

Abstract
A new strategy for the synthesis of (±)-aspidophytine has been developed and is based on a Rh(ll)-catalyzed cyclization/dipolar-cycloaddition sequence. The resulting [3+2]-cycloadduct undergoes an efficient Lewis acid mediated cascade that rapidly provides the complete skeleton of aspidophytine. The synthesis also features a mild decarbomethoxylation reaction.
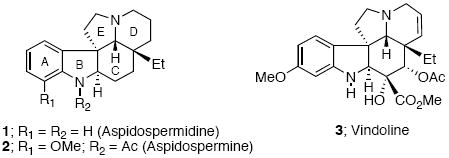
The Aspidosperma alkaloids occupy a central place in natural product chemistry because of their wide range of complex structural variation and diverse biological activity.1 This family of indole alkaloids contains over 250 members that share in their molecular structure a common pentacyclic ABCDE framework, with the C-ring being of critical importance because all six stereocenters and most of the functionalities are located in this ring.2 Individual members differ mainly in functionality and stereochemistry. Over the years, efficient and elegant routes to this molecular framework have been developed.3,4
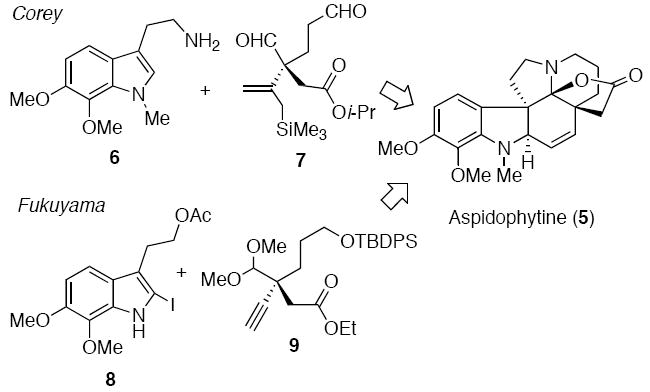
In 1973, Cava and Yates reported on the structural determination of haplophytine (4), a dimeric indole alkaloid isolated from the leaves of Haplophyton cimicidum.5,6 Acid cleavage of haplophytine (4) led to aspidophytine (5),7 a lactonic aspidospermine type of alkaloid which has been suggested to not only be a biosynthetic precursor of 4 but also a possible intermediate to be used in its synthesis.8,9 Because of its intriguing structure, aspidophytine has attracted the attention of two major research groups. In 1999 Corey et al8 and four years later Fukuyama et al9 accomplished the synthesis of aspidophytine utilizing completely different strategies. The Corey approach hinged on a creative cascade reaction between dialdehyde 7 and indole 6, synthesized from vanillin acetate in 10 steps (Scheme 1). The Fukuyama group used their signature radical cascade chemistry10 to construct indole 8 from vanillin acetate (11 steps), followed by a Sonogashira coupling11 with alkyne 9 and then an effective amine-aldehyde condensation cascade to furnish the aspidosperma skeleton9.
Scheme 1.
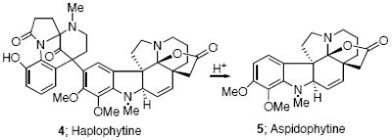
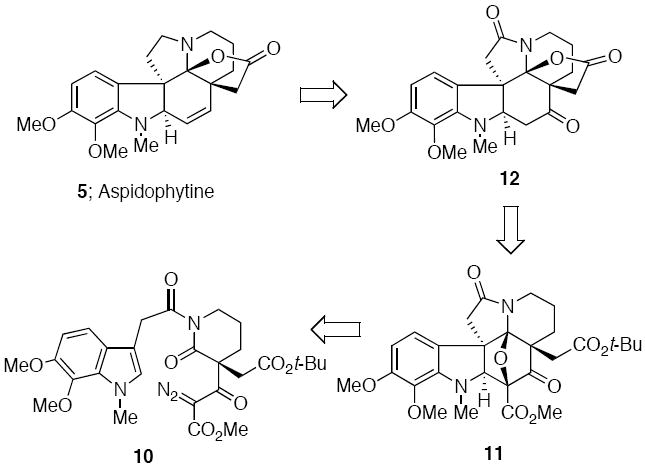
Our approach to aspidophytine was guided by a longstanding interest in developing new applications of the Rh(II)-cyclization/cycloaddition cascade for the synthesis of complex natural products, particularly alkaloids.12 The generation of onium ylides by a transition-metal promoted cyclization reaction has emerged in recent years as an important and efficient method for the assembly of ring systems that are difficult to prepare by other means.13,14 In earlier work from our laboratory we had described the formation of push-pull dipoles from the Rh(II)-catalyzed reaction of α-diazo imides and noted that a smooth intramolecular 1,3-dipolar cycloaddition occurred across both alkenyl and heteroaromatic π-bonds to provide novel pentacyclic compounds in good yield and in a stereocontrolled fashion.15 Our plan for the synthesis of aspidophytine is shown in retrosynthetic format in Scheme 2 and is centered upon the construction of the key cycloadduct 11 by making use of α-diazo imide 10. The successful completion of this synthesis demonstrates the utility of this cascade methodology for the construction of complex indole-containing natural products.
