleotides were stored at −80°C until further use.
Exhaustion of Kvβ2 bound NADPH and addition of synthesized NADPH or NADPD
The Kvβ2 protein purified from bacteria remains tightly bound to NADPH, which is not removed by dialysis. Hence, for kinetic isotope effect studies, NADPH bound to Kvβ was removed by oxidizing it to NADP+, which dissociates from the protein more readily than NADPH. For this, Kvβ2 purified from bacteria was incubated with 4-nitrobenzaldehyde (600 μM) and the disappearance of NADPH was monitored at 360 nm. After driving the reaction to completion, the reaction mixture was transferred to a dialysis cassette (10,000 Da cutoff; Pierce) and dialyzed against 0.2 M potassium phosphate (pH7.4) at 4°C for 16-20 h. Synthesized NADPH or NADPD was added to Kvβ2 and the nucleotide was allowed to bind to the protein for 30 min at 4°C. Excess NADPH/D was removed by equilibrium dialysis. The progress of the reaction was monitored at each step by measuring changes in absorbance at 360 nm.
Kinetic isotope effects
To study kinetic isotope effects, 150 μM synthesized NADPH or NADPD was mixed with 20 μM of Kvβ2 in 0.2 M potassium phosphate, pH7.4 at 37°C and enzyme reaction was started by adding varying concentrations (0.1 – 2 mM) of the substrate. Oxidation of the NADPH/D was monitored at 340 nm for 15 min at 37°C. For calculating initial velocity, only the linear part of the reaction was used. Kinetic isotope effects on single turnover reactions were calculated from the rate of oxidation of NADPH or NADPD bound to Kvβ2 measured from changes in absorbance at 360 nm at 37°C for 30 min after the addition of the substrate.
Product identification
The reduction of phospholipid aldehyde 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphatidylcholine (POVPC) to 1-palmitoyl-2-(5)-hydroxyvaleroyl-sn-glycero-3-phosphorylcholine (PHVPC) by Kvβ2 was followed by electrospray mass spectrometry (ESI+/MS) analysis as described earlier (23). The reaction containing 35 μM Kvβ2, 150 μM NADPH and 80 μM POVPC was incubated at 37°C for 3 h. Additional NADPH was added to the reaction mixture at 45 min intervals. The reaction was stopped after 3 h and phospholipids were extracted by using the Bligh and Dryer (24) procedure. For ESI+/MS analysis, a MicroMass ZMD 2000 mass spectrophotometer (Waters-Micromass, Milford, MA) was used. The injection solvent was 2:1 methanol/chloroform (v/v) containing 1.0% acetic acid in positive ionization mode and 2:1 methanol/chloroform (v/v) containing 10 mM ammonium hydroxide in the negative ionization mode. Samples were injected into the spectrometer using a Harvard syringe pump at a flow rate of 25 μl/ min. The ESI+/MS operating parameters were: capillary voltage 3.38 kV, cone voltage 25 V, extractor voltage 9V, RF lens voltage 0.9 V, source block, and desolvation temperatures 100 and 200 °C, respectively. Nitrogen was used as the nebulizer gas at a flow rate of 3.4 liters/h. Spectra were acquired at a rate of 275 atomic mass units per s over the mass range of 2–1000 atomic mass units and were averaged over a period of 5 min or 100 scans (23).
HPLC Analysis
Reagent 4-hydroxy-trans-2-nonenal (4-HNE), 1,4-dihydroxynonenal (DHN) were synthesized as described before (25). The 4-oxo-2-nonenol was synthesized by the reduction of 4-oxo-2-nonenal (4-ONE) by aldose reductase (25). Reagent 4-HNE, 4-ONE, DHN and 4-oxo-2-nonenol were separated by HPLC using a Varian reverse phase ODS C18 column pre-equilibrated with 0.1% aqueous trifluoroacetic acid. The compounds were eluted using a gradient consisting of solvent A (0.1% aqueous trifluoroacetic acid) and solvent B (100% acetonitrile) at a flow rate of 1 ml/min. The gradient was established such that solvent B reached 26% in 30 min, and was held at this value for 10 min. In the next 10 min, solvent B reached 60%. In an additional 5 min, it reached 100% and was held at this value for 10 min. Under these conditions, DHN, 4-oxo-2-nonenol, 4-HNE, and 4-ONE eluted with a retention time of 31, 37, 43, and 53 min, respectively.
Analysis of reduced product by Gas Chromatography/Mass Spectrometry
HPLC purified peak II was extracted in dichloromethane and dried under nitrogen. The samples were derivatized with 50 μl of BSTFA-TMS (N,O-Bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane) for 5 min at 80 °C. An aliquot (1 μl) of the mixture was utilized for gas chromatography/electron impact ionization - mass spectrometry (GC/EI-MS) analysis. GC/EI-MS analysis was performed using a HP-6890/HP5973 GS/MS system (Hewlett Packard; Palo Alto, CA, USA). Compounds were separated on a bonded phase capillary column (DB-5MS, 30 m × 0.25 mm × 0.25-μm film thickness from J&W Scientific, Folson, CA, USA). The GC injection port and interface temperature were set at 280 °C. Injections were made in the splitless mode with the inlet port purged for 1 min following injection. The GC oven temperature was held initially at 100 °C for 1 min, and then increased at a rate of 10 °C/min to 280 °C, and then held there for 5 min. Under these conditions, the retention time of 4-oxo-2-nonenol was 8.06 min.
Hammett Plot analysis
The kinetic constants, Km and kcat were obtained with the series of benzaldehyde compounds. For each para-substituted benzaldehyde, values for electronic (σ+), steric (υ) and hydrophobic (Π) parameters were obtained from published literature (26-28). The log kcat or the log Km or the log kcat/Km values were plotted against σ+ and the correlation coefficients were obtained from multivariate weighted analysis as described before (22;29) for the following equation using SAS 9.1:
| (2) |
where σ+ is the Hammett constant for electronic effects, Π is the Hansch constant for the hydrophobic interactions, and υ is the steric constant; ρ, A, B and C are parameter coefficients.
Data Analysis
Steady-state kinetic constants kcat and Km were calculated by fitting the Michaelis-Menten equation directly in the hyperbolic form to the initial velocities using Marquardt-Levenberg algorithm. For nonlinear regression analysis, Sigmaplot 9.0 software was used. Single turnover traces were analyzed using a single-exponential decay equation:
| (3) |
Calculated pseudo-first order rate constants, ktrans, were then plotted against substrate concentration and analyzed using nonlinear regression. Kinetic isotope effects were calculated from the initial velocities obtained with NADPH and NADPD at various substrate concentrations using the following equations:
| (4) |
| (5) |
where A is the concentration of aldehyde substrate, Fi is the fraction of deuterium in the NADPH/NADPD cofactor, Ekcat/Km and Ekcat are the isotope effects minus 1 on kcat/Km and kcat, respectively. Equation 5 assumes that Ekcat/Km = Ekcat. Multivariate regression analysis was performed using SigmaPlot 9.0. Data are presented as mean ± SEM. Intergroup comparisons were made using the unpaired Students t test and P < 0.05 were considered to be statistically significant.
Results
Kvβ2-mediated catalysis
To determine whether the purified Kvβ2 protein was enzymatically active, fundamental properties of its catalysis were determined. For this, the pH and ionic dependence of the reaction and the dependence of the reaction velocity on the concentration of the protein and its substrates were studied. In addition, we also established the identity of the acid-base catalyst and authenticated the formation of reduced products generated by the protein. In the first series of experiments, the rate of NADPH reduction was measured using 4-nitrobenzaldehdye (4-NB) as a model AKR substrate. When incubated with 1 mM 4-NB, a time-dependent decrease in A340 was observed, indicating oxidation of NADPH to NADP+ (Fig. 1A). The loss of absorbance was linear for the first 10 min. The linear portion of the progress curve was used for all initial velocity measurements. The initial rate of NADPH reduction showed an increase with an increase in protein concentration (Fig. 1B), indicating that the reaction was dependent on Kvβ2 and not a spurious outcome of reaction conditions. No saturation of the reaction rate was observed when the protein concentration was increased from 10 to 50 μM. Hence, in all future experiments, a concentration of 15-25 μM protein was used.
