

Abstract
Matricellular proteins play a unique role in the skeleton as regulators of bone remodeling, and the matricellular protein osteonectin (SPARC, BM-40) is the most abundant non-collagenous protein in bone. In the absence of osteonectin, mice develop progressive low turnover osteopenia, particularly affecting trabecular bone. Polymorphisms in a regulatory region of the osteonectin gene are associated with bone mass in a subset of idiopathic osteoporosis patients, and these polymorphisms likely regulate osteonectin expression. Thus it is important to determine how osteonectin gene dosage affects skeletal function. Moreover, intermittent administration of parathyroid hormone (PTH) (1-34) is the only anabolic therapy approved for the treatment of osteoporosis, and it is critical to understand how modulators of bone remodeling, such as osteonectin, affect skeletal response to anabolic agents. In this study, 10 week old female wild type, osteonectin-haploinsufficient, and osteonectin-null mice (C57Bl/6 genetic background) were given 80 μg/kg body weight/day PTH(1-34) for 4 weeks. Osteonectin gene dosage had a profound effect on bone microarchitecture. The connectivity density of trabecular bone in osteonectin-haploinsufficient mice was substantially decreased compared with that of wild type mice, suggesting compromised mechanical properties. Whereas mice of each genotype had a similar osteoblastic response to PTH treatment, the osteoclastic response was accentuated in osteonectin-haploinsufficient and osteonectin-null mice. Eroded surface and osteoclast number were significantly higher in PTH-treated osteonectin-null mice, as was endosteal area. In vitro studies confirmed that PTH induced the formation of more osteoclast-like cells in marrow from osteonectin-null mice compared with wild type. PTH treated osteonectin-null bone marrow cells expressed more RANKL mRNA compared with wild type. However, the ratio of RANKL:OPG mRNA was somewhat lower in PTH treated osteonectin-null cultures. Increased expression of RANKL in response to PTH could contribute to the accentuated osteoclastic response in osteonectin-/- mice, but other mechanisms are also likely to be involved. The molecular mechanisms by which PTH elicits bone anabolic vs. bone catabolic effects remain poorly understood. Our results imply that osteonectin levels may play a role in modulating the balance of bone formation and resorption in response to PTH.
Keywords: PTH; Skeletal remodeling; Matricellular protein; Bone matrix; SPARC, BM-40
Introduction
The “matricellular” protein family consists of extracellular matrix glycoproteins that play a predominant role in the modulation of cell-matrix interactions, as well as contributing to matrix organization. These proteins are expressed at high levels in development and in response to injury, generally induce cell de-adhesion, and interact with cell surface receptors, extracellular matrix, growth factors, cytokines and proteases [1]. Recent studies demonstrate that matricellular proteins play a unique role in the skeleton as regulators of bone remodeling. Matricellular proteins important in bone include osteopontin, bone sialoprotein, tenascin C, thrombospondins 1 and 2, and osteonectin or SPARC (secreted protein acidic and rich in cysteine; BM-40). These proteins interact with the extracellular matrix, regulating bone matrix deposition, assembly, and mineralization. Selected matricellular proteins have been shown to interact with growth factors, cytokines and proteases, modulating the binding of growth factors to receptors and promoting the activation of metalloproteinases [reviewed in 2, 3]. Further, the interaction of matricellular proteins with cell surface molecules, including integrins and their associated proteins, modulates signaling from down stream effectors [2, 4, 5]. The process of bone remodeling allows the skeleton to respond to mechanical and physiologic stresses, and matricellular proteins play an important role in skeletal remodeling induced by fracture repair, unloading, and estrogen depletion [2].
Although widely expressed in mammalian tissues, the matricellular protein osteonectin is the most abundant noncollagenous protein in bone [3, 6]. In the absence of osteonectin, mice develop progressive low turnover osteopenia. Trabecular bone volume is dramatically decreased, while cortical bone has compromised matrix quality [7, 8]. In vivo, osteonectin-null mice have decreased osteoblast and osteoclast numbers and surface, as well as decreased bone formation rate. Further, osteonectin suppresses adipogenesis, and in vitro studies indicate that osteonectin promotes osteoblastic commitment, differentiation, and survival [3, 9, 10].
We found that marrow stromal cells from wild type and osteonectin-null mice were not different in their ability to produce cAMP in response to parathyroid hormone (PTH) in vitro, however this response to PTH was attenuated in cultured osteonectin-null osteoblastic cells [10]. Nonetheless, the role of osteonectin in PTH-stimulated bone remodeling in vivo remains unknown. Since intermittent administration of the amino-terminal fragment of PTH(1-34) is the only anabolic therapy approved for the treatment of osteoporosis, it is critical to understand how modulators of bone remodeling, such as osteonectin, may affect skeletal response to anabolic agents [11].
Our previous studies of osteonectin-null mice showed that osteonectin is critical for normal bone remodeling. However, the impact of osteonectin-haploinsufficiency on skeletal biology has not yet been determined. It is known that osteonectin expression is decreased in osteoblasts from patients with osteogenesis imperfecta [12, 14]. Further, haplotypes consisting of 3 single nucleotide polymorphisms (SNPs) in the 3′ untranslated region (UTR) of the osteonectin gene have been associated with bone mass in a subset of idiopathic osteoporosis patients [14]. The 3′ UTR regulates mRNA stability, trafficking, and translation, and it is likely that these 3′ UTR SNPs differentially regulate osteonectin expression [15, 16]. Therefore, it is important to determine how osteonectin gene dosage affects skeletal function. In this study, we characterized the response of wild type (+/+), osteonectin-haploinsufficient (+/-) and osteonectin-null (-/-) mice to intermittent treatment with PTH(1-34). The goal of this study was to determine the impact of osteonectin gene dosage on the response of the skeleton to an anabolic therapy.
Methods and Materials
Animals
The osteonectin-/- mice used in these studies were back crossed 8 times into the C57/Bl6 genetic background [10]. Wild type mice of similar genetic background were also maintained. Osteonectin+/- mice were generated by crossing wild type and osteonectin-/- animals. Mice were maintained under standard non-barrier conditions and had access to mouse chow and water ad libitum. Female mice received intraperitoneal injections of calcien (10 mg/kg) and demeclocycline (30 mg/kg) 10 and 3 days prior to euthanasia, respectively [17]. Femurs and vertebrae were dissected and fixed in 70% ethanol. Studies were performed only on females because females experience greater age-related bone loss than males [18]. All studies were approved by the Institutional Animal Care and Use Committee at the University of Connecticut Health Center.
For the intermittent PTH administration protocol, 10 week old female mice were injected subcutaneously with 80 μg/kg/day rhPTH(1-34) (PTH) (Bachem, Torrance, CA) or vehicle alone (2% heat-inactivated osteonectin-/- mouse serum in acidified saline [0.1N]). Mice were treated 5 days per week, for 4 weeks [19]. Mice were weighed weekly, and the dose of PTH was adjusted as required.
Bone Mineral Density (BMD)
Whole body BMD and BMC (bone mineral content) (excluding the skull) at baseline and after 4 weeks of PTH treatment was measured by DEXA (PIXImus, GE-Lunar, Madison, WI). Using the “region of interest” tool with the PIXImus software, BMD and BMC for the diaphysial region of the left femur, and for L3+L4 vertebrae were also determined. 7-8 mice per group were analyzed.
microCT Analysis
Trabecular morphometry within the metaphyseal region of distal femurs and centrum of the third lumbar vertebrae (L3), and cortical morphometry of femora at mid-diaphysis, was quantified using conebeam micro-focus X-ray computed tomography (μCT40, Scanco Medical AG, Bassersdorf, Switzerland). Serial tomographic images were acquired at 55 kV and 145 μA, collecting 1000 projections per rotation at 300 msec integration time. Three-dimensional 16-bit grayscale images were reconstructed using standard convolution back-projection algorithms with Shepp and Logan filtering, and rendered within a 12.3 mm field of view at a discrete density of 578,704 voxels/mm3 (isometric 12 μm voxels). Segmentation of bone from marrow and soft tissue was performed in conjunction with a constrained Gaussian filter to reduce noise, applying a hydroxyapatite-equivalent density threshold of 470 mg/cm3. Volumetric regions for trabecular analysis were selected within the endosteal borders to include the central 80% of vertebral height and secondary spongiosa of femoral metaphyses located 960 μm (6% of length) from the growth plate and extending 1 mm proximally. Trabecular morphometry was characterized by measuring the bone volume fraction, trabecular thickness, trabecular number, trabecular spacing, and trabecular connectivity density, calculated as a function of the three-dimensional Euler number normalized to volume. Cortical morphometry in the femur was quantified within a 600 μm long segment (50 serial sections) extending distally from the diaphyseal mid-point between proximal and distal growth plates. Cross-sectional measurements included average periosteal area (cortical bone + marrow), endosteal area (marrow), and cortical bone area. 7-8 mice per group were analyzed.
Histomorphometry
Undecalcified femurs were embedded in methyl methacrylate. 5 μm thick longitudinal sections were cut on a microtome (Polycut S, Leika Heidelberg, Germany) and stained with toluidine blue (pH 6.4). Static parameters of bone structure, formation and resorption were measured at the distal metaphyses (magnification × 250), 195 μm from the growth plate, in a total of 20 fields using an OsteoMeasure morphometry system (Osteometrics, Atlanta, USA). The ratio of osteoblast number to osteoclast number (ObN/BPm/OcN/BPm) was calculated for each mouse, and group means are reported. Dynamic bone parameters were obtained from unstained 10 μm sections examined by fluorescent light microscopy (Nikon, Tokyo, Japan). The mineral apposition rate was expressed in micrometers per day, and bone formation rate was expressed per unit of bone surface. The terminology and units used are those recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research. 4-8 mice per group were analyzed.
In vitro osteoclast-like cell formation
Marrow cells were harvested from the femur and tibia of female mice at 6-8 weeks of age. Cells were plated at 1.55 × 105 cells/cm2 and cultured in αMEM containing 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals, Norcross, GA) for up to 6 days. Cells were re-fed with fresh medium and test compounds on day 3 [20]. Cells were treated with 0, 1, or 10 nM rhPTH(1-34) or with 0, 1, 10 or 30 ng/ml RANKL (receptor activator of NF-kB ligand; recombinant mouse; R & D Systems, Minneapolis, MN) in the presence of 30 ng/ml M-CSF (macrophage colony stimulating factor; recombinant mouse; Sigma, Saint Louis, MO). Formation of osteoclast-like (OCL) cells was determined by staining for TRAP (tartrate-resistant acid phosphatase) activity (leukocyte acid phosphatase kit; Sigma). TRAP-positive cells containing more than 3 nuclei were scored as OCL. RNA was isolated from cultures using Nucleospin columns (BD Biosciences, Palo Alto, CA), and levels of RNA for RANKL, OPG (osteoprotegerin), and 18S rRNA were determined by qRT-PCR (Applied Biosystems, ABI Prism 7300, Foster City, CA). The relative standard curve method was used to determine the quantity of target transcripts, and mRNA levels for RANKL and mOPG were normalized to 18S. For determination of OCL formation, 6 replicate wells per group were used, and each experiment was repeated at least twice, with comparable results. For determination of RNA levels, 3 replicates were used, and the experiment was performed twice, with comparable results.
Data analysis
Data are presented as mean ± SEM. Data were analyzed by Student’s t test or one-way ANOVA with Bonferroni post-hoc test as appropriate.
Results
BMD and BMC
At 10 weeks of age (i.e. base line), osteonectin-/- mice had significantly lower whole body BMD compared with osteonectin+/- or wild type mice (44.2 ± 0.3 vs. 45.9 ± 0.4 and 46.8 ± 0.4 mg/cm2, respectively; p ≤ 0.01). Wild type and osteonectin+/- mice treated with PTH for 4 weeks had a 13% increase in whole body BMD, whereas PTH treated osteonectin-/- mice had whole body BMD values similar to those observed in vehicle treated mice (Figure 1A). However, significant increases in bone mineral content (BMC) were observed in both wild type and osteonectin-/- mice treated with PTH (Figure 1B). Examination of cortical bone in the diaphysis of the femur showed that PTH treatment increased BMD and BMC in mice of each genotype, although the greatest fold change was observed in osteonectin-/- mice (Figure 1C and D). There was a trend toward a PTH-induced increase in BMD and BMC in the cortical and trabecular bone of vertebrae in mice of each genotype (Figure 1E and F).
Figure 1.

Percent change in BMD and BMC (vs. 10 week baseline) in vehicle and PTH treated wild type, osteonectin+/- and osteonectin-/- mice. A. Whole body BMD; B. Whole body BMC; C. Femoral diaphysis BMD; D. Femoral diaphysis BMC; E. Vertebral (L3+L4) BMD; E. Vertebral (L3+L4) BMC. * = significantly different from corresponding vehicle treated, p ≤ 0.05.
Vertebrae
At 10 weeks of age, osteonectin-/- mice had significantly lower trabecular bone volume compared with osteonectin+/- or wild type mice (Figure 2B, Table 1). From 10 to 14 weeks of age, trabecular bone volume did not significantly change in vehicle treated wild type and osteonectin+/- mice. In contrast, osteonectin-/- mice lost vertebral bone volume during this interval, due to a decrease in trabecular number (Table 1, Figure 2B and D). Trabecular connectivity density decreased in vehicle treated mice of each genotype between 10 and 14 weeks of age (Figure 2E). However bone microarchitecture was most strikingly compromised in the absence of osteonectin, as connectivity density was decreased by ∼50% in osteonectin-/- mice. Overall, these data indicate that the young osteonectin-/- mice lose trabecular bone faster than wild type mice.
Figure 2.