Scheme 2.
Construction of the required α-diazo imide 10 entailed the synthesis of two building blocks, the substituted 2-(indol-3-yl)acetic acid 16 and the diazo lactam 20. The preparation of 16 was carried out in five steps in 36% overall yield starting from aniline 13 and is summarized in Scheme 3. Commercially available aniline 13 was iodinated using IC1 (73%) and the resulting iodoaniline 14 was subsequently alkylated with methyl bromocrotonate to give the secondary amine 15 in 75% yield based on recovered starting material. Intramolecular Heck cyclization (90%) of 15 afforded the expected indole ring16 which was easily converted to 16 by N-methylation with CH3I (80%) followed by a subsequent hydrolysis step (94%).
Scheme 3.
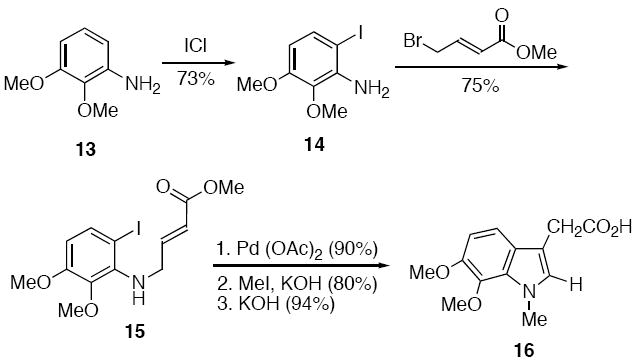
Preparation of the diazo lactam unit 20 commenced with commercially available δ-valerolactam 17 which was deprotonated with excess base and allowed to react with t-butyl bromoacetate to give lactam 18 in 80% yield (Scheme 4). The ethyl ester portion of 18 was converted to the methyl 3-oxopropanoate group using a modified Masamune procedure17 to furnish β-keto ester 19 in 54% overall yield. Finally, the requisite α-diazo lactam 20 was easily obtained using standard diazo transfer conditions18 and was isolated in 96% yield.
Scheme 4.