Figure 1. Optimal conditions for catalysis of aldehyde reduction by Kvβ2.

Recombinant rat Kvβ2 was purified from E.coli and the catalytic activity was measured in reaction mixtures containing 0.15 mM NADPH and 1 mM 4-nitrobenzaldehyde (4-NB). Time course of change in absorbance at 340 nm (A340) was determined at 10 (dashed line) and 50 μM (solid line) concentrations of the protein in 0.2 M potassium phosphate, pH 7.4, at 37 °C (A) and the initial velocity for 10 min was calculated at different protein concentration (B), pH (C) and ionic strength (D). Data are presented as mean ± SEM (n = 3). Line in panel B is the best fit of linear equation to the data (R2=0.99). Lines in panels C and D are the best fits of (C), three parameter normal logarithmic distribution (R2=0.97) and (D), hyperbolic (R2=0.96) function (y=y0+a.x/(b+x)) to the data.
To optimize activity assays, the pH-dependence of the reaction was investigated. When the enzyme activity was measured in reaction mixture of pH 6.5 to 7.9, maximal activity was observed between 7.2-7.4. The enzyme activity declined at pH >7.5 or < 6.5 and the pH-dependence conformed to a bell-shaped curve (Fig. 1C). In all subsequent measurements, a pH of 7.4 was used. Next, using a pH of 7.4, the ionic dependence of catalysis was measured. The catalytic activity of the protein was insensitive to changes in ionic strength, when the concentration of phosphate in the reaction mixture was varied between 0.1 and 0.25 M, however, a significant decrease in the enzyme activity was observed at phosphate concentrations < 50 mM (Fig. 1D). At 0.15 M potassium phosphate (pH 7.4), the maximal activity was ∼2.5 mU/mg protein, and the turnover number was between 0.08 and 0.3 min−1. At conditions optimal for NADPH oxidation, no oxidation of NADH was observed (data not shown). Moreover, inclusion of oxidized cofactor NADP+ (40 μM) or the reduced product p-nitrobenzyl alcohol (40 μM) did not influence the initial velocity of the reaction with 4-NB, suggesting that neither product inhibition nor reverse reaction occurs to an appreciable extent during initial velocity measurements. Collectively, these data provide optimal values of protein concentration, pH and ionic strength for measuring Kvβ catalysis and are consistent with the view that Kvβ2 is an enzymatically active protein that preferentially catalyzes aldehyde reduction. These results also show that the catalytic reduction of aldehydes by Kvβ2 is specific for NADPH and is not limited or affected by product inhibition.
To establish specificity of catalysis, a site-directed Kvβ2 mutant was generated in which Tyr-90 was replaced with phenylalanine. Although previous studies show that D85N and K118M substitutions decrease Kvβ2-mediated reduction of 4-CB, neither of these residues act as acid-base catalyst and could indirectly affect reduction by preventing substrate or NADPH binding (18). The residue corresponding to Tyr-90, on the other hand, is widely accepted to be a direct participant in acid-base catalysis and substitution of this residue with phenylalanine abolishes catalytic activity in other AKRs (21;30;31). The absorbance spectrum of purified Kvβ2:Y90F was similar to that of the WT protein, with absorbance peaks at 275 and 360 nm (Fig. 2 A), indicating that the mutant protein remains tightly-bound to NADPH. To quantitatively determine whether the Y90F mutation affects NADPH binding, KdNADPH of the WT and mutant proteins was determined by fluorometric titrations. As shown in Fig. 2B, addition of different concentrations of NADPH led to a similar decrease in the fluorescence of WT and Y90F Kvβ2. The KdNADPH for Kvβ2 WT was 0.2 ± 0.005 μM, which is similar to that determined for Kvβ2:Y90F (0.13 ± 0.011 μM), indicating that substitution of Tyr-90 with Phe does not significantly alter NADPH binding (Table 1). Nevertheless, the catalytic activity of the protein was profoundly affected. As shown in Fig. 2 C, significant NADPH oxidation by 4-NB was observed with the WT protein, whereas NADPH oxidation by Kvβ2:Y90F was near background levels. The turnover rate of WT Kvβ2 with 4-NB was reduced from 0.12 to 0.009 min−1 by the Y90F mutation (Table 1). From these results, we conclude that the aldehyde reductase activity of Kvβ2 is inherent to the protein and could not be ascribed to a contaminating protein. Moreover, the catalytic activity of the protein appears to be due to acid-base catalysis provided by Tyr-90, which corresponds to the proton donor identified in other AKRs.
Figure 2. Cofactor binding and catalytic activity of Kvβ2:Y90F.

(A) Absorbance spectra of WT (solid line) and Y90F (dashed line) Kvβ2 purified from bacteria. Peak absorbance was observed at 275 nm with a shoulder at 360 nm indicating NADPH bound to the protein. The inset shows SDS-PAGE of purified WT and Y90F Kvβ2 protein; (B) NADPH-dependent changes in the fluorescence of WT (filled circles) and Y90F (open circles) Kvβ2 (15 μg) measured in 0.2 M potassium phosphate, pH 7.4 using an excitation wavelength of 280 nm and emission wavelength of 340 nm. Data are shown as discrete points and curves are best fits of eq. [1] to the data (R2=0.98 for WT, and 0.95 for Y90F). Changes in fluorescence were normalized to the value of the initial fluorescence of the protein; and (C) time-dependent changes in absorbance at 340 nm in a reaction mixture containing 0.15 mM NADPH and 1 mM 4-nitrobenzaldehyde and WT (solid line) or Y90F (dashed line) Kvβ2.
Table 1.
Specific activity and NADPH binding of WT and Y90F:Kvβ2.
| KdNADPH (μM) |
Specific activity (min−1) | |||
|---|---|---|---|---|
| 4-nitrobenzaldehyde (1 mM) |
4-nitroacetophenone (1 mM) |
|||
| 1. | Kvβ2:WT | 0.2 ± 0.005 | 0.118 ± 0.02 | 0.050 ± 0.008 |
| 2. | Kvβ2:Y90F | 0.13 ± 0.011 | 0.009 ± 0.002* | 0.006 ± 0.003* |
Values of KdNADPH and specific activity represent a mean of 3 independent measurements.
P < 0.05 versus WT.
Substrate Specificity
To determine whether Kvβ reductase activity is limited to aldehydes or whether ketone compounds are also reduced, the enzyme activity with series of acetophenones was measured. Of the several acetophenones tested, only 4-nitroacetophenone (4-NAP) showed appreciable activity. The catalytic efficiency (kcat/Km) of the protein with 4-NAP (0.46 min−1.mM−1) was similar to that observed with 4-NB (0.59 min−1.mM−1), indicating that Kvβ2 catalyzes the reduction of ketones as well as aldehydes. The protein also displayed high catalytic activity with 9,10-phenanthrenequinone (Km = 0.24 ± 0.14 mM; kcat = 0.19 ± 0.07 min−1 and kcat/Km = 0.79) and the endogenous carbonyl methylglyoxal (Km = 12 ± 1.0 mM; kcat = 0.32 ± 0.04 min−1 and kcat/Km = 0.03). Measurable catalytic activity was also detected with several other carbonyls (Table 2) such as 4-oxo-trans-2-nonenal, phenylglyoxal, succinic semialdehyde, and the phospholipid aldehyde 1-palmitoyl-2-oxovaleroyl phosphatidylcholine (POVPC). Low activity and poor affinity of the protein for these carbonyls, however, precluded accurate estimates of kinetic constants. Several other compounds which are generally good substrates for AKR enzymes were also tested. These include DL-glyceraldehyde, glucose, xylose, D-glucuronic acid, D-fructose, 5α-androstene 3α,17β diol, and cortisone. We found little or no activity with these compounds at conditions optimal for other substrates. From these data we conclude that Kvβ2 reduces both aldehydes and ketones with equal affinity.
