

SUMMARY
Dot1 (Disruptor of telomeric silencing-1) is a histone H3 lysine 79 methyltransferase that contributes to the establishment of heterochromatin boundary and has been linked to transcription elongation. We found that histone H4 N-terminal domain, unlike other histone tails, interacts with Dot1 and is essential for H3 K79 methylation. Furthermore, we show that the heterochromatin protein Sir3 inhibits Dot1-mediated methylation and that this inhibition is dependent on lysine 16 of H4. Sir3 and Dot1 bind the same short basic patch of histone H4 tail, and Sir3 also associates with the residues surrounding H3 K79 in a methylation-sensitive manner. Thus, Sir3 and Dot1 compete for the same molecular target on chromatin. ChIP analyses support a model in which acetylation of H4 lysine 16 displaces Sir3, allowing Dot1 to bind and methylate H3 lysine 79, which in turn further blocks Sir3 binding/spreading. This draws a detailed picture of the succession of molecular events occurring during the establishment of telomeric heterochromatin boundaries.
INTRODUCTION
Covalent modifications of residues on histone proteins have been the focus of intense study over the past 10 years, and their importance has been highlighted during gene regulation/transcription, the establishment of euchromatin/heterochromatin, and the maintenance of genome integrity. Histone modifications can affect chromatin structure and/or have a signaling role. They can be recognized by specific protein domains present in many types of nuclear protein/complexes with distinct roles in chromatin structure, genome expression, and stability. Different residue-specific modifications have also been shown to affect others within the same histone protein or between different histones in chromatin. Various combinations of these histone marks are thought to form a signature that specifies local chromatin states for accurate regulation/ function (reviewed in Kouzarides, 2007; Millar and Grunstein, 2006).
Several protein complexes harboring histone acetyltransferase/deacetylase (HAT/HDAC), methyltransferase/demethylase (HMT/HDM), kinase/phosphatase, or ubiquitylase/deubiquitylase activities have been characterized, many of them also containing specific histone mark-binding modules or even more than one modifying enzyme (Kouzarides, 2007; Lee and Workman, 2007; Shilatifard, 2006). While histone acetylation is generally linked to transcription, methylation of specific lysine residues on histones can correlate with transcription activity, i.e., H3K4, H3K36, and H3K79; or silencing/heterochromatin, i.e., H3K9, H3K27, and H4K20 (Kouzarides, 2007; Millar and Grunstein, 2006; Shilatifard, 2006).
While most histone lysine methyltransferases contain a SET domain, Disruptor of telomeric silencing-1 (Dot1/ KMT4) is an exception. Two features of this enzyme distinguish it from other known HMTs. First is its substrate requirement for chromatin, not free histones, and second is its modification of a lysine residue within the globular region of histone H3, away from the usual modification platforms that are the N- and C-terminal domains (Feng et al., 2002; Lacoste et al., 2002; Ng et al., 2002; van Leeuwen et al., 2002). Dot1 is responsible for all H3K79 methylation (mono-, di-, and tri-) in budding yeast, a mark predicted to be at the surface of the nucleosome (Ng et al., 2002; van Leeuwen et al., 2002). Deletion of DOT1 leads to telomeric silencing and meiotic checkpoint defects (San-Segundo and Roeder, 2000; Singer et al., 1998). The majority of H3K79 is methylated in yeast (van Leeuwen et al., 2002), a large portion of which is linked to chromatin on transcribed coding regions and regulated by the Paf1 complex and histone H2B monoubiquitylation on lysine 123 (reviewed in Shilatifard, 2006). Dot1-dependent methylation of H3K79 is also important for DNA damage response since the Tudor domain of Rad9/53BP1 recognizes this mark on chromatin surrounding DNA double-stranded breaks (DSBs), an interaction required for G1/S checkpoint response (Huyen et al., 2004; Wysocki et al., 2005). Crystal structures of the yeast and human Dot1 proteins have been obