

Abstract
Transcriptional activation of protective genes is mediated by a cis-acting element called the antioxidant responsive element (ARE). The transcription factor Nrf2 (NF-E2-related factor 2) binds to the ARE. Activation of this pathway protects cells from oxidative stress-induced cell death. Increased oxidative stress is associated with neuronal cell death during the pathogenesis of multiple chronic neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis. We hypothesize that Nrf2-ARE activation is a novel neuroprotective pathway that confers resistance to a variety of oxidative stress-related neurodegenerative insults. In recent studies, primary neuronal cultures treated with chemical activators of the Nrf2-ARE pathway displayed significantly greater resistance to oxidative stress induced neurotoxicity. Similar cultures generated from ARE-hPAP reporter mice demonstrated selective activation of the Nrf2-ARE pathway in astrocytes suggesting that Nrf2 activation in astrocytes somehow confers resistance to naïve neurons. Further, in chemical models of neurodegeneration, Nrf2 knockout mice are significantly more sensitive to mitochondrial complex I and II inhibitors. Combining these observations with the results implying that the astrocyte is central to Nrf2-ARE mediated neuroprotection, we transplanted Nrf2-overexpressing astrocytes into the mouse striatum prior to lesioning with malonate. This procedure led to dramatic protection against malonate-induced neurotoxicity. Translating this to other chemical and genetic models of neurodegeneration will be discussed.
Keywords: Nrf2, oxidative stress, neurodegenerative diseases
The Nrf2-ARE pathway
The antioxidant responsive element (ARE) is an enhancer element that initiates the transcription of a battery of genes encoding phase II detoxification enzymes1,2 and factors essential for neuronal survival3. The ARE is activated through the binding of its transcription factor, Nrf2 (NF-E2-related factor 2)4,5. The mechanism by which this binding is induced is still emerging, but likely includes contributions from the repressor of Nrf2, Keap1, heterodimeric binding partners, and ubiquitin/proteasome-mediated regulation of Nrf2 half-life4,6,7,8,9,10. Importantly, in in vitro and in vivo models, this system has been shown to be effective at blocking neurotoxicity resulting from glutathione depletion, lipid peroxidation, intracellular calcium overload, excitotoxins, and disruption of the mitochondrial electron transport chain11,12,13,14,15. In addition to the typical induction of detoxification enzymes, Nrf2-ARE activation results in increased cellular energetics and redox potential, inhibitory neurotransmitter signaling, and metabolic processes16,17,18,19,12,13,20. It should be noted that ARE-driven genes are preferentially activated in astrocytes21,19,13 and that this Nrf2-ARE activation in glial cells not only precludes oxidative damage in astrocytes but also confers protection to neighboring neurons12,13,14,15.
Increased levels of GSH may be a major component of the protection observed by Nrf2 activation. GSH is synthesized by the consecutive action of two enzymes, glutamate-cysteine ligase and glutathione synthetase. Glutamate-cysteine ligase catalytic (GCLC) and modifier (GCLM) subunits make up the rate-limiting enzyme complex for GSH synthesis. Interestingly, the expression of both GCLC and GCLM is increased by Nrf2 activation. The preferential activation of Nrf2 in astrocytes21,19,13 leads to more efficient GSH synthesis and higher GSH content in astrocytes than neurons22. Increased production and secretion of GSH by astrocytes is known to improve the antioxidant status of co-cultured neurons and protect them from oxidative insults22,23,12. Several studies have demonstrated that secreted GSH can protect neurons by acting as an antioxidant in the extracellular compartment and/or boosting GSH levels in neurons by increasing the availability of precursors for GSH synthesis22,23,24.
The Nrf2-ARE pathway in cultured neurons and astrocytes
Experiments using primary cortical neuronal and astrocyte culture systems in our laboratory were designed to better understand the protective potential of the Nrf2-ARE pathway. The use of ARE-human placental alkaline phosphatase (hPAP) reporter mice21 and the Nrf2 KO mice25 have been important tools in these studies. In primary cortical neuronal cultures from ARE-hPAP mice (E15), tBHQ (tert-butylhydroquinone), a prototypical Nrf2-ARE activator, increased hPAP activity 30 to 50 fold and indicated that the hPAP activity was induced primarily in the astrocyte21,13. These mixed cortical cultures consist of approximately 30% astrocytes and 70% neurons. This Nrf2-ARE activation in astrocytes provided significant neuroprotection from glutamate and hydrogen peroxide induced neuronal cell death12,13.