Table 2.
Kvβ2 specific activity with biologic carbonyls.
| Substrate | Structure | Specific activity (min−1) | |
|---|---|---|---|
| 1. | Phenylglyoxal (100 μM) | 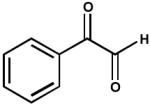 |
0.014 ± 0.0006 |
| 2. | Methylglyoxal (12 mM) | 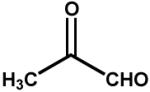 |
0.32 ± 0.04 |
| 3. | 3-deoxyglucosone (5 mM) | 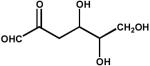 |
0.039 ± 0.01 |
| 4. | 2-carboxybenzaldehyde (10 mM) |
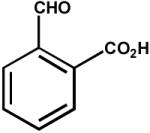 |
0.010 ± 0.005 |
| 5. | Acrolein (100 μM) | 0.010 ± 0.003 | |
| 6. | 4-oxo-nonenal (100 μM) | 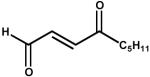 |
0.028 ± 0.004 |
| 7. | 4-hydroxy-nonenal (100 μM) | 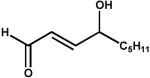 |
--a |
| 8. | Succinic semialdehyde (25 mM) | 0.011 ± 0.004 | |
| 9. | 12-oxoETE (10 μM) | 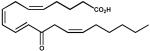 |
0.044 ± 0.007 |
| 10. | PGJ2 (100 μM) | 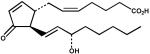 |
0.086 ± 0.019 |
| 11. | PGD2 (100 μM) | 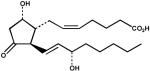 |
--a |
| 12. | PGF2α (100 μM) | 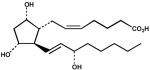 |
--a |
| 13. | 9, 10 Phenanthrenequinone (200 μM) |
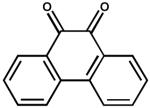 |
0.19 ± 0.07 |
| 14. | POVPC (100 μM) | 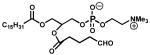 |
0.078 ± 0.016 |
| 15. | 5α-androstan-17β-ol-3-one (100 μM) |
 |
--a |
| 16. | Cortisone (100 μM) |  |
--a |
Activity less than two fold over the background.
To establish whether Kvβ2 catalysis results in the appearance of a reduced alcohol product, the protein was incubated with POVPC, which is a good substrate of AKR1B1 and other related AKRs (32). ESI+/MS analysis of POVPC revealed a predominant ion at m/z 594.6 (Fig. 3A). To examine reduction, the phospholipid aldehyde was incubated with 35 μM Kvβ2 for 180 min, after which the phospholipid was extracted using the Bligh and Dryer (24) procedure and re-analyzed by mass spectrometry. The phospholipid mixture recovered after incubation with Kvβ2 showed the appearance of an additional ion with m/z 596.4, consistent with reduction of POVPC to PHVPC. The total conversion of POVPC to PHVPC was 18 ± 2% (n=4), which corresponds to the amount of NADPH oxidized. These observations confirm the AKR nature of the Kvβ2 active site and substantiate its catalytic activity leading to the generation of a reduced product.
Figure 3. Kvβ2-catalyzed reduction of phospholipid aldehyde.

Electrospray mass spectra of 1-palmitoyl-2-oxovaleroyl phosphatidyl choline (POVPC) incubated without (A) and with (B) Kvβ2. The formation of the reduced product - 1-palmitoyl-2-oxovaleroyl phosphatidyl choline (PHVPC) is evident by the appearance of a new ion with m/z = 596.4. For reduction, POVPC (80 μM) was incubated without or with Kvβ2 (35 μM) and NADPH (0.15 mM) for 180 min at 37°C. The 2Da increase in m/z was assigned the reduction of the aldehyde at the sn-2 position to an alcohol (as indicated).
Because Kvβ2 can reduce both aldehydes and ketones, we asked which of these groups is preferentially reduced by Kvβ2 in a keto-aldehyde? To test this, the protein was incubated with 4-ONE, which contains a keto group at C-4 and an aldehyde at C-1. As shown in Scheme 1, reduction of this compound at C-1 will result in the formation of 4-oxo-nonenol, whereas reduction at C-4 will form 4-HNE. Further reduction of 4-HNE and 4-oxo-nonenol would lead to DHN formation. The Kvβ-derived product of 4-ONE was first analyzed by HPLC. As shown in Fig. 4, an appropriate separation protocol was developed in which the retention times of 4-HNE, DHN, 4-ONE, and 4-oxo-nonenol were measurably different. Incubation with Kvβ2 shifted the retention time of 4-ONE from 53 min to 37 min, which corresponds to the retention time of 4-oxo-nonenol. No products eluted with a retention time corresponding to 4-HNE or DHN, indicating that Kvβ2 reduces 4-ONE to 4-oxo-nonenol. To further confirm the structural identity of the product, the Kvβ2 derived peak at 37 min was derivatized and analyzed by GC-MS. On GC, the product displayed a retention time of 8.06, which was identical to that of reagent 4-oxo-nonenol. On the mass spectrum of the product (Fig. 4 C), the derivatized molecular ion with 228 m/z (M) was identified, along with major fragments with m/z 213 (M-CH3), 157 (M-C5H11), 129 (M-C6H11O1). This fragmentation pattern corresponds to 4-oxo-nonenol, but not 4-HNE, 4-ONE or DHN (data not shown), indicating that the keto-group at C-4 position remains unreduced whereas the aldehyde group at C-1 position is reduced; leading to the formation of 4-oxo-2-nonenol. These data indicate that Kvβ prefers aldehydes to ketones and that carbonyls with high electron density at oxygen (e.g., C-1 of 4-ONE) are reduced more efficiently than less polarized carbonyls (e.g., C-4 of 4-ONE or C-1 of 4-HNE).
Scheme 1. Potential products of 4-oxo-2-nonenal (ONE) reduction.

Reduction of 4-oxo-2-nonenal at C-1 aldehyde would lead to the formation of 4-oxo-2-nonenol, whereas reduction of the C-4 keto group should yield 4-hydroxy-2-nonenal (HNE). Additional reduction of HNE or 4-oxo-2-nonenol would form 1,4-dihydroxy-2-nonenal (DHN). Note: Reduction in the presence of Kvβ2 led to the appearance of only 4-oxo-2-nonenol and no HNE or DHN formation was observed (see, Fig. 4). The reduction of the C-1 aldehyde to an alcohol is indicated by a dotted box.
Figure 4. Identification of Kvβ2-derived product of 4-oxo-2-nonenal (4-ONE).

Panel A shows the separation of reagent 3H-1,4-dihydroxynonenol (DHN; 3000 cpm), 4-oxo-2-nonenol, 4-hydroxy-2-nonenal (4-HNE) and 4-ONE, 40 nmoles each on ODS C18 reverse phase HPLC column. Y-axis on the right side shows the radioactivity profile of 3H-DHN whereas Y-axis on the left side shows OD224. Peaks I-IV eluting with retention times of 31, 37, 43 and 53 min are due to reagent DHN, 4-oxo-2-nonenol, 4-HNE and 4-ONE, respectively. Panel B shows the HPLC traces of the Kvβ2-catalyzed reduction product of 4-ONE. Reagent 4-ONE (100 nmoles) was incubated with 0.6 mg Kvβ2 and 0.3 mM NADPH in 0.2 M potassium phosphate for 2h at 37 °C and the product was separated on HPLC. Panel C: inset shows a representative gas chromatogram of derivatized peak II (from panel B), eluting at 8.06 min and panel C shows the electron impact ionization-mass spectrum of the siliated metabolite of the GC peak eluting at 8.06 min. Ion with m/z 228 represents moleclular ion (M) of 4-oxo-2-nonenol. Ions with m/z 213, 157 and 129 respectively, correspond to M-CH3, M-C5H11 and M-C6H11O fragments of 4-oxo-2-nonenol.