Trabecular bone parameters in vertebrae of vehicle and PTH treated wild type, osteonectin+/- and osteonectin-/- mice. A. Representative microCT images from 14 week old mice. Percent change (vs. 10 week baseline) in B. Trabecular bone volume; C. Trabecular thickness; D. Trabecular number; E. Trabecular connectivity density. * = significantly different from corresponding vehicle treated, p ≤ 0.02. # = significantly different from vehicle treated wild type, p ≤ 0.02.
Table 1.
μCT analysis of trabecular bone parameters in vertebrae (mean ± SEM). of N = 7-8 mice per group
| Wild type | Haploinsufficient | Null | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | 10 wk | 14 wk Vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH |
| Bone volume fraction (BV/TV) | 23.2 ± 0.9 | 24.3 ± 1.3 | 30.0 ± 0.8 b | 18.3 ± 0.8 | 20.2 ± 0.7 c | 26.2 ± 0.7 b | 14.2 ± 0.3 a | 10.7 ± 0.4 c, a | 13.9 ±0.5 b |
| Trabecular thickness (μm) | 48.2 ± 0.7 | 51.8 ± 0.7 | 52.2 ± 0.8 | 44.8 ± 1.7 | 50.5 ± 1.1 | 52.5 ± 1.1 | 45.0 ± 0.5 | 46.3 ± 0.8 c | 50.9 ± 1.1 b |
| Trabecular number (per mm) | 5.01 ± 0.90 | 4.58 ± 0.14 | 5.39 ± 0.17 b | 4.61 ± 0.07 | 4.07 ± 0.10 c, a | 4.80 ± 0.79 | 3.86 ± 0.03 | 3.14 ±0.05 c, a | 3.36 ± 0.09 |
| Trabecular spacing (μm) | 193 ± 3 | 213 ± 8 | 187 ± 6 b | 211 ± 4 | 241 ± 7 c | 200 ± 3 b | 252 ± 6 | 315 ± 6 c, a | 289 ± 7 b |
| Connectivity density (per mm3) | 201 ±11 | 154 ± 9 a | 481 ± 18 b, a | 165 ± 3 | 130 ±6 a, c | 292 ± 28 b | 144 ± 7 | 63 ± 3 c, a | 86 ± 5 b |
significantly different from corresponding 10 week, p ≤ 0.02
significantly different from corresponding vehicle treated, p ≤ 0.02
significantly different from vehicle treated wild type, p ≤ 0.02
PTH treatment caused a modest (1.2 to 1.3 fold), but significant increase in vertebral trabecular bone volume in mice of each genotype between 10 to 14 weeks of age (Table 1). This resulted in a 30-40% net gain in bone volume for PTH treated wild type and osteonectin+/- mice, due primarily to increases in trabecular number (Figures 2B and 2D, Table 1). However, a net increase in trabecular bone volume during this interval was not seen in PTH treated osteonectin-/- mice, since the vehicle-treated animals lost a substantial amount of bone, and PTH treatment only abrogated this loss (Figure 2B, Table 1). Further, PTH treatment dramatically improved connectivity density in wild type and osteonectin+/- mice, increasing this parameter compared with vehicle treated mice and with the 10 week baseline. In contrast, connectivity density was decreased by 14 weeks in osteonectin-/- mice, regardless of PTH treatment (Figure 2E, Table 1).
Femur - Trabecular Bone
In the femur, mice of each genotype had a decrease in trabecular number from 10 to 14 weeks of age (Figure 3C, Table 2). However, this effect was more pronounced in osteonectin+/- and osteonectin-/- mice, contributing to decreases in trabecular bone volume (Figure 3A, Table 2). The trabecular connectivity density was unchanged between 10 and 14 weeks in wild type mice, but this parameter was decreased nearly 50% in osteonectin+/- and osteonectin-/- mice (Figure 3D). Overall, data from femur and vertebrae confirm that young osteonectin+/- and osteonectin-/- mice lose trabecular bone faster than wild type mice.
Figure 3.

Trabecular bone parameters in femur of vehicle and PTH treated wild type, osteonectin+/- and osteonectin-/- mice. Percent change (vs. 10 week baseline) in A. Trabecular bone volume; B. Trabecular thickness; C. Trabecular number; D. Trabecular connectivity density. * = significantly different from corresponding vehicle treated, p ≤ 0.02. # = significantly different from vehicle treated wild type, p ≤ 0.02.
Table 2.
μCT analysis of trabecular bone parameters in femur (mean ± SEM). N = 7-8 mice per group
| Wild type | Haploinsufficient | Null | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | 10 wk | 14 wk Vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH |
| Bone volume fraction (BV/TV, %) | 9.0 ±0.5 | 8.7 ± 0.7 | 17.0 ± 2.2 b | 11.4 ± 0.01 | 8.3 ± 0.3 a | 17.6 ± 1.9 b | 9.5 ± 1.0 | 7.6 ± 0.7 | 11.6 ± 1.1 b |
| Trabecular thickness (μm) | 44.6 ± 1.1 | 46.9 ± 1.0 | 56.5 ± 2.1 b | 45.2 ± 1.84 | 49.3 ± 1.9 | 59.5 ± 1.9 b | 43.0 ± 0.6 | 48.4 ± 1.5 | 56.2 ± 1.7 b |
| Trabecular number (per mm) | 4.30 ± 0.08 | 4.01 ± 0.06 a | 4.51 ± 0.16 b | 4.66 ± 0.13 | 3.95 ± 0.08 a | 4.59 ± 0.15 b | 4.24 ± 0.08 | 3.53 ± 0.16 c, a | 3.41 ± 0.08 |
| Trabecular spacing (μm) | 233 ± 5 | 247 ± 4 | 220 ± 7 | 214 ± 7 | 253 ± 5 | 223 ± 8 | 237 ± 5 | 289 ± 15 a | 305 ± 7 |
| Connectivity density (per mm3) | 79 ± 5 | 81 ± 7 | 183 ± 26 b | 114 ± 15 | 58 ± 6 c, a | 162 ± 16 b | 96 ± 14 | 52 ± 6c | 133 ± 26 b |
significantly different from corresponding 10 week, p ≤ 0.02
significantly different from corresponding vehicle treated, p ≤ 0.02
significantly different from vehicle treated wild type, p ≤ 0.03
Although PTH increased trabecular bone volume in the femur of mice of each genotype, the net increase in trabecular bone volume with PTH was significantly diminished in osteonectin-/- mice compared with wild type (p<0.05) (Figure 3A, Table 2). As seen in the vertebrae, PTH protected wild type and osteonectin+/- mice from loss of trabecular number from 10 to 14 weeks of age, but this effect was not seen in osteonectin-/- mice (Figure 3C, Table 2). Similarly, PTH did not significantly improve connectivity density in osteonectin-/- mice (Figure 3D, Table 2)
Next, histomorphometry was used to analyze bone remodeling and structural parameters in the femur, comparing vehicle and PTH treated mice at 14 weeks of age (Table 3). 10 week old mice were not examined by histomorphometry in this study. In mice of each genotype, PTH increased trabecular bone volume and trabecular number in by 1.3 to 1.6 fold, and increased osteoblast number and surface by 1.6 to 2 fold (Table 3). However, significant differences in mineral apposition rate (MAR) and bone formation rate (BFR) were not detected in PTH treated mice of any genotype (Figure 4A, Table 3). Rather, a significant increase in osteoid volume and mineralization lag time was observed in PTH treated mice of each genotype, and the fold increase in these parameters was highest in osteonectin-/- mice (Figure 4B and 4C, Table 3). Thus, PTH treatment increased osteoblast number and matrix synthesis, but this matrix was not mineralizing appropriately.
Table 3.
Histomorphometric analysis of trabecular bone parameters in femur (mean ± SEM). N = 4-8 mice per group
| Wild type | Haploinsufficient | Null | ||||
|---|---|---|---|---|---|---|
| Parameter | 14 wk vehicle | 14 wk PTH | 14 wk vehicle | 14 wk PTH | 14 wk vehicle | 14 wk PTH |
| Structure | ||||||
| Bone volume (BV/TV, %) | 7.14 ± 0.49 | 11.70 ± 0.85 a | 6.40 ± 0.51 | 9.85 ± 0.74 | 4.20 ± 0.46 b | 5.99 ± 0.62 a |
| Trabecular thickness (μm) | 29.14 ± 1.45 | 33.48 ± 1.70 | 26.63 ± 1.11 | 32.55 ± 1.78 | 26.82 ± 1.45 | 29.90 ± 2.01 |
| Trabecular number (per mm) | 2.43 ± 0.15 | 3.47 ± 0.17 a | 2.40 ± 0.14 | 3.05 ± 0.22 | 1.52 ± 0.13 b | 1.97 ± 0.13 |
| Trabecular spacing (μm) | 379 ± 27 | 259 ± 16 | 407 ± 24 | 313 ± 29 | 678 ± 73 b | 503 ± 39 a |
| Formation | ||||||
| Osteoblast surface (ObS/BS, %) | 19.87 ± 2.74 | 33.00 ± 2.23 a | 18.14 ± 2.54 | 29.75 ± 1.57 a | 15.87 ± 2.01 | 26.33 ± 1.97 a |
| Osteoblast number (NOb/BPm, /mm2) | 12.71 ± 1.86 | 21.33 ± 1.68 a | 11.45 ± 1.66 | 16.93 ± 0.62 | 10.51 ± 1.52 | 19.91 ± 1.71 a |
| Mineral apposition rate (MAR, μm/day) | 0.41 ± 0.01 | 0.42 ± 0.03 | 0.44 ± 0.05 | 0.50 ± 0.04 | 0.41 ± 0.06 | 0.36 ± 0.05 |
| Mineralizing surface (MS/BS, %) | 11.11 ± 1.13 | 13.70 ± 1.17 | 12.33 ± 1.65 | 10.99 ± 2.14 | 10.38 ± 2.21 | 11.78 ± 0.95 |
| Bone formation rate (BFR/BS, μm3/μm2/day) | 0.038 ± 0.008 | 0.050 ± 0.009 | 0.063 ± 0.013 | 0.053 ± 0.011 | 0.031 ± 0.008 | 0.059 ± 0.014 |
| Mineralization Lag Time (MLT, days) | 3.95 ± 0.49 | 11.29 ± 1.64 a | 5.17 ± 1.19 | 12.70 ± 3.49 a | 3.62 ± 0.98 | 14.51 ± 1.29 a |
| Osteoid Volume (OV/BV, %) | 2.01 ± 0.35 | 3.47 ± 0.21 a | 2.52 ± 0.39 | 4.07 ± 0.52 a | 1.85 ± 0.34 | 4.90 ± 0.61 a |
| Resorption | ||||||
| Eroded surface (%) | 9.39 ± 0.87 | 11.20 ± 1.90 a | 6.31 ± 1.04 | 11.99 ± 1.11 a | 4.71 ± 0.86 | 9.72 ± 1.30 a |
| Osteoclast surface (OcS/BS, %) | 1.79 ± 0.27 | 2.91 ± 0.36 | 1.85 ± 0.45 | 3.40 ± 0.48 | 1.54 ± 0.27 | 3.05 ± 0.40 |
| Osteoclast number (NOc/BPm, /mm2) | 0.83 ± 0.13 | 1.15 ± 0.12 | 0.79 ± 0.37 | 1.39 ± 0.78 a | 0.69 ± 0.11 | 1.40 ± 0.19 a |
significantly different from corresponding vehicle treated, p ≤ 0.05
significantly different from vehicle treated wild type, p ≤ 0.05
Figure 4.

Histomorphometric evaluation of trabecular bone parameters in femur of vehicle and PTH treated wild type, osteonectin+/- and osteonectin-/- mice. A. Mineral appostition rate; B. Mineralization lag time; C. Osteoid volume; D. Ratio of osteoclast number to osteoblast number; E. Eroded surface. * = significantly different from vehicle treated, p ≤ 0.05.
In contrast to effects on bone formation parameters, the impact of PTH on bone resorption parameters was distinctly different with osteonectin genotype. PTH increased eroded surface ∼2 fold in osteonectin+/- and -/- mice, compared with a 1.2 fold increase in wild type (Figure 4E, Table 3). PTH had similar effects on osteoclast number. Indeed, the ratio of osteoblasts to osteoclasts was increased in PTH-treated wild type mice, but not in osteonectin+/- and -/- mice, due to primarily to increases in osteoclast number (Figure 4D and Table 3).
Femur - Cortical Bone
At 10 weeks of age, overall cortical bone area was similar in mice of each genotype (Table 4). However, osteonectin-/- mice had decreased periosteal and endosteal dimensions compared with wild type animals (p<0.05 vs wild type). Between 10 and 14 weeks of age, bone area in vehicle treated wild type and osteonectin+/- mice was significantly increased, but a significant increase in this parameter was not seen in osteonectin-/- mice (Table 4). At 14 weeks of age, vehicle treated osteonectin-/- mice continued to display lower periosteal and endosteal area, contributing to the lower bone area compared with wild type animals (Table 4).
Table 4.