We have also shown that astrocytes overexpressing Nrf2 can protect neurons from oxidative insult. Cortical neuronal cultures were prepared from Nrf2 KO mice. As expected, these mixed cultures contained approximate 20–30% astrocytes as determined by GFAP immunostaining. Cultures were infected with 50 multiplicity of infection (MOI) of adenoviral vectors containing either CMV-driven GFP (Ad-GFP) or the combination of CMV-driven GFP and CMV-driven Nrf2 (Ad-Nrf2). At this MOI, nearly all of the astrocytes were infected, whereas virtually no other cell types were infected13. Dramatic neuronal protection against hydrogen peroxide was observed when the Nrf2 KO cultures were treated with Ad-Nrf2 (Fig. 1). The loss of beta(III)-tubulin staining (a neuronal marker) in cultures exposed to hydrogen peroxide was completely rescued by replacing Nrf2 in the astrocytes. This suggested that the astrocytes are somehow conferring protection to the naïve neurons in the culture.
Fig. 1. Nrf2 KO Primary Cortical Neurons are Protected by Nrf2 Overexpressing Astrocytes.
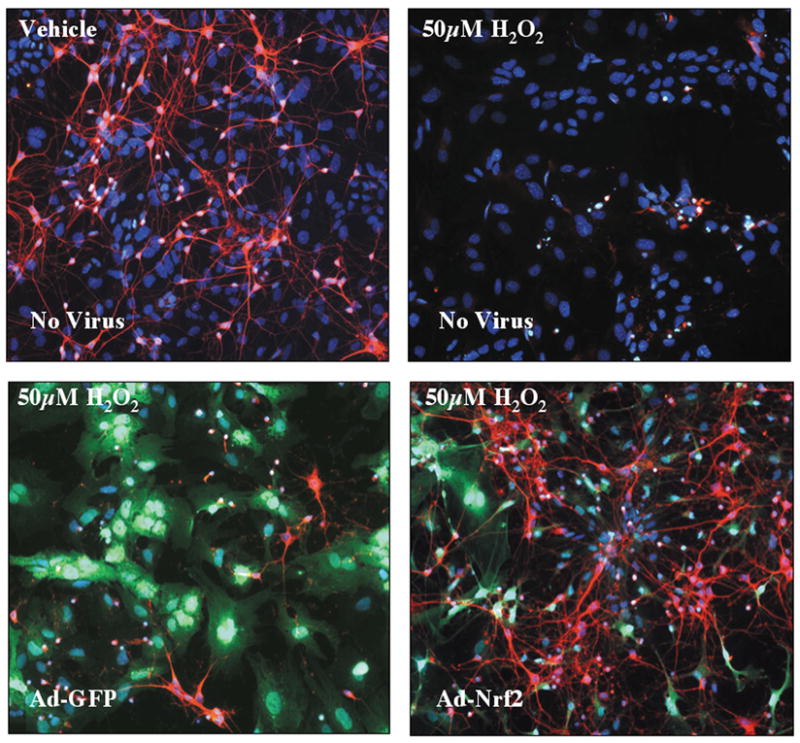
Mixed primary cortical cultures were prepared and treated with vehicle or 50 pM H2O2 (top panels). H2O2 killed virtually all neurons (beta-III tubulin labeled with Texas-Red in all panels). Cortical cultures were also infected with Ad-GFP or Ad-Nrf2 48hr before H2O2 treatement. Adenovirus preferentially infected astrocytes (GFP in green) and pretreatment with Ad-Nrf2 virus protects neurons from H2O2 insult (bottom right). Hoechst 33258 (blue). Reprinted from Journal of Neuroscience, Volume 24, Kraft et al., Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult, 1101-12, 2004 with permission from Journal of Neuroscience.
Genetic changes in cortical astrocytes were analyzed by microarray analysis19. There was an orchestrated change in different gene clusters as shown in Figure 2. Numerous genes encoding GSH-dependent enzymes are increased by Nrf2 activation and this is coupled with changes leading to increased GSH production and recycling (Fig. 2A). Another cluster of genes encoding proteins very important for cellular defense against oxidative stress and proper management of free iron are increased (Fig. 2B). Finally, genes that encode proteins that utilize NADPH as an electron donor to function are increased and these changes are coupled with another set of genes that are responsible for the increased production of NADPH (Fig. 2C). Together, these coordinately regulated gene clusters strongly support the hypothesis that Nrf2-dependent gene expression is central to efficient detoxification of reactive metabolites and reactive oxygen species.