Reaction sequence
To determine the reaction sequence of the catalytic cycle, initial velocity of the reaction was measured at different 4-NB and NADPH concentrations. For these experiments, 4-NB was chosen as the model substrate because of all the carbonyls examined, highest kcat/Km values and low Km values were observed with 4-NB. Double reciprocal plots of initial velocity vs. substrate concentration were linear and showed no signs of product or substrate inhibition. Double reciprocal plots at different NADPH concentration conformed to a pattern of straight lines intersecting at the ordinate (Fig. 5). This pattern is consistent with a rapid-equilibrium ordered kinetic mechanism in which NADPH binds first to the enzyme followed by the carbonyl substrate (Scheme 2). To determine kinetic constants, the following relationship was used:
| (6) |
where A is the concentration of NADPH, and B is the concentration of 4-NB, VM is Vmax, Kb is the Michaelis constant for 4-NB; and Kia is the binding constant of NADPH. Best fit of the equation 6 to the data yielded Kia = 107 ± 33 μM, Kb = 490 ± 119 μM, and kcat (VM/Et) = 0.15 ± 0.015 min−1. When the equation for a steady-state ordered mechanism containing the term KaB in the denominator of equation 6 was fitted to the data an undefined Ka was obtained. A replot of slopes (KmB/Vm apparent) (determined from Fig. 5) versus 1/NADPH was linear with slope of KbKia/Vm and an intercept on the ordinate axis of Kb/Vm. The intercept on the 1/[NADPH] axis was equal to −1/Kia. Values obtained from each curve separately (slope replot; Fig.5; inset) are similar with those determined by global analysis (i.e., by fitting equation 6 to the data). These data confirm that Kvβ2 follows a rapid-equilibrium ordered scheme in which NADPH binding precedes substrate binding (Scheme 2).
Figure 5. Initial velocity profile of aldehyde reduction by Kvβ2.

Double reciprocal plots of initial velocity were derived from measuring the rates of change of absorbance at 340 nm at different concentrations of 4-nitrobenzaldehyde at 75 (filled circle), 100 (open circle) and 200 (filled inverted triangle) μM NADPH. Lines represent best fit of equation 6 to the data (R2=0.97). Inset shows a replot of individual slopes of the double reciprocal plot versus 1/[NADPH].
Scheme 2. Catalytic cycle for NADPH-dependent carbonyl reduction by Kvβ2.

Sequential binding of the protein to NADPH and the carbonyl substrate results in the formation of a ternary complex in which the carbonyl is reduced to its corresponding alcohol. The release of products is also shown to be ordered. The microscopic rate constants for each reaction step in the forward and reverse direction are indicated as kn and k−n, respectively.
Kinetic Isotope Effects
To understand the mechanism of Kvβ2 catalysis in greater detail we investigated the rate-determining step. For this, the primary kinetic isotope effects were determined by using NADPH and NADPD as cofactors. NADPH or NADPD was synthesized as described under Material and Methods and initial velocity was measured at different concentrations of the aldehyde substrate. In the first series of experiments, purified Kvβ2 was used for activity measurements directly, using 4-NB or 4-NAP as a variable substrate. Both equations 4 and 5 were used to fit the data obtained (Fig. 6). Equation 5, however, gave a better fit to the data than equation 4 as determined by F values and the standard errors in the values of the kinetic constants. The best-fit values of kinetic constants obtained from equation 5 are shown in Table 3. Similar values of kinetic isotope effects were obtained with either 4-NB or 4-NAP as the varied substrate (Table 3). High values of primary kinetic isotope effect indicate that the chemical step is rate-limiting in Kvβ2-catalyzed reduction. The lack of isotope effect on KmB suggests that Km is not a true steady-state kinetic parameter, but a dissociation constant for aldehyde binding to the E:NADPH binary complex. The reverse reaction, i.e., oxidation of 4-nitrobenzyl alcohol by NADP+ was also investigated. However, with 4-nitrobenzyl alcohol as the substrate, no measurable activity under multiple turnover conditions was detected. This precluded determination of Haldane relationship and calculation of several microscopic constants including k−3 which is the rate constant for alcohol oxidation (Scheme 2).
Figure 6. Primary deuterium kinetic isotope effects on Kvβ2-catalyzed aldehyde reduction.

Initial velocity plots of the reaction at different concentrations of 4-nitrobenzaldehyde (A; 4-NB) and 4-nitroacetophenone (B; 4-NAP) with 0.15 mM NADPH or NADPD as indicated are shown. For each measurement, the Kvβ2 protein was mixed either with NADPH or NADPD (0.15 mM) and the reaction was initiated by the addition of the substrate. Time-dependent changes in total sample absorbance (multiple turnover mode) were measured. The values of initial velocity are normalized to the concentration of the total Kvβ2 protein in the cuvette. Data are shown as discrete points and curves are best fits of eq. [5] to the data (R2=0.99 for 4-NB and 0.97 for 4-NAP).
Table 3.
Kinetic constants and primary kinetic isotope effects (KIE) on Kvβ2-mediated carbonyl reduction.
| Substrate | Km (μM) |
kcat (min−1) |
kcat/Km (mM−1.min−1) |
KIE |
|---|---|---|---|---|
| 4-nitroacetophenone | 209 ± 49 | 0.092 ± 0.006 | 0.46 ± 0.09 | 1.69 ± 0.1 |
| 4-nitrobenzaldehyde | 283 ± 31 | 0.16 ± 0.030 | 0.59 ± 0.17 | 2.07 ± 0.4 |
| 4-nitrobenzaldehydea | ---b | ---b | ---b | 3.13 ± 0.2 |
The value of each parameter is a representative of 2-5 separate determinations using different preparations of the protein. For each determination, the initial velocity was measured at 6-9 different concentrations of the substrate.
Data from single turnover experiments.
Not determined.
Single-turnover Measurements
Transient kinetic measurements, such as single turnover allow determination of some of the microscopic rate constants associated with enzyme catalytic mechanism. The Kvβ2 binds tightly to NADPH and forms a binary complex. Free NADPH in solution absorbs at 340 nm, whereas binding to the β2 subunit causes a red shift of this peak to 360 nm. Hence for single turnover experiments, we monitored changes in absorbance at 360 nm to measure the activity of the β2:NADPH binary complex with 4-NB.
To measure isotope effects under single turnover conditions the Kvβ2 protein was preloaded with NADPD. Because Kvβ2 purified from bacteria is tightly bound to NADPH (Fig. 7A), which is not removed by dialysis, NADPH bound to Kvβ2 was first oxidized to NADP+ in a single turnover reaction using 4-NB as the substrate. As shown in Fig. 7A, incubation with 4-NB led to a progressive decrease in A360, indicating that all of the NADPH bound to the protein was oxidized to NADP+. The unutilized 4-NB and the 4-nitrobenzyl alcohol generated from the reaction were removed by equilibrium dialysis, and the protein was reloaded with either NADPH or NADPD by dialysis with these cofactors. This led to an increase in the absorbance at 360 nm, indicating that NADP+ bound to the protein was completely replaced by NADPH or NADPD. Analysis of the absorbance at 275 nm and 360 nm using an extinction coefficient of 60,880 for the protein and 6,200 M−1cm−1 for NADPH indicated that approximately 90-100% of the protein was saturated with NADPH/D (Fig. 7A). These preparations were then used for subsequent kinetic isotope studies under single turnover conditions.
Figure 7. Kinetic isotope effects on single turnover reactions of Kvβ2.