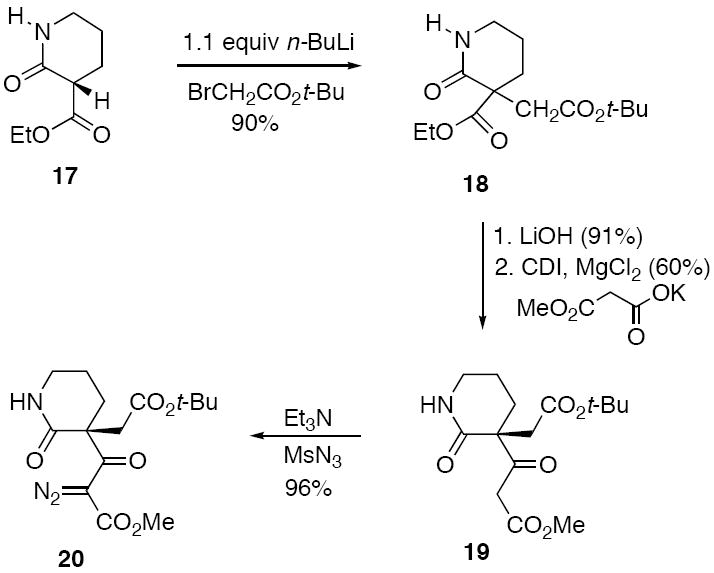
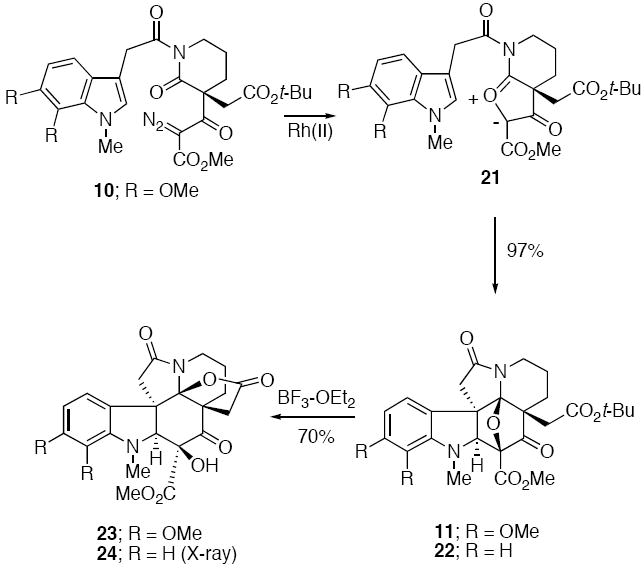
At this point we joined the two synthesized fragments by treating acid 16 with (COC1)2 and allowing the resulting acid chloride to react with the stable N-H diazo lactam in the presence of 4A° molecular sieves. The desired α-diazo imide 10 was obtained in 92% yield. Formation of the push-pull dipole 21 was achieved by reaction of 10 with Rh2(OAc)4, which afforded a rhodium carbenoid species that readily underwent cyclization onto the neighboring imido carbonyl to form the carbonyl ylide dipole 21.12
Subsequent intramolecular cycloaddition across the tethered indolyl group furnished cycloadduct 11 in 97% yield.19 The acid lability of cycloadduct 11 was exploited in the next step of the synthesis. Treatment of 11 with a Lewis acid such as BF3·OEt2 resulted in cleavage of the oxabicyclic ring and formation of a transient N-acyl iminium ion which was captured by the adjacent carbonyl group of the t-butyl ester (Scheme 5). Loss of isobutylene from the resulting oxonium ion resulted in the isolation of 23 in 70% yield. The relative stereochemistry of 23 was assigned on the basis of its spectroscopic properties which were essentially identical to the related ring opened product 24 (R=H) whose structure was confirmed by a single-crystal X-ray analysis.20
Scheme 5.
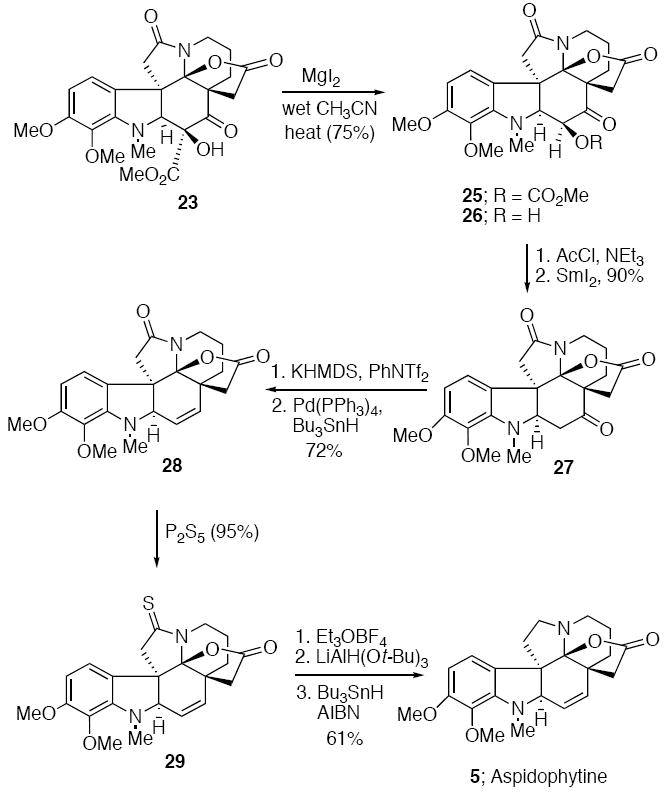
The completion of the synthesis of aspidophytine (5) from 23 is outlined in Scheme 6. First, the carbomethoxy and hydroxyl groups next to the keto group were sequentially removed so as to produce compound 27. Treatment of 23 with Mgl2 in refluxing acetonitrile containing a small quantity of water resulted in the formation of alcohol 26 in 75% yield. While a cursory analysis of the conditions suggests a Krapcho dealkoxycarbonylation reaction,21 the isolation of carbonate 25 from the reaction is more consistent with an unexpected carbomethoxy group migration22 to the adjacent hydroxyl group followed by further reaction of the carbonate with halide ion and loss of CO2 to give 26.23 Acetylation of 26 with acetyl chloride/NEt3 afforded the corresponding acetate which was subsequently reduced using SmI2 to