μCT analysis of cortical bone parameters in femur (mean ± SEM). N = 7-8 mice per group
| Wild type | Haploinsufficient | Null | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | 10 wk | 14 wk vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH | 10 wk | 14 wk vehicle | 14 wk PTH |
| Periosteal area (mm2) | 1.65 ± 0.06 | 1.65 ± 0.23 | 1.70 ± 0.03 | 1.60 ± 0.04 | 1.60 ± 0.31 | 1.75 ± 0.03 b | 1.46 ± 0.04 | 1.47 ± 0.03 c | 1.60 ± 0.02 b |
| Endosteal area (mm2) | 0.88 ± 0.03 | 0.85 ± 0.02 | 0.81 ± 0.02 | 0.85 ± 0.03 | 0.79 ± 0.02 | 0.84 ± 0.02 b | 0.71 ± 0.03 | 0.68 ± 0.02 c | 0.73 ± 0.03 b |
| Bone area (mm2) | 0.68 ± 0.03 | 0.76 ± 0.01 a | 0.82 ± 0.02 b | 0.68 ± 0.02 | 0.76 ± 0.02 a | 0.86 ± 0.02 b | 0.68 ± 0.01 | 0.73 ± 0.01 c | 0.82 ± 0.02 b |
significantly different from corresponding 10 week, p ≤ 0.04
significantly different from corresponding vehicle treated, p ≤ 0.02;
significantly different from vehicle treated wild type, p ≤ 0.02
PTH significantly increased cortical bone area in mice of each genotype by 21-26% (Figure 5A, Table 4). However, the mechanisms underlying these changes in bone area were different with genotype. In wild type mice, there was a trend toward increased periosteal area and decreased endosteal area with PTH treatment. In osteonectin+/- mice, PTH caused a significant increase in periosteal area, and a trend toward decreased endosteal area. In contrast, PTH significantly increased periosteal area, with a trend toward increased endosteal area in osteonectin-/- mice. The increase in endosteal area with PTH treatment suggests higher osteoclastic activity in cortical bone of osteonectin-/- mice.
Figure 5.
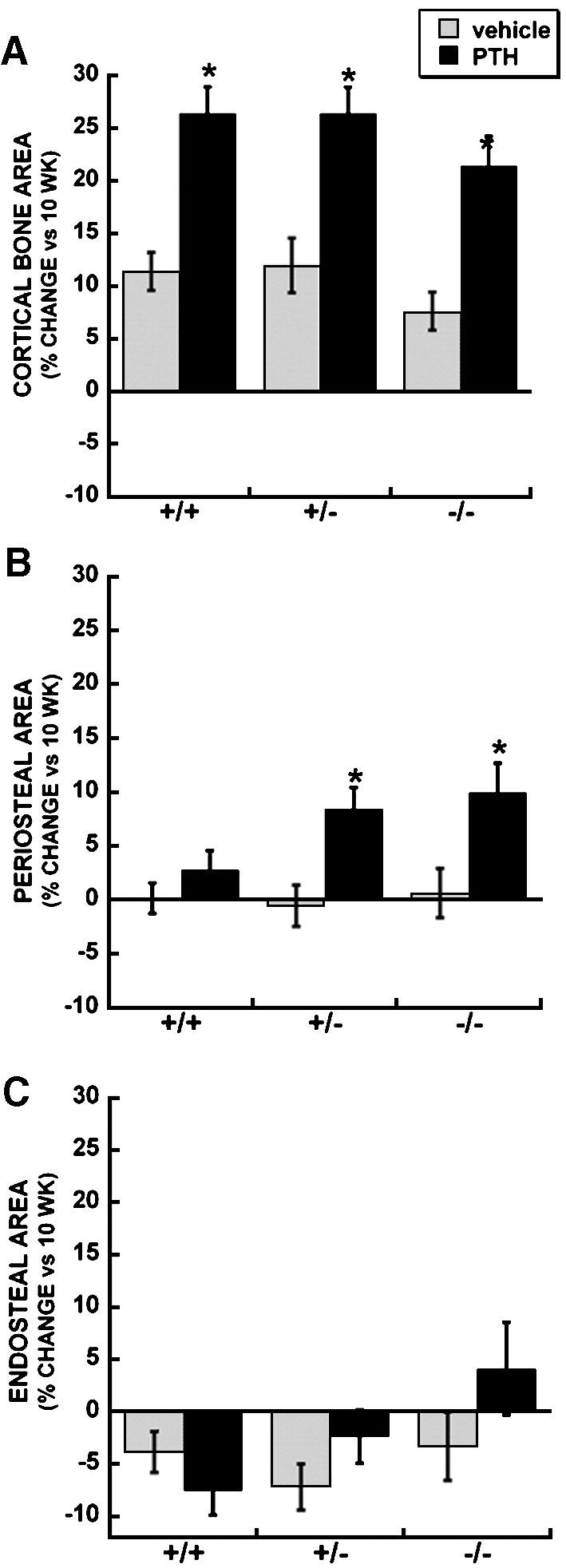
Cortical bone parameters in femur of vehicle and PTH treated wild type, osteonectin+/- and osteonectin-/- mice. Percent change (vs. 10 week baseline) in A. Bone area; B. Periosteal area; C. Endosteal area. * = significantly different from corresponding vehicle treated, p ≤ 0.02.
Osteoclast formation in vitro
The preceding in vivo data suggest that osteonectin-null mice may have increased osteoclastic activity in the basal state and in response to intermittent PTH treatment. To confirm that osteonectin-/- mice make greater numbers of osteoclastic cells in response to PTH, bone marrow from wild type, osteonectin+/- and -/- mice was cultured with increasing doses of PTH for 6 days, and multinucleated, TRAP-positive cells were scored (Figure 6). Treatment with 1 nM PTH caused a 5 fold increase in the formation of osteoclast-like cells in marrow of all genotypes (although the increase was significant only for osteonectin-/-). However, treatment of osteonectin+/- and -/- marrow with 10 nM PTH resulted in the generation of significantly greater numbers of osteoclastic cells compared with wild type, ∼9 and ∼12 fold respectively (Figure 6).
Figure 6.
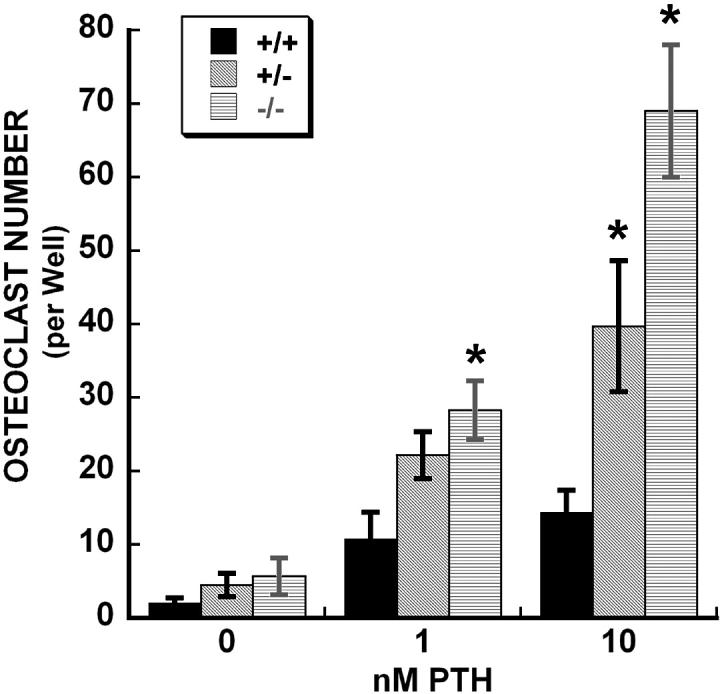
Formation of osteoclast-like cells in bone marrow from wild type, osteonectin+/- and osteonectin-/- mice cultured in vitro with increasing doses of PTH for 6 days. * = significantly different from corresponding wild type, p ≤ 0.05.