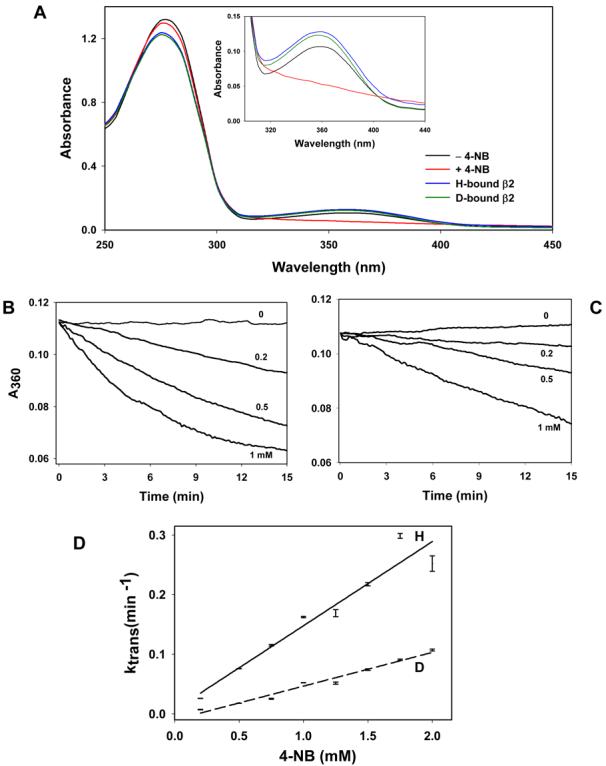
(A) Replacement of intrinsically bound NADPH with NADPD. The NADPH bound to Kvβ2 (black) was oxidized by incubating the protein with 1 mM 4-nitrobenzaldehyde which led to a progressive decline in absorbance of the protein at 360 nm (inset; red line), indicating oxidation of NADPH to NADP+. The reaction mixture was then dialyzed for 16-20 h at 4°C against 0.2 M potassium phosphate, pH7.4. The protein was then incubated with synthesized NADPH or NADPD for 30 min at 4°C and dialyzed for 16-20 h. Reloading with NADPH (blue) or NADPD (green) led to an increase in A360, indicating re-formation of the protein-nucleotide binary complex; Time-dependent decrease in A360 of NADPH (B) and NADPD (C) -bound Kvβ2 incubated with the indicated concentrations of 4-NB; and (D) The values of the pseudo-first order rate constant (ktrans), obtained by fitting a single exponential equation to the progress curve data shown in panels B and C, plotted as function of 4-NB concentration. Data are shown as discrete points ± SEM and the lines are best fit of Eq. (5) to the data (R2=0.96); solid line, NADPH; dotted line, NADPD.
Absorbance at 360 nm decreased with time upon addition of 4-NB (0.2 – 2mM) to the NADPH- or NADPD-loaded enzyme (Fig. 7 B & C). Experimental time courses for NADPH/D oxidation were best described by a single exponential function. Values of ktrans increased linearly with 4-NB concentration and showed no significant saturation within the concentration range of 4-NB that could be practically achieved (Fig. 7D). From these experiments a kinetic isotope effect of 3.13 ± 0.2 was obtained (Table 3)., indicating that the chemical step is rate-limiting in single turnover conditions.
Structure-activity relationship
To understand the catalytic mechanism further, Hammett plots with catalytic efficiencies (kcat/Km) and turnover numbers (kcat) were generated with a series of para-substituted benzaldehydes (Fig. 8A and B). A plot of log kcat/Km versus σ+ displayed a linear relationship (Fig. 8A), suggesting that the reaction mechanism does not change within this series, and that substitutent effects was primarily electronic in nature. A Hammett plot with kcat/Km showed a positive ρ coefficient, the value (1.95 ± 0.36) of which is similar to that reported for other AKRs (22;29). This fit was only marginally improved by incorporating hydrophobicity or the steric constants (Supplemental Table 1), indicting that these characteristics make only minor contributions to benzaldehyde binding. A plot of kcat versus σ+ gave a slope of 0.04 ± 0.02 (Fig. 8B). Among the series tested, the only exception was 4-NB. Reasons for the anomalous behavior of 4-NB are unclear but may relate to specific interaction of the nitro group with the active site residues of the protein. Log Km displayed a linear inverse relationship with σ+ with a correlation coefficient (ρ) of −1.85 ± 0.52 (Fig. 8C) and no significant contribution of steric or hydrophobic features of the benzaldehydes was evident (Supplemental Table 1).
Figure 8. Structure-activity relationships for Kvβ2-mediated reduction of substituted benzaldehydes.

Relationship between log (A) kcat/Km, (B) kcat and (C) Km of para-substituted benzaldehydes and the Hammett constant - σ+. The enzyme activity was determined under multiple turnover condition with 0.15 mM NADPH. Data are shown as discrete points and the lines are best weighted fits of a linear equation to the data (R=0.79-0.93). Calculated slope values are listed on the upper right hand corner of the panel. Results of multivariate regression analysis are shown in supplemental Table 1.
Discussion
The β-subunits of voltage-gated (Kv) potassium channels belong to the AKR superfamily of enzymes (13). These proteins display 20-26 % sequence identity with other AKRs, and the architecture of their active site resembles that of other AKR oxidoreductases (12). Nevertheless, the catalytic properties of Kvβ are largely unknown. We, therefore, studied the substrate-specificity and the kinetic behavior of Kvβ2 in order to identify features of catalysis that are unique to Kvβ proteins and those that they share with other AKRs. Our results show that Kvβ2 displays significant enzyme activity and that it utilizes NADPH to catalyze the reduction of both aldehydes and ketones. As expected of an enzyme reaction, the rate of reduction was increased with an increase in protein and substrate concentration; the reaction showed distinct pH and ionic strength dependence; and the initial velocity was saturated at high substrate concentration. Importantly, catalysis, but not NAPDH binding, was prevented by substituting tyrosine-90 with phenylalanine, indicating that, as in other AKRs (21;30;31), this residue in Kvβ2 participates in general acid-base catalysis and that carbonyl reduction is not due to contamination by other proteins in the preparation or a direct covalent reaction with NADPH, but that it is a controlled conversion catalyzed at the Kvβ2 active site.
Substrate-specificity studies show that the protein was active with several aldehydes and ketones. Highest activity was observed with aromatic substrates. Comparable activity was observed with benzaldehydes and acetophenone as well as 9,10 phenanthroquinone. Most AKRs display high catalytic activity with 4-nitrobenzaldehyde (Km 2 μM (AKR1B1) (36) – 3mM (AKR7A2) (37)) and methylglyoxal (Km 8 μM (AKR1B1) – 1.2 mM (AKR1A1) (38)) with kcat ranging from 24 to 360 min−1 (36;39). 4-nitroacetophenone and 9, 10 phenanthrenequinone are typical model substrates for the AKR1C family members, which have substantial catalytic activity with ketone as well as aldehyde compounds. These enzymes possess lower kcat values (2-190 min−1), however Km values typically lie in the low micromolar range (1-200 μM) (31;37). Several physiological aldehydes such as lipid peroxidation products 4-HNE and POVPC are specific substrates for the AKR1B enzymes (32;40). Our results show that Kvβ catalyzes the reduction of a similar range of aldehydes and ketones with Km values in the range observed with other AKRs. On the basis of substrate specificity showing that it could catalyze the reduction of both aldehydes and ketones, we propose that Kvβ2 should be considered a general AKR that catalyzes the reduction of both aldehydes and ketones. Nonetheless, when both aldehyde and ketone groups were presented to the enzyme in the same substrate (e.g., in 4-ONE), the enzyme reduced the aldehyde, indicating that it prefers more electronegative carbonyls.
The physiological substrate of Kvβ2 remains unknown. The protein, however, displayed high catalytic activity with several carbonyls that are generated endogenously from enzymatic and non-enzymatic oxidation of lipids. High activity was observed with 2 enzymatically generated carbonyls, 12-oxo-ETE and PGJ2. 12-oxo-ETE is leukotriene B4 receptor agonist (42) and is derived from the oxidation of arachidonic acid by 12-lipoxygenase (43), whereas PGJ2 is a precursor of 15-deoxy Δ12,14-PGJ2, which is thought to be a natural ligand of peroxisome proliferator-activated receptor (PPAR-γ) (44). Carbonyl compounds generated from non-enzymatic oxidation of phospholipids such as 4-HNE, 4-ONE, and POVPC were also reduced by Kvβ2. These aldehydes are reactive chemicals and they affect a variety of signaling pathways leading to changes in vasodilation (45), myocyte excitability (46) and cell adhesion (47). Steroid-based natural carbonyl compounds such as 5α-androstene-3,17-dione, 4-androstene-3,17-dione and cortisone, on the other hand, were minimally reduced under conditions that were optimal for the reduction of other carbonyls. Whether any of these carbonyls are reduced by the protein in vivo remains unclear. Nonetheless, the preference of Kvβ2 for hydrophobic aldehydes raises the possibility that the function of this protein is related to the reduction of membrane-derived oxidized lipids.
Our kinetic analyses suggest that Kvβ2 follows rapid-equilibrium kinetics in which the bond-breaking step is rate-limiting (Scheme 2). This scheme is indicated by the lack of isotope effect of Km and the convergence of double reciprocal initial velocity plots at different substrate concentrations on the ordinate. This plot also indicates that the enzyme follows an obligate ordered reaction sequence in which coenzyme binding precedes substrate binding. Significant primary kinetic isotope effects were observed on kcat, indicating that chemistry is rate-limiting. Single and multiple turnovers displayed similar isotope effects (Table 3), although the slightly higher values for single (3.13) versus multiple (2.07) turnover may be due to kinetic steps that contribute to multiple, but not single, turnover reactions. These steps include those leading to NADPH binding (k1, Scheme 2) or NADP+ (k5) release. They appear to be nearly as slow as the bond-breaking step (k3) and could thus contribute to the rate of the overall reaction cycle. The value of the microscopic rate constant for the chemical step (k3) can be estimated from the following relationship:
| (7) |
If we assume that the value of Dk3 = 6.5, as calculated from NADPH-dependent reduction of D-xylose by AKR1B1 (51) and consistent with the kinetics of Candida tenuis xylose reductase (29), then calculations using formula (7) show that k3 = 0.82 ± 0.15 min−1 for 4-NB, and 0.73 ± 0.05 min−1 for 4-NAP. However, if we use a value of 3.1 estimated by Jin et al. for the intrinsic isotope effect on AKR1C2-catalyzed reduction of 5α-DHT by NADPH (33) k3 for 4-NB is 0.31±0.06 min−1 . Both these values are in agreement with the estimate for k3 obtained from our single turnover experiments. Fitting of multiple single turnover traces obtained with various 4-NB concentrations (Fig. 6) using Dynafit predicts that k3 is between 0.3 and 0.8 min−1. This prediction suggests that 20 to 50 % of the overall rate limitation is due to the hydride-transfer step with the remaining contributed by NADP+ or product release. A slow release of NADP+ has been identified to be the rate-limiting step in the AKR1B1-catalyzed aldehyde reduction (51), and is believed to be due to the slow movement of the NADPH binding loop (51;52). This possibility is consistent with the crystal structure of Kvβ2, which shows that the cofactor is held tightly at the active site (12). Thus NADP+ release, which requires extensive conformational rearrangement, could contribute to the inequality of isotope effects between single and multiple turnover reactions, and by implication, to the overall rate of the catalytic cycle.
Our data exclude cooperative or allosteric effects on catalysis and are consistent with our previous work showing a lack of cooperativity in NADPH or NADP+ binding to Kvβ2 (14). Native Kvβ proteins are, however, arranged as a tetramer around the T1 domain of Kvα (53) and Kvβ2 purified from bacteria (used for the kinetic experiments described here) is a tetramer in solution (14), indicating that cooperative behavior is possible. Nevertheless, no cooperatively was observed. Perhaps in solution, as in crystal structure with T1 (53), the active site of Kvβ faces the solvent and is minimally affected by other β monomers in the complex. Nonetheless, the possibility of allosteric effects in the regulation of Kv current by the β-subunits in situ cannot be ruled out.
The catalytic mechanism for carbonyl reduction by Kvβ2 appears to be similar to that observed for other AKRs. Catalytic inactivation by the Y90F mutation suggests that this residue play a similar role in Kvβ2 as it does in other AKRs (21;30;48). In addition to tyrosine (Tyr- 48 in AKR1B1), an active site histidine residue (His-110 in AKR1B1) has also been suggested to play an important role in AKR catalysis and substrate binding (21;30;49). This residue is located at the center of the hydrogen bonding network that facilitates substrate binding and polarization of the substrate carbonyl (48;50). The imidazole of this histidine in AKR2B5 plays an important role in precise positioning of the carbonyl group for catalysis (50). Significantly, His-110 is absent in Kvβ and is replaced by an asparagine residue. This shift in charge is likely to prevent tight binding of the carbonyl at the active site and may be responsible for the low catalytic activity of Kvβ2 (0.06 to 0.2 min−1 versus 2 to 600 min−1 for other AKRs). Low catalytic rates of Kvβ2 could thus be due to loose positioning and partial charge delocalization at the carbonyl cause by the sole tyrosine phenolate residue rather than a complex between a tyrosine and histidine as in other AKRs. Indeed, replacement of His-113 in AKR2B5 with alanine causes a 600- to1000-fold decrease in the catalytic activity of the protein (50).
Despite the absence of an active site histidine, the binding of aldehydes to Kvβ active site was similar to other AKRs. Our previous experiments with AKR1A1 show a strong dependence of kcat/Km as well as kcat on variations in substituents on the aromatic ring of benzaldehyde substrate (22). Similar results have been reported for AKR2B5 (29). Indeed, the ρ value obtained from Hammett plot of kcat/Km for Kvβ2 (1.95) was comparable to the ρ values obtained for AKR1A1 (1.95) and AKR2B5 (1.4). These data suggest that the active sites of AKRs are similar and that electron-withdrawing groups stabilize the formation of the ternary complex in the catalytic cycle of these proteins. Moreover, a negative correlation of Km with σ+ in Kvβ2 indicates that the binding of benzaldehydes is strongly dependent on the electron-withdrawing potential of para subsitutents. The ρ value for Km of Kvβ2 (−1.85) is, however, higher than that of AKR1A1 (−0.95) (22), indicating that electronic features play a more important role in substrate binding to Kvβ2 (AKR6) than in AKR1A1. The importance of electronic feature is further underscored by the observation that Kvβ2 was active with PGJ2, which contains a carbonyl polarized by α,β-unsaturation, but not PGD2, (Table 2) which is identical to PGJ2 except that it lacks the conjugated system and therefore has a less polarized carbonyl. Likewise, the more electrophilic C-9 aldehyde 4-ONE was a substrate, but the less polarized 4-HNE, despite a similar structure, was not reduced. Hence, a polarized carbonyl with a partial negative charge on the oxygen appears to favor substrate binding. Furthermore, correlation between substrate binding and σ+ within the benzaldehyde series suggests that a negative charge develops on the substrate carbonyl during ground state interaction, which may be indicative of the presence of a positively charged group that forms a hydrogen bond with the substrate (Scheme 3; Complex 1). The identity of this group remains to be established, but Tyr-90 is a strong possibility and binding to this residue could increase polarization of the enzyme carbonyl to facilitate acid-base catalysis (Scheme 3).
Scheme 3. Mechanism of NADPH-dependent carbonyl reduction catalyzed by Kvβ2.