give 27 in 90% yield from both steps. The carbonyl group of 27 was transformed into the corresponding enol triflate which, upon treatment with Pd(Ph3P)4 and n-Bu3SnH according to Corey’s experimental conditions,8 gave 28 (72%). Treatment of 28 with P2S5 furnished thiolactam 29 in 95% yield.24 Although not investigated in detail, our attempts to reduce 29 with Raney-Ni were not successful. Instead, S-ethylation of thiolactam 29 with Meerwein’s reagent followed by LiAlH(Ot-Bu)3/n-Bu3SnH reduction25 provided (±)-aspidophytine (5) in 61% yield from 29. Confirmation of the structure was obtained by comparison of the spectral data with that of an authentic sample of aspidophytine provided by Professor Fukuyama.
Scheme 6.
In conclusion, a concise total synthesis of (±)-aspidophytine was achieved featuring a Rh(II)-cyclization/cycloaddition cascade of a suitably substituted α-diazo imide as the key step. We are currently refining this strategy and further applying the methodology toward other aspidosperma alkaloids.
Supplementary Material
Spectroscopic data and experimental details for the preparation of all new compounds together with an ORTEP drawing for compounds 24 and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We appreciate the financial support provided by the National Institutes of Health (GM 059384) and the National Science Foundation (CHE-0450779). We wish to thank Professor Tohru Fukuyama and Dr. Hidetoshi Tokuyama (University of Tokyo) for an authentic sample of aspidophytine for comparison purposes.
References
- 1.Saxton JE. In: The Alkaloids: Chemistry and Biology. Cordell GA, editor. Vol. 51. Academic Press; San Diego, CA: 1998. pp. 1–197. [Google Scholar]
- 2.Saxton JE. Indoles, Part 4: The Monoterpenoid Indole Alkaloids. Wiley; Chichester: 1983. Herbert RB. In: The Monoterpenoid Indole Alkaloids. Saxton JE, editor. Chapter 1 Wiley; Chichester: 1994. Supplement to Vol 25, part 4 of The Chemistry of Heterocyclic Compounds Toyota M, Ihara M. Nat Prod Rep. 1998:327. and references cited therein
- 3.For the first synthesis of aspidospermine and vindoline, see: Stork G, Dolfini JE. J Am Chem Soc. 1963;85:2872.Ando M, Buchi G, Ohnuma T. J Am Chem Soc. 1975;97:6880.