A principal mechanism by which PTH stimulates osteoclastogenesis is by increasing the expression of RANKL and decreasing expression of OPG [21, 22]. To determine whether marrow cells from wild type and osteonectin-/- mice exhibit differences in the regulation of RANKL and OPG by PTH, RNA was extracted from bone marrow cells cultured with increasing doses of PTH for 6 days. We chose to only examine wild type and osteonectin-/- cells because the osteonectin+/- cells appear to have an intermediate phenotype. RNA levels for RANKL, OPG, and 18S rRNA were determined by real time RT-PCR. Basal levels of OPG and RANKL mRNA, normalized for 18S, were not significantly different between wild type and osteonectin-/- marrow cultures (for OPG: 2.83 ± 0.85 and 1.58 ± 0.31 respectively, for RANKL: 1.04 ± 0.17 and 0.63 ± 0.04 respectively). PTH decreased the expression of OPG mRNA in wild type cells at all doses tested, but this inhibitory effect was diminished osteonectin-/- cultures (Table 5). In contrast, a significant increase in RANKL mRNA was only observed in wild type cells treated with the highest dose of PTH, whereas PTH induced higher levels of RANKL mRNA in osteonectin-/- marrow cultures at all doses tested. PTH treatment dramatically increased the ratio of RANKL:OPG mRNA in both wild type and osteonectin-/- marrow cells. However, this increase was less pronounced in osteonectin-/- cells, because OPG mRNA levels remained comparatively high in this group (Table 5).
Table 5.
OPG and RANKL mRNA levels (relative to 0 nM PTH) in bone marrow cells cultured for 6 days with up to 10 nM PTH (mean ± SEM). N = 3
| Wild type | Null | |||||
|---|---|---|---|---|---|---|
| mRNA | 0 nM PTH | 1 nM PTH | 10 nM PTH | 0 nM PTH | 1 nM PTH | 10 nM PTH |
| OPG | 100 ± 30 | 14 ± 1 a | 9 ± 0.5 a | 100 ± 19 | 41 ± 2 | 30 ± 2 |
| RANKL | 100 ± 16 | 175 ± 26 | 360 ± 19 a | 100 ± 6 | 255 ± 27 a | 540 ± 37 a, b |
| RANKL:OPG (absolute ratio) | 0.41 ± 0.07 | 4.49 ± 0.64 a | 15.27 ± 0.70 a | 0.43 ± 0.07 | 2.46 ± 0.14 a | 7.19 ± 0.11 a, b |
significantly different from corresponding 0 nm PTH, p ≤ 0.02
significantly different from corresponding wild type, p ≤ 0.01
To determine whether osteonectin-/- bone marrow cells were more responsive to RANKL, cells were cultured with 0, 1, 10 or 30 ng/ml RANKL in the presence of 30 ng/ml M-CSF for 5 days, and formation of osteoclast-like cells was quantified. Osteoclast formation was not detected in the absence of RANKL, but 1 ng/ml RANKL stimulated osteoclast formation in all cultures. Compared with 1 ng/ml, the 10 ng/ml dose of RANKL significantly increased osteoclast-like cell number by 1.4 fold in both wild type and osteonectin-/- cells (p<0.01 vs 1 ng/ml). However, the 30 ng/ml RANKL dose did not significantly increase osteoclast formation relative to the 10 ng/ml dose in any of the cultures. Since osteonectin-/- bone marrow cells do not appear to be differentially responsive to RANKL, we determined whether wild type and osteonectin-/- marrow may contain a different number of osteoclastic precursors. For these experiments, 50,000 marrow cells were cultured for 5 days with 30 ng/ml each RANKL and M-CSF, doses known to drive all potential osteoclast precursors to form osteoclast-like cells [23]. We found that wild type marrow produced 195 ± 15 osteoclastic cells, whereas osteonectin-/- marrow produced 239 ± 10, suggesting, at most, a ∼20% increase in osteoclast precursors in the mutant marrow (p=0.03).
Discussion
This study provides 3 novel insights into the function of the matricellular protein osteonectin in the skeleton. First, we find that although young (i.e. 10-14 week old) osteonectin+/- mice have trabecular bone volume and cortical bone parameters similar to that of wild type mice, osteonectin gene dosage has a profound effect on bone microarchitecture. The connectivity density of trabecular bone in the vertebrae and femur of osteonectin+/- mice is substantially decreased compared with that of wild type mice, suggesting compromised mechanical properties (Figures 2 and 3). Second, we found that young osteonectin+/- and osteonectin-/- mice lose trabecular bone faster than wild type mice (Figures 2 and 3). This is note worthy because healthy, young adult women and men exhibit substantial, early-onset trabecular bone loss, and the underlying mechanisms remain unexplained [24]. It is possible that osteonectin levels may play a role in this phenomenon. Third, we demonstrate that whereas wild type, osteonectin+/-, and osteonectin-/- mice display a similar osteoblastic response to intermittent PTH treatment, the osteoclastic response to PTH is accentuated in osteonectin+/- and -/- mice (Figure 4). This observation highlights a previously unknown function of osteonectin, to regulate osteoclast formation in vivo and in vitro. Overall, these data indicate that osteonectin impacts response to bone remodeling stimuli.
In our earlier characterization of the skeletal phenotype of osteonectin-/- mice, we concluded that the low turnover osteopenia observed in vivo resulted primarily from defects in bone formation rate, since osteoclastic parameters were decreased in conjunction with osteoblastic parameters [7]. Further, our in vitro studies support the idea that osteonectin-/- osteoblasts do not fully differentiate and function normally [3, 8]. Although we previously showed that osteonectin-/- osteoblasts synthesize less cAMP in response to stimulation with PTH, it is important to consider that the biologic response of bone cells to PTH is regulated at multiple levels and mediated by multiple signaling pathways. Whereas coupling of PTH1R (PTH/PTH related peptide receptor) to Gαs leads to the stimulation of adenylyl cyclase and down stream signaling via protein kinase C, its coupling to Gαq leads to stimulation of phospholipase C, and its coupling to Gα12/Gα13 leads to stimulation of phospholipase D [25, 26]. Further, responsiveness to PTH is modulated by expression level of the PTHR1 itself and by the recycling of the receptor, regulated by G protein-coupled receptor kinases and β-arrestins [27-29]. The molecular mechanisms by which PTH regulates bone formation and resorption in osteonectin-/- mice warrant additional study.
Using intermittent PTH as a remodeling challenge in vivo, our data indicate that the osteoblastic response to PTH is comparable or even increased in osteonectin-/- mice (Tables 3 and 4). Despite this response, osteonectin-/- mice fail to obtain a significant increase in BMD with intermittent PTH, owing to increased osteoclastic activity. In trabecular bone of the femur, the fold increase in osteoclast number and eroded surface with PTH treatment is greater in osteonectin+/- and -/- mice (Figure 4). In cortical bone of osteonectin-/- mice, PTH treatment significantly increased periosteal area, further indicating more osteoclastic activity (Figure 5). The decrease in trabecular number and the increase in trabecular spacing with age in normal female C57Bl/6 mice indicate complete removal of trabecular plates, a process requiring aggressive bone resorption (Tables 1 and 2) [18]. This process occurs at a faster rate in the osteonectin+/- and -/- mice, further suggesting increased osteoclastic response in these mutant animals.