In complex 1, Tyr-90 is shown poised to form a hydrogen bond with the substrate carbonyl which results in carbonyl polarization, accelerating the hydride transfer of the pro-R hydrogen from the nicotinamide ring of NADPH to the carbonyl carbon of the substrate. Complex 2 shows a non-charged transition state in which the polarization at the carbonyl is quenched by the proton transfer from the protein tyrosine and a concerted hydride transfer to the carbonyl carbon. The reduced carbonyl then dissociates from the acid-base catalyst and a net charge develops on the oxidized cofactor and the tyrosinate anion (Complex 3).
In contrast to binding, the bond-breaking step (= kcat) was relatively insensitive to the polarizing effects of the substituents. In this regard, Kvβ2 is different from AKR1A1 which gives a ρ value of 0.8 for the Hammett plot of kcat (22). The most straightforward explanation for the lack of substituent effects on kcat is that the charge developed on the carbonyl in the transition state is small. Given, however, that acid-base catalysis by other AKRs involves polarization of the substrate carbonyl; we suggest that in Kvβ the charge develops in the ground state of the carbonyl group. This polarization leads to the development of a slightly negative carbonyl O1 and a slightly electron-deficient C1 (Scheme 3; Complex 1). During transition state these charges are rapidly compensated by a proton transfer to the O1 and hydride transfer to C1 (Scheme 3; Complex 2). Because these events occur in concert, no net charge develops during the transition state. This mechanism appears to be similar to that of AKR2B5, which also shows no significant substitutent effects (29), but differs from AKR1A1 catalysis. Substituent effects on AKR1A1 are more consistent with a stepwise hydride and proton transfer leading to the formation of a charged transition state. Overall, it appears that in Kvβ2-mediated catalysis, the lack of hydrogen bonding (due to the absence of an active site histidine) prevents stabilization of a transition state in which the carbonyl is hydrogen bonded before the proton transfer occurs. Therefore, the reaction proceeds with a concerted, rather than stepwise hydride and proton transfer (Scheme 3; Complex 2). We speculate that this mechanism may be particularly relevant to the regulation of Kvβ catalysis by membrane voltage and for the ability of Kvβ to alter Kv inactivation.
It has been suggested that the reason why Kv channels associate with auxiliary subunits possessing oxidoreductase activity relates to two possibilities (54). The first possibility is that the Kvβ proteins are redox sensors that couple changes in the intracellular redox state with membrane excitability. This possibility is consistent with Kvβ-dependent changes in Kv inactivation in cells subjected to hypoxia (55). Based on the high NADPH binding affinity of Kvβ and the observation that Kvβ-mediated Kv inactivation is removed by NADP+, but not NADPH, we had suggested that changes in intracellular oxygen or intermediary metabolism could affect Kv currents by altering redox state of nucleotides bound to Kvβ (16). The redox state of the nucleotide bound to the protein could also be changed by catalysis and, in view of the high catalytic activity of the protein with several endogenous carbonyls, it appears likely that changes in endogenous NADPH-reducible endogenous carbonyls could regulate Kvβ activity, and in turn the inactivation or the voltage-dependence of Kv channels. Furthermore, because the dissociation constants of Kvβ for NADPH and NADP+ (0.1 to 0.6 μM) are far below the range of physiological changes in pyridine nucleotide concentration, the protein under most conditions is likely to respond to changes in the concentration of the carbonyl substrate. Thus any oxygen- or metabolism-dependent changes in substrate carbonyl concentration will rapidly alter the catalytic rate of Kvβ, leading to cofactor oxidation and changes in Kv function (15;16).
A second scenario is that voltage-dependent changes in Kv channel could regulate Kvβ catalysis by affecting the configuration of Kvβ active site. We suggest that a mechanism in which catalysis is rate-limiting is ideally suited for such a voltage-sensitive oxidoreductase. Because catalysis or the rate of interconversion of the ternary complexes could be regulated by a single step (independent of cofactor or substrate concentration) voltage changes could efficiently modulate the overall rate of the reaction. Clearly, further experiments are required to fully evaluate and assess the physiological implications of the kinetic behavior of Kvβ proteins.
Supplementary Material
Acknowledgements
The authors thank Yuting Zheng for generating the cDNA construct, Xioaping Li for assistance in protein purification, Dr. Yonis Ahmed for his advice on Hammett plot analysis, David Hoetker for ESI-MS experiments and Dan Riggs for assistance with HPLC assays.
† This work was partly supported by NIH grants HL-544771, HL-59378, ES-11860 (to A.B.), HL-089372 (to O.A.B.), and fellowship from the American Heart Association 0425439B (to S.M.T.).
Abbreviations and Textual Footnotes
- 4-NB
4-nitrobenzaldehyde
- 4-NAP
4-nitroacetophenone
- 4-HNE
4-hydroxy-trans-2-nonenal
- DHN
1,4-dihydroxy-trans-2-nonene
- 4-ONE
4-oxo-trans-2-nonenal
- Kv
voltage-gated potassium channel
- Kvβ
voltage-gated potassium channel β-subunit
- AKR
aldo-keto reductase
Footnotes
Supporting Information Available
Table S1 with parameters obtained from the multivariate regression analysis of Hammett plots as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 2.Dodson PD, Forsythe ID. Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004;27:210–217. doi: 10.1016/j.tins.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Hille B. Ionic Channels of Excitable Membranes. Sinauer; Sutherland, MA: 1992. [Google Scholar]
- 4.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 5.Krick S, Platoshyn O, Sweeney M, Kim H, Yuan JX. Activation of K+ channels induces apoptosis in vascular smooth muscle cells. Am. J. Physiol. 2001;280:C970–C979. doi: 10.1152/ajpcell.2001.280.4.C970. [DOI] [PubMed] [Google Scholar]
- 6.Ekhterae D, Platoshyn O, Krick S, Yu Y, McDaniel SS, Yuan JX. Bcl-2 decreases voltage-gated K+ channel activity and enhances survival in vascular smooth muscle cells. Am. J. Physiol. 2001;281:C157–C165. doi: 10.1152/ajpcell.2001.281.1.C157. [DOI] [PubMed] [Google Scholar]
- 7.Ekhterae D, Platoshyn O, Zhang S, Remillard CV, Yuan JX. Apoptosis repressor with caspase domain inhibits cardiomyocyte apoptosis by reducing K+ currents. Am. J. Physiol. 2003;284:C1405–C1410. doi: 10.1152/ajpcell.00279.2002. [DOI] [PubMed] [Google Scholar]
- 8.George CK, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farber GK, Petsko GA. The evolution of α/β barrel enzymes. Trends Biochem. Sci. 1990;15:228–234. doi: 10.1016/0968-0004(90)90035-a. [DOI] [PubMed] [Google Scholar]
- 10.Bachur NR. Cytoplasmic aldo-keto reductases: a class of drug metabolizing enzymes. Science. 1976;193:595–597. doi: 10.1126/science.959821. [DOI] [PubMed] [Google Scholar]
- 11.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel β subunit. Cell. 1999;97:943–952. doi: 10.1016/s0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- 13.McCormack T, McCormack K. Shaker K+ channel β subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell. 1994;79:1133–1135. doi: 10.1016/0092-8674(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 14.Liu SQ, Jin H, Zacarias A, Srivastava S, Bhatnagar A. Binding of pyridine nucleotide coenzymes to the β-subunit of the voltage-sensitive K+ channel. J. Biol. Chem. 2001;276:11812–11820. doi: 10.1074/jbc.M008259200. [DOI] [PubMed] [Google Scholar]