- 4.For some select methods to synthesize the pentacyclic framework of aspidospermidine (1), see: Camerman A, Camerman N, Kutney JP, Piers E, Trotter J. Tetrahedron Lett. 1965:637.Harley-Mason J, Kaplan M. J Chem Soc Chem Commun. 1967:915.Laronze J-Y, Laronze-Fontaine J, Lévy J, Le Men J. Tetrahedron Lett. 1974:491.Ban Y, Yoshida K, Goto J, Oishi T. J Am Chem Soc. 1981;103:6990.Gallagher T, Magnus P, Huffman J. J Am Chem Soc. 1982;104:1140.Wenkert E, Hudlicky T. J Org Chem. 1988;53:1953.Mandal SB, Giri VS, Sabeena MS, Pakrashi SC. J Org Chem. 1988;53:4236.Meyers AI, Berney D. J Org Chem. 1989;54:4673.Node M, Nagasawa H, Fugi K. J Org Chem. 1990;55:517.Le Menez P, Kunesch N, Lui S, Wenkert E. J Org Chem. 1991;56:2915.Desmaële D, d’Angelo J. J Org Chem. 1994;59:2292.Wenkert E, Lui S. J Org Chem. 1994;59:7677.Forns P, Diez A, Rubiralta M. J Org Chem. 1996;61:7882. doi: 10.1021/jo9609900.Schultz AG, Pettus L. J Org Chem. 1997;62:6855.Callaghan O, Lampard C, Kennedy AR, Murphy JA. J Chem Soc Perkin Trans 1. 1999:995.Iyengar R, Schildknegt K, Aubé J. Org Lett. 2000;2:1625. doi: 10.1021/ol005913c.Toczko MA, Heathcock CH. J Org Chem. 2000;65:2642. doi: 10.1021/jo991599s.Patro B, Murphy JA. Org Lett. 2000;2:3599. doi: 10.1021/ol006477x.Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628. doi: 10.1021/ja017863s.Banwell MG, Smith JA. J Chem Soc Perkin Trans 1. 2002:2613.Marino JP, Rubio MB, Cao G, de Dios A. J Am Chem Soc. 2002;124:13398. doi: 10.1021/ja026357f.Gnecco D, Vázquez E, Galindo A, Terán JL, Bernès S, Enríquez RG. Arkivoc. 2003;11:185.Tanino H, Fukuishi T, Ushiyama M, Okada K. Tetrahedron. 2004;60:3273.Banwell MG, Lupton DW. Org Biomol Chem. 2005;3:213. doi: 10.1039/b415977b.
- 5.Yates P, MacLachlan FN, Rae ID, Rosenberger M, Szabo AG, Willis CR, Cava MP, Behforouz M, Lakshmikantham MV, Ziegler W. J Am Chem Soc. 1973;95:7842. [Google Scholar]
- 6.(a) Saxton JE. Alkaloids. 1965;8:673. [Google Scholar]; (b) Cheng P-T, Nyburg SC, MacLachlan FN, Yates P. Can J Chem. 1976;54:726. [Google Scholar]
- 7.(a) Cava MP, Talapatra SK, Nomura K, Weisbach JA, Douglas B, Shoop EC. Chem Ind (London) 1963:1242. [Google Scholar]; (b) Cava MP, Talapatra SK, Yates P, Rosenberger M, Szabo AG, Douglas B, Raffuaf RF, Shoop EC, Weisbach JA. Chem Ind (London) 1963:1875. [Google Scholar]; (c) Rae ID, Rosenberger M, Szabo AG, Willis CR, Yates P, Zacharias DE, Jeffrey GA, Douglas B, Kirkpatrick JL, Weisbach JA. J Am Chem Soc. 1967;89:3061. [Google Scholar]
- 8.He F, Bo Y, Altom JD, Corey EJ. J Am Chem Soc. 1999;121:6771. [Google Scholar]
- 9.(a) Sumi S, Matsumoto S, Tokuyama H, Fukuyama T. Org Lett. 2003;5:1891. doi: 10.1021/ol034445e. [DOI] [PubMed] [Google Scholar]; (b) Sumi S, Matsumoto K, Tokuyama H, Fukuyama T. Tetrahedron. 2003;59:8571. doi: 10.1021/ol034445e. [DOI] [PubMed] [Google Scholar]
- 10.(a) Fukuyama T, Chen X, Peng G. J Am Chem Soc. 1994;116:3127. [Google Scholar]; (b) Tokuyama H, Kaburagi Y, Chen X, Fukuyama T. Synthesis. 2000:429. [Google Scholar]; (c) Tokuyama H, Watanabe M, Hayashi Y, Kurokawa T, Peng G, Fukuyama T. Synlett. 2001:1403. [Google Scholar]; (d) Kobayashi S, Peng G, Fukuyama T. Tetrahedron Lett. 1999;40:1519. [Google Scholar]; (e) Kobayashi S, Ueda T, Fukuyama T. Synlett. 2000:883. [Google Scholar]
- 11.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467. [Google Scholar]
- 12.For some leading references, see: Padwa A, Hornbuckle SF. Chem Rev. 1991;91:263.Padwa A, Weingarten MD. Chem Rev. 1996;96:223. doi: 10.1021/cr950022h.Padwa A. Top Curr Chem. 1997;189:121.Padwa A. Pure & Appl Chem. 2004;76:1933.Padwa A, Brodney MA, Lynch SM, Rashatasakhon P, Wang Q, Zhang H. J Org Chem. 2004;69:3735. doi: 10.1021/jo049808i.