Fig. 2. Orchestrated regulation of Nrf2-dependent genes in astrocytes.
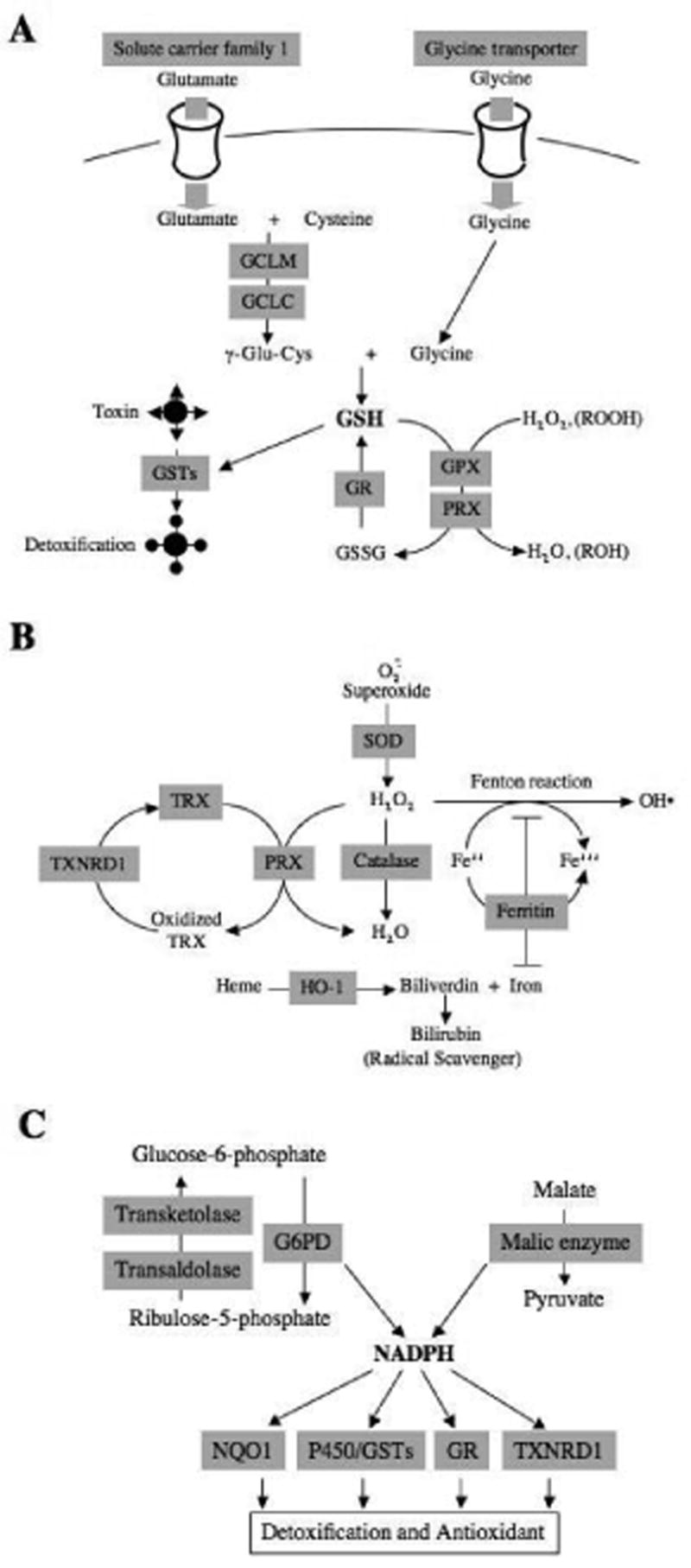
Nrf2-dependent genes identified by oligonucleotide microarray analysis are depicted in shaded squares. Genes are categorized based on function and metabolic pathway. Genes are related to (A) glutathione homeostasis, (B) detoxification of H2O2 and iron homeostasis, and (C) NADPH homeostasis. Reprinted from Journal of Biological Chemistry, Volume 278, Lee et al., Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis, 12029–12038, 2003 with permission from Journal of Biological Chemistry.