- 15.Tipparaju SM, Liu SQ, Barski OA, Bhatnagar A. NADPH binding to β-subunit regulates inactivation of voltage-gated K+ channels. Biochem. Biophys. Res. Commun. 2007;359:269–276. doi: 10.1016/j.bbrc.2007.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tipparaju SM, Saxena N, Liu SQ, Kumar R, Bhatnagar A. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am. J. Physiol Cell Physiol. 2005;288:C366–C376. doi: 10.1152/ajpcell.00354.2004. [DOI] [PubMed] [Google Scholar]
- 17.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J. Biol. Chem. 2006;281:15194–15200. doi: 10.1074/jbc.M513809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bahring R, Milligan CJ, Vardanyan V, Engeland B, Young BA, Dannenberg J, Waldschutz R, Edwards JP, Wray D, Pongs O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvbeta subunits. J Biol. Chem. 2001;276:22923–22929. doi: 10.1074/jbc.M100483200. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 21.Barski OA, Gabbay KH, Grimshaw CE, Bohren KM. Mechanism of human aldehyde reductase: characterization of the active site pocket. Biochemistry. 1995;34:11264–11275. doi: 10.1021/bi00035a036. [DOI] [PubMed] [Google Scholar]
- 22.Bhatnagar A, Liu SQ, Srivastava SK. Structure-activity correlations in human kidney aldehyde reductase-catalyzed reduction of para-substituted benzaldehyde by 3-acetyl pyridine adenine dinucleotide phosphate. Biochim. Biophys. Acta. 1991;1077:180–186. doi: 10.1016/0167-4838(91)90056-6. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava S, Spite M, Trent JO, West MB, Ahmed Y, Bhatnagar A. Aldose reductase-catalyzed reduction of aldehyde phospholipids. J. Biol. Chem. 2004;279:53395–53406. doi: 10.1074/jbc.M403416200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bligh EG, Dyer WJ. A Rapid method of total lipid extraction and purification. Can. J. of Biochem. and Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr., DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charton M. Steric Effects .8. Racemization of Chiral Biphenyls. Journal of Organic Chemistry. 1977;42:2528–2529. [Google Scholar]
- 27.Hansch C. a. L. A. Exploring QSAR: Fundamentals and Application in Chemistry and Biology. American Chemical Society; 1995. [Google Scholar]
- 28.Page MI. The Chemistry of Enzyme Action. Elsevier Science; New York: 1984. [Google Scholar]
- 29.Mayr P, Nidetzky B. Catalytic reaction profile for NADH-dependent reduction of aromatic aldehydes by xylose reductase from Candida tenuis. Biochem. J. 2002;366:889–899. doi: 10.1042/BJ20020080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohren KM, Grimshaw CE, Lai CJ, Harrison DH, Ringe D, Petsko GA, Gabbay KH. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021–2032. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- 31.Pawlowski JE, Penning TM. Overexpression and mutagenesis of the cDNA for rat liver 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase. Role of cysteines and tyrosines in catalysis. J Biol. Chem. 1994;269:13502–13510. [PubMed] [Google Scholar]
- 32.Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, Bhatnagar A, Srivastava S. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem. J. 2007;405:95–105. doi: 10.1042/BJ20061743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin Y, Penning TM. Multiple steps determine the overall rate of the reduction of 5alpha-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: implications for the elimination of androgens. Biochemistry. 2006;45:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wermuth B, Monder C. Aldose and aldehyde reductase exhibit isocorticosteroid reductase activity. Eur J Biochem. 1983;131:423–426. doi: 10.1111/j.1432-1033.1983.tb07280.x. [DOI] [PubMed] [Google Scholar]
- 35.Sokolova O, Accardi A, Gutierrez D, Lau A, Rigney M, Grigorieff N. Conformational changes in the C terminus of Shaker K+ channel bound to the rat Kvβ2-subunit. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12607–12612. doi: 10.1073/pnas.2235650100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohren KM, Page JL, Shankar R, Henry SP, Gabbay KH. Expression of human aldose and aldehyde reductases. Site-directed mutagenesis of a critical lysine 262. J Biol Chem. 1991;266:24031–24037. [PubMed] [Google Scholar]
- 37.O'Connor T, Ireland LS, Harrison DJ, Hayes JD. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem J. 1999;343:t-504. [PMC free article] [PubMed] [Google Scholar]
- 38.Vander JD, Robinson B, Taylor KK, Hunsaker LA. Reduction of trioses by NADPH-dependent aldo-keto reductases. Aldose reductase, methylglyoxal, and diabetic complications. J Biol Chem. 1992;267:4364–4369. [PubMed] [Google Scholar]
- 39.Barski OA, Papusha VZ, Kunkel GR, Gabbay KH. Regulation of aldehyde reductase expression by STAF and CHOP. Genomics. 2004;83:119–129. doi: 10.1016/s0888-7543(03)00213-1. [DOI] [PubMed] [Google Scholar]
- 40.Srivastava S, Chandra A, Bhatnagar A, Srivastava SK, Ansari NH. Lipid peroxidation product, 4-hydroxynonenal and its conjugate with GSH are excellent substrates of bovine lens aldose reductase. Biochem. Biophys. Res. Commun. 1995;217(3):741–746. doi: 10.1006/bbrc.1995.2835. [DOI] [PubMed] [Google Scholar]
- 41.Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- 42.Wang S, Gustafson E, Pang L, Qiao X, Behan J, Maguire M, Bayne M, Laz T. A novel hepatointestinal leukotriene B4 receptor. Cloning and functional characterization. J. Biol. Chem. 2000;275:40686–40694. doi: 10.1074/jbc.M004512200. [DOI] [PubMed] [Google Scholar]
- 43.Yoshimoto T, Takahashi Y. Arachidonate 12-lipoxygenases. Prostaglandins Other Lipid Mediat. 2002;68-69:245–262. doi: 10.1016/s0090-6980(02)00034-5. [DOI] [PubMed] [Google Scholar]
- 44.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 45.Tsakadze NL, Srivastava S, Awe SO, Adeagbo AS, Bhatnagar A, D'Souza SE. Acrolein-induced vasomotor responses of rat aorta. Am. J. Physiol Heart Circ. Physiol. 2003;285:H727–H734. doi: 10.1152/ajpheart.00269.2003. [DOI] [PubMed] [Google Scholar]
- 46.Bhatnagar A. Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ. Res. 1995;76:293–304. doi: 10.1161/01.res.76.2.293. [DOI] [PubMed] [Google Scholar]
- 47.Leitinger N, Watson AD, Faull KF, Fogelman AM, Berliner JA. Monocyte binding to endothelial cells induced by oxidized phospholipids present in minimally oxidized low density lipoprotein is inhibited by a platelet activating factor receptor antagonist. Adv. Exp. Med. Biol. 1997;433:379–382. doi: 10.1007/978-1-4899-1810-9_82. [DOI] [PubMed] [Google Scholar]
- 48.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3 α-hydroxysteroid dehydrogenase reveals a “push-pull” mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37(10):3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 49.Tarle I, Borhani DW, Wilson DK, Quiocho FA, Petrash JM. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J Biol. Chem. 1993;268:25687–25693. [PubMed] [Google Scholar]
- 50.Kratzer R, Kavanagh KL, Wilson DK, Nidetzky B. Studies of the enzymic mechanism of Candida tenuis xylose reductase (AKR 2B5): X-ray structure and catalytic reaction profile for the H113A mutant. Biochemistry. 2004;43:4944–4954. doi: 10.1021/bi035833r. [DOI] [PubMed] [Google Scholar]
- 51.Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Human aldose reductase: rate constants for a mechanism including interconversion of ternary complexes by recombinant wild-type enzyme. Biochemistry. 1995;34:14356–14365. doi: 10.1021/bi00044a012. [DOI] [PubMed] [Google Scholar]
- 52.Kubiseski TJ, Hyndman DJ, Morjana NA, Flynn TG. Studies on pig muscle aldose reductase. Kinetic mechanism and evidence for a slow conformational change upon coenzyme binding. J Biol. Chem. 1992;267:6510–6517. [PubMed] [Google Scholar]
- 53.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 54.Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309(5736):897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 55.Perez-Garcia MT, Lopez-Lopez JR, Gonzalez C. Kvβ1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to Kv4.2 but not to Shaker channels. J. Gen. Physiol. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.