Our in vitro data indicate that PTH treated osteonectin-/- bone marrow cells express more RANKL mRNA compared with wild type, although the relative ratio of RANKL:OPG mRNA is actually lower in PTH treated osteonectin-/- marrow cultures (Table 6). Increased expression of RANKL in response to PTH could contribute to the increased osteoclast formation and activity in osteonectin-/- mice. However, other mechanisms, possibly modulating the amplitude of response to RANKL signaling, are likely to be involved [30].
Although osteonectin mRNA it is expressed by some myelo-macrophage cell lines and lymphoma cells, to our knowledge, osteonectin has not been reported to be expressed by osteoclasts [31]. Thus, other mechanisms whereby osteoclast formation is increased in osteonectin-/- mice may involve the direct interaction of extracellular osteonectin with osteoclasts or osteoclast precursors, and the effect of the osteonectin-null mutation of the phenotype of marrow stromal cells and osteoblasts, which support osteoclast development.
Our in vitro data suggest a modest increase in osteoclast precursors in the marrow of osteonectin-/- mice. It is note worthy that osteonectin can interact with pre-B cells, which have the potential to influence osteoclast formation and activity [31]. Abnormalities in immune function have been described in osteonectin-/- mice in the C57Bl/6 genetic background, particularly in the B cell compartment [32, 33]. Osteoblasts support B cell commitment and differentiation [34]. In the bones of osteonectin-/- mice, B cell differentiation and targeting may be abnormal, contributing to increased osteoclastic commitment and activity.
Osteoblasts and marrow stromal fibroblasts regulate osteoclast formation through membrane-associated proteins as well as soluable proteins, and one cell surface molecule important for osteoclast differentiation is VCAM-1 (vascular cell adhesion molecule-1; CD106) [35, 36]. Recently, osteonectin was shown to interact with VCAM-1 and cause cytoskeletal rearrangement in endothelial cells [33]. It is possible that the interaction of osteonectin with VCAM-1 on stromal cells and osteoblasts in normal bone marrow may prevent the interaction of VCAM-1 with α4β1 integrin on osteoclast precursors, potentially attenuating osteoclastogenesis [36].
There are a limited number of studies describing the role of matricellular proteins in bone remodeling. Unlike osteonectin, other matricellular proteins important in the skeleton, including osteopontin, bone sialoprotein and thrombospondin 2, are documented integrin binding proteins [2]. When osteopontin-null mice were treated with intermittent PTH(1-34), the increase in whole body BMD was accentuated due to enhanced of bone formation rate and decreased osteoclast formation [37]. Similarly, in response to ovariectomy and skeletal unloading, osteopontin-null mice display decreased bone resorption compared with wild type, and the mutant mice do not lose bone [38, 39]. Thrombospondin-2-null mice also fail to lose bone in response to ovariectomy, due to increased osteoblastogenesis and decreased bone resorption [40]. Further, these mice display altered cortical bone formation response when stimulated with mechanical loading [41]. The response of other matricellular protein-mutant mice to bone remodeling challenges has not yet been reported, and the results obtained from our study of osteonectin-/- mice appear to be unique.
The skeleton is a dynamic environment, consisting of bone matrix and an array of cells including osteoblasts, osteocytes, osteoclasts, stromal cells, and cells in the hematopoietic niche. Proteins that modulate the function, growth and survival of each of these cellular components have the capacity to modify the balance between bone formation and resorption. In vivo studies strongly suggest that matricellular proteins have the potential to regulate both arms of the bone remodeling process, and our data indicate that osteonectin levels alter the response of the skeleton to intermittent PTH therapy. The molecular mechanisms by which PTH elicits bone anabolic vs. bone catabolic effects remain poorly understood [11]. Our data imply that osteonectin levels may play a role in modulating the balance of formation and resorption in response to PTH.
Acknowledgements
We thank Dr B. Kream (University of Connecticut Health Center) and Dr. R.C. Pereira (University of California, Los Angeles) for thoughtful discussions of the data. We thank Dr. C Pilbeam (University of Connecticut Health Center) for assistance with the qRT-PCR analysis.
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, AR44877 (A. Delany).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- [1].Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opinion Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- [2].Alford AI, Hankenson KD. Matricellular proteins: extracellular modulators of bone development, remodeling, and regeneration. Bone. 2006;38:749–757. doi: 10.1016/j.bone.2005.11.017. [DOI] [PubMed] [Google Scholar]
- [3].Robey PG, Boskey AL. Extracellular matrix and biomineraliztion of bone. In: Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. American Society for Bone and Mineral Research; Washington DC: 2003. pp. 38–45. [Google Scholar]
- [4].Barker TH, Baneyx G, Cardo-Vila m, Workman GA, Weaver M, Menon PM, Dedhar S, Rempel SA, Arap W, Pasqualini R, Vogel V, Sage EH. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase. J Biol Chem. 2005;280:36483–36493. doi: 10.1074/jbc.M504663200. [DOI] [PubMed] [Google Scholar]
- [5].Kessler C, Delany AM. Increased notch 1 expression and attenuated stimulatory G protein coupling to adenylyl cyclase in osteonectin-null osteoblasts. Endocrinology. 2007;148:1666–1674. doi: 10.1210/en.2006-0443. [DOI] [PubMed] [Google Scholar]
- [6].Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107:1049–1054. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Delany AM, Amling M, Priemel M, Howe C, Baron R, Canalis E. Osteopenia and decreased bone formation in osteonectin-null mice. J Clin Invest. 2000;105:915–923. doi: 10.1172/JCI7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wild type controls. J Bone Miner Res. 2003;18:1005–1011. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- [9].Bradshaw AD, Graves DC, Motamed K, Sage EH. SPARC-null mice exhibit increased adiposity without significant differences in overall body weight. Proc Natl Acad Sci USA. 2003;100:6045–6050. doi: 10.1073/pnas.1030790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E. Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology. 2003;144:2588–2596. doi: 10.1210/en.2002-221044. [DOI] [PubMed] [Google Scholar]