- 13.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. John Wiley & Sons; New York: 1998. [Google Scholar]
- 14.Boger and co-workers have recently described an alternate approach to onium ylides based on a [4+2]-cycloaddition of 1,3,4-oxadiazoles followed by a thermal extrusion of nitrogen and have elegantly exploited this chemistry for the construction of a variety of aspidosperma alkaloid targets, see: Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539. doi: 10.1021/ol051975x. and references cited therein
- 15.(a) Padwa A, Price AT. J Org Chem. 1995;60:6258. [Google Scholar]; (b) Padwa A, Price AT. J Org Chem. 1998;63:556. doi: 10.1021/jo971424n. [DOI] [PubMed] [Google Scholar]; (c) Mejía-Oneto JM, Padwa A. Org Lett. 2004;6:3241. doi: 10.1021/ol048915w. [DOI] [PubMed] [Google Scholar]; (d) Padwa A, Lynch SM, Mejía-Oneto JM, Zhang H. J Org Chem. 2005;70:2206. doi: 10.1021/jo047834a. [DOI] [PubMed] [Google Scholar]
- 16.(a) Mori M, Chiba K, Ban Y. Tetrahedron Lett. 1977:1037. [Google Scholar]; (b) Odle R, Blevins B, Ratcliff M, Hegedus LS. J Org Chem. 1980;45:2709. [Google Scholar]; (c) Aubert KM, Heathcock CH. J Org Chem. 1999;64:16. doi: 10.1021/jo9815397. [DOI] [PubMed] [Google Scholar]
- 17.Brooks DW, Lu DL, Masamune S. Angew Chem Int Ed Engl. 1979;18:72. [Google Scholar]
- 18.(a) Regitz M. Chem Ber. 1966;99:3128. [Google Scholar]; (b) Regitz M, Hocker J, Liedhegener A. Org Synth. 1973;5:179. [Google Scholar]
- 19.In contrast to previous finding,15 the exo-cycloadducts 11 and 22 were the exclusive products isolated from the Rh(II)-catalyzed reaction. We assume that the bulky t-butyl ester functionality blocks the endo-approach thereby resulting in cycloaddition taking place from the less congested exo-face.
- 20.The authors have deposited coordinates for structure 24 with the Cambridge Crystallographic Data Centre. The coordinates can be obtained, on request, from the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, B2 1EZ, UK.
- 21.For a review of dealkoxycarbonylations, see: Krapcho AP. Synthesis. 1982:805–893.
- 22.For some related examples, see: Rubin MB, Inbar S. Tetrahedron Lett. 1977:1037.Davis FA, Clark C, Kumar A, Chen B-C. J Org Chem. 1994;59:1184.
- 23.Structure 25 was established on the basis of a X-ray crystal structure analysis. A more detailed study of this unusual rearrangement is currently underway in our laboratory.
- 24.Curphey TJ. J Org Chem. 2002;67:6461. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 25.Raucher S, Klein P. Tetrahedron Lett. 1980;21:4061. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic data and experimental details for the preparation of all new compounds together with an ORTEP drawing for compounds 24 and 25. This material is available free of charge via the Internet at http://pubs.acs.org.