To further explore this cell-specific effect, primary cortical neuronal cultures were treated with tBHQ in the presence and absence of dominant-negative Nrf2 adenovirus (Ad-DN-Nrf2). DN-Nrf2 blocks Nrf2 signaling and the Ad-DN-Nrf2 selectively infected the astrocytes13. This allowed us to evaluate the effect of neutralizing Nrf2 in astrocytes of this mixed culture system. Nrf2-ARE activation was significantly attenuated and the protective effects of tBHQ were reversed validating that the neuroprotective effects of Nrf2-mediated ARE activation reside in the astrocyte. In addition, the selective infection of astrocytes (GFP-positive cells) allowed for fluorescent activated cell sorting (FACS) of the mixed cultures and subsequent microarray analysis of astrocytes (GFP-positive pool) and neurons (GFP-negative pool) from cultures treated with tBHQ. Ninety-seven genes were increased and 37 decreased in astrocytes by tBHQ treatment and in the presence of Ad-DN-Nrf2, 82% and 100% of the changes were significantly attenuated, respectively. Strikingly, only 4 of the increased genes and 9 of the decreased genes changed in both astrocytes and neurons. These data show a very cell-type specific change in genetic profiles following tBHQ treatment and that the changes occurring in the neuron are dependent on Nrf2-ARE activation in the astrocyte. Genetic changes of particular interest are shown in Figure 3.
Fig. 3. Nrf2 activation and enhanced metabolic coupling between astrocytes and neurons.

Primary cortical neuronal cultures were infected with Ad-GFP (50 MOI) prior to treatment with tBHQ. At this MOI the adenovirus selectively infected astrocytes allowing for FAC sorting to be performed on the cultures. Post-sort analysis of the isolated cell populations showed < 0.01% contamination on the neuronal GFP-negative pool with astrocytes and < 5% contamination of the astrocyte GFP-positive pool with neurons. RNA was isolated from the sorted pools and analyzed by microarray analysis (Kraft et al, 2004). A hypothetical diagram of some of the pathways changed with tBHQ treatment in the astrocytes (GFP-positive) and the neurons (GFP-negative) based upon microarray analysis is shown above. The protective cellular shield reflects genes that are consistently changed with activation of Nrf2 to combat oxidative stress and toxicants. The majority of these genes change in the astrocyte. Other genes involved in energy metabolism were also changed. Solid arrowheads indicate pathways with one or more enzymes upregulated by tBHQ treatment (eg. More than five enzymes converting glucose to pyruvate are increased by tBHQ in the astrocyte population). Hollow arrowheads on solid lines indicate known interactions that are not changed by tBHQ. Arrows with stippled lines indicate a proposed interaction between neurons and glia that is not yet proven. Files containing all of the microarray data are available for download at http://www.pharmacy.wisc.edu/facstaff/sciences/JohnsonGroup/microdata.cfm. Reprinted from Molecular Biology and Pharmacology of Tissue Repair, Volume 1302, Johnson et al., The Nrf2-ARE pathway: A potential therapeutic target for neurodegenerative diseases, 143–153, 2007 with permission from Elsevier.
Similar genetic changes as shown in Figure 2 were noted here as well. In addition, a very interesting cluster of genes associated with energy production were also up regulated by tBHQ in the astrocyte of these mixed cortical cultures including the majority of the ten genes catalyzing the glycolysis of glucose to pyruvate (Fig. 3). Glucose metabolism is essential for calcium homeostasis in the brain via the synthesis of ATP. In addition, pyruvate can be further metabolized to lactate, which is the preferred energy substrate for neurons and helps to maintain synaptic transmission. The increases in astrocytic glycogen synthesis (glucan branching enzyme), gluconeogenesis (GPI), and neuronal creatine production (Gatm and creatine kinase) may also indicate an enhanced maintenance of neuronal activity by tBHQ.
Astrocytes are known to couple neuronal activity (e.g. glutamate release) to energy production and glutathione synthesis. Further metabolism of pyruvate to Acetyl CoA leads to an increased synthesis of cholesterol (via squalene epoxidase and lanosterol synthase) and fatty acids for phospholipids and energy storage (via malic enzyme, fatty acid synthase, Scd1 and 2). It is hypothesized that neurons require glia-derived cholesterol to form efficient synaptic connections. Thus, aspects of astrocytic metabolism regulated by this inducible system likely contribute to neuronal protection.