- [11].Canalis E, Guistina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. New England J Med. 2007;357:905–916. doi: 10.1056/NEJMra067395. [DOI] [PubMed] [Google Scholar]
- [12].Fedarko NS, Gehron Robey P, Vetter UK. Extracellular matrix stoichiometry in osteoblasts from patients with osteogenesis imperfecta. J Bone Miner Res. 1995;10:1122–1129. doi: 10.1002/jbmr.5650100718. [DOI] [PubMed] [Google Scholar]
- [13].Fedarko NS, Moerike M, Brenner R, Gehron Robey P, Vetter U. Extracellular matrix formation by osteoblasts from patients with osteogenesis imperfecta. J Bone Miner Res. 1992;7:921–930. doi: 10.1002/jbmr.5650070809. [DOI] [PubMed] [Google Scholar]
- [14].Delany AM, McMahon DJ, Powell JS, Greenberg DA, Kurland ES. Osteonectin/SPARC polymorphisms in Caucasian men with idiopathic osteoporosis and healthy male controls. Osteoporosis Int. doi: 10.1007/s00198-007-0523-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jansen RP. mRNA localization: message on the move. Nat Cell Biol. 2001;4:247–256. doi: 10.1038/35067016. [DOI] [PubMed] [Google Scholar]
- [16].Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- [17].Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–665. doi: 10.1210/en.2004-0766. [DOI] [PubMed] [Google Scholar]
- [18].Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22:1197–1207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- [19].Bouxsein ML, Pierroz DD, Glatt V, Goddard DS, Cavat F, Rizzoli R, Ferrai SL. B-arrestin2 regulates the differential response of cortical and trabecular bone to intermittent PTH in female mice. J Bone Miner Res. 2005;20:635–643. doi: 10.1359/JBMR.041204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee S-K, Kadono Y, Okada F, Jacquin C, Koczon-Jaremko B, Gronowicz G, Adams DL, Aguila HL, Lorenzo JA. T lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res. 2006;21:1704–1712. doi: 10.1359/jbmr.060726. [DOI] [PubMed] [Google Scholar]
- [21].Lee S-K, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–3561. doi: 10.1210/endo.140.8.6887. [DOI] [PubMed] [Google Scholar]
- [22].Huang JC, Sakata T, Pfleger LL, Bencsik M, Halloran BP, Bilke DD, Nissenson RA. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235–244. doi: 10.1359/JBMR.0301226. [DOI] [PubMed] [Google Scholar]
- [23].Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. 2006;21:67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- [24].Riggs BL, Melton LJ, III, Robb RA, Camp JJ, Atkinson EJ, McDaniel L, Amin S, Rouleau PA, Khosla S. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J Bone Miner Res. 2008;23:205–214. doi: 10.1359/JBMR.071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gensure RC, Gardella TJ, Juppner H. Parathyroid hormone and parathyroid homrone-related peptide, and their receptors. Biochem Biophys Res Comm. 2005;328:666–678. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- [26].Singh ATK, Gilchrist A, Voyno-Yasenetskaya T, Radeff-Huang JM, Stern PH. Gα12/Gα13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology. 2005;146:2171–2175. doi: 10.1210/en.2004-1283. [DOI] [PubMed] [Google Scholar]
- [27].Wang L, Liu S, Quarles LD, Spurney RF. Targeted overexpression of G protein-coupled receptor kinase-2 in osteoblasts promotes bone loss. Am J Physiol Endocrinol Metab. 2005;288:E826–E834. doi: 10.1152/ajpendo.00422.2004. [DOI] [PubMed] [Google Scholar]
- [28].Chauvin S, Bencsik M, Bambino T, Nissenson RA. Parathyroid hormone receptor recycling: role of receptor dephosphorylation and β-arrestin. Mod Endocrinol. 2002;16:2720–2732. doi: 10.1210/me.2002-0049. [DOI] [PubMed] [Google Scholar]
- [29].Ferrari SL, Pierroz DD, Glatt V, Goddard DS, Bianchi EN, Lin FT, Manen D, Bouxsein ML. Bone response to intermittent parathyroid hormone is altered in mice null for β-arrestin2. Endocrinology. 2005;146:1854–1862. doi: 10.1210/en.2004-1282. [DOI] [PubMed] [Google Scholar]
- [30].Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50–65. doi: 10.1111/j.0105-2896.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- [31].Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134:771–782. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rempel SA, Hawley RC, Gutierrez JA, Mouzon E, Bobbitt KR, Lemke N, Schultz CR, Schultz LR, Golembieski W, Koblinski J, VanOsdol S, Miller CG. Splenic and immune alterations in the Sparc-null mouse accompany a lack of immune response. Genes and Immunity. 2007;8:262–274. doi: 10.1038/sj.gene.6364388. [DOI] [PubMed] [Google Scholar]
- [33].Kelly KA, Allport JR, Yu AM, Sinh S, Sage EH, Gerszten RE, Weissleder R. SPARC is a VCAM-1 counter-ligand that mediates leukocyte transmigration. J Leuko Biol. 2007;81:748–756. doi: 10.1189/jlb.1105664. [DOI] [PubMed] [Google Scholar]
- [34].Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, Joe GJ, Hexner E, Choi Y, Taichman RS, Emerson SG. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109:3706–3712. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
- [35].Feuerbach D, Feyen JH. Expression of the cell-adhesion molecule VCAM-1 by stromal cells is necessary for osteoclastogenesis. FEBS Lett. 1997;402:21–24. doi: 10.1016/s0014-5793(96)01495-0. [DOI] [PubMed] [Google Scholar]
- [36].Michigami T, Shimizu N, Williams PJ, Niewolna M, Dallas SL, Mundy GR, Yoneda T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast stimulating activity. Blood. 2000;96:1953–1960. [PubMed] [Google Scholar]
- [37].Kitahara K, Ishijima M, Rittling SR, Tsuji K, Kurosawa H, Nifuji A, Denhardt DT, Noda M. Osteopontin deficiency induces parathyroid hormone enhancement of cortical bone formation. Endocrinology. 2003;144:2132–2140. doi: 10.1210/en.2002-220996. [DOI] [PubMed] [Google Scholar]
- [38].Yoshitake H, Rittling SR, Denhardt DT, Noda M. Osteopontin-deficient mice are resistant to ovariectomy-induced bone loss. Proc Natl Acad Sci. 1999;96:8156–8160. doi: 10.1073/pnas.96.14.8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ishijima M, Tsuji K, Rittling SR, Yamashita T, Kurosawa H, Denhardt DT, Nifuji A, Noda M. Resistance to unloading-induced three-dimensional bone loss in osteopontin-deficient mice. J Bone Miner Res. 2002;17:661–667. doi: 10.1359/jbmr.2002.17.4.661. [DOI] [PubMed] [Google Scholar]
- [40].Hankenson KD, James IE, Apone S, Stroup GB, Blake SM, Liang X, Lark MW, Bornstein P. Increased osteoblastogenesis and decreased bone resorption protect against ovariectomy-induced bone loss in thrombospondin-2-null mice. Matrix Biology. 2005;24:362–370. doi: 10.1016/j.matbio.2005.05.008. [DOI] [PubMed] [Google Scholar]
- [41].Hankenson KD, Ausk BJ, Bain SD, Bornstein P, Gross TS, Srinivasan S. Mice lacking thrombospondin 2 show atypical pattern of endocoritcal loading and periosteal bone formation I response to mechanical loading. Bone. 2006;38:310–316. doi: 10.1016/j.bone.2005.08.027. [DOI] [PubMed] [Google Scholar]