The Nrf2-ARE pathway in models of Neurodegeneration
Through the use of multiple neurotoxic models, it has been shown that neurons in Nrf2 KO mice are more sensitive to the insults. The Nrf2 KO mice were more sensitive to the mitochondrial complex II inhibitors 3-nitropropionic acid (3-NP) and malonate14,26. These chemicals are used as models of Huntington’s disease. Similar increased sensitivity has been seen in models of Parkinson’s disease using 6-hydroxydopamine15 or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) 27,unpublished data, cerebral ischemia28,29, intracerebral hemorrhage30,31, traumatic brain injury32, and an epileptic model using kainic acid33.
These observations in combination with the culture data discussed earlier suggest that the lack of Nrf2 in astrocytes may be a common contributing factor to this differential sensitivity. Can we translate the in vitro observations to the in vivo situation? Three approaches are currently underway to address this question: 1) direct injection viruses containing Nrf2 into different brain regions; 2) the transplantation of astrocytes and/or neuroprogenitor cells (NPC) overexpressing Nrf2; and 3) development of a transgenic mouse overexpressing Nrf2 specifically in astrocytes (GFAP-Nrf2). Initial studies doing intrastriatal transplants of GFP and Nrf2 overexpressing primary astrocytes into mouse brain were carried out. Five weeks post-transplantation the mice were lesioned with malonate14. The extent of neuroprotection afforded by the astrocytes containing Nrf2 was highly significant (Fig. 4). These experiments have been successfully repeated using NPC overexpressing Nrf2, however, the number of NPC needed to show similar protection was 5-times less than the primary astrocytesunpublished data.
Fig. 4. Transplantation of Nrf2 overexpressing astocytes protects against malonate lesioning.
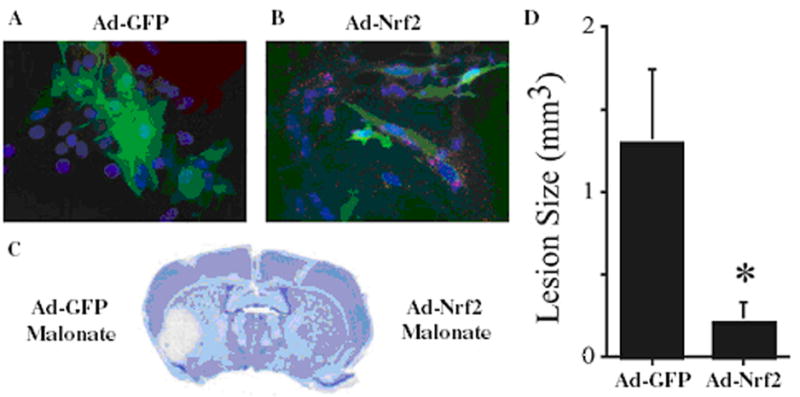
ARE-hPAP+ astrocytes were infected with Ad-GFP or Ad-Nrf2-GFP. GFP expression (green; A,B) and hPAP histochemistry (red stippling; B) were visualized to show the extent of infection (> 95% of cells) and activation Nrf2-ARE pathway by Nrf2 and not GFP. Astrocytes were lifted and transplanted in the striatum. Mice were lesioned 5 weeks post-transplant with malonate (n=4). Lesions were visualized by cresyl violet (C) and the volume was quantified (D). *p<0.05 compared to hemispheres receiving GFP-infected astrocytes. Reprinted from Proceeding of the National Academy of Sciences USA, Volume 102, Calkins et al., Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription, 244-9, 2005 with permission from the National Academy of Sciences USA.
GFAP-Nrf2 mice have been generated and crossed with the ARE-hPAP reporter mice. There is dramatic Nrf2-ARE activation in primary astrocytes (100-fold) and different regions of the central nervous system. The greatest change was 12,768-fold in the cerebellum as well as larger changes in cortex (1128-fold), brainstem (740-fold) and spinal cord (84-fold). No difference in hPAP activity was detected in the liver, lung, spleen, or muscle of the GFAP-Nrf2 mice relative to the non-transgenic controls demonstrating that increased Nrf2 expression is isolated to the central nervous system. Preliminary studies suggest that the GFAP-Nrf2 mice have increased resistant to malonate and MPTP toxicity unpublished data. The effectiveness of these approaches in genetic models of neurodegenerative disease is the next step.
These data suggest that activation of Nrf2 in astrocytes can confer significant protection to neurons in vivo and that chemical/drugs that activate this pathway may have efficacy in blocking neuronal cell death. Indeed, some current work shows that treatment with chemical activators of the Nrf2-ARE pathway prior to the toxic insults can reduce cellular damage in the Nrf2 wildtype mice but not the Nrf2 KO26,27,28,31,32. These include tBHQ26,28, 3H-1,2-dithiole-3-thione27 and sulforaphane31,32. Other potential activators the Nrf2-ARE pathway in brain that have not been tested in Nrf2 KO mice to show specificity include NEPP compounds35, carnosic acid36 and triterpenoids37. Most of these studies have activated the Nrf2-ARE pathway prior to injury. However, in the sulforaphane studies31,32, this Nrf2-ARE activator was administered 30 minutes after intracerebral hemorrhage31, and 15 minutes after traumatic brain injury32. In both cases, damage was attenuated and the protective effect was Nrf2-dependent. These studies imply that Nrf2 activation after the injury has been initiated can also be efficacious. To what level, how broadly, and in what cell type the Nrf2-ARE pathway is activated in the brain by these chemical remains to be determined.
Discussion and Future Directions
The data with respect to the Nrf2-ARE pathway and neuroprotection has increased considerably over the past few years and has now been replicated in multiple laboratories using different chemical or acute models of neurodegeneration. Continued work on how this pathway is involved in or mitigates neurodegeneration needs to be done in genetic models of Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis. The data generated in these models is critical to show that Nrf2-ARE pathway activation is a valid target for the treatment of neurodegeneration.
The role of the astrocyte and the mechanisms underlying the neuroprotective components that confer resistance to neurons are also important to determine. It has been shown, based on in vitro data, that GSH secretion from the astrocyte is increased following Nrf2-ARE activation. This effect is hypothesized to be the primary factor leading to neuroprotection of cortical12 and motor neurons34 in culture. However, since in vitro and in vivo systems are very different, this hypothesis needs to be examined in the animal.
Numerous chemicals, drugs and natural products have been shown to activate the Nrf2-ARE pathway. A focussed effort to find those that cross the blood-brain-barrier efficiently, activate the pathway in astrocytes, and have efficacy against neurotoxicity is underway. Clearly, a greater understanding of how the Nrf2-ARE pathway is intimately involved with the pathogenesis of neurodegenerative diseases and its significant neuroprotective properties could be pivotal in halting the progression of multiple neurological diseases through the development of new therapeutic approaches targeting this pathway.
Acknowledgments
The work discussed and ongoing studies are supported by NIEHS R01 ES10042, ES08089, Amyotrophic Lateral Sclerosis Association, Packard Center for ALS Research, and Hereditary Disease Foundation.
References
- 1.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265(24):14648–53. [PubMed] [Google Scholar]
- 2.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266(18):11632–9. [PubMed] [Google Scholar]
- 3.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel R, Johnson JA. Nrf2, a Multi-organ Protector? FASEB J. 2005;19(9):1061–6. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 4.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1996;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93(25):14960–5. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 7.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 9.Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 11.Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278(39):37948–56. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 12.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24(5):1101–12. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102(1):244–9. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144C:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- 17.Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE) Mol Med. 1995;1:827–837. [PMC free article] [PubMed] [Google Scholar]
- 18.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 19.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004;37:433–441. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- 22.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 23.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dringen R, Gutterer JM, Gros C, Hirrlinger J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J Neurosci Res. 2001;66:1003–1008. doi: 10.1002/jnr.10042. [DOI] [PubMed] [Google Scholar]
- 25.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93(24):13943–8. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280(24):22925–36. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 27.Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 28.Shih AY, Li P, Murphy TH. A Small-Molecule-Inducible Nrf2-Mediated Antioxidant Response Provides Effective Prophylaxis against Cerebral Ischemia In Vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147(1):53–9. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43(3):408–14. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38(12):3280–6. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 32.Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27(38):10240–8. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem. 2006;98(6):1852–65. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- 34.Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97(3):687–96. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 35.Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci U S A. 2006;103(3):768–73. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2007;104(4):1116–31. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6(1):154–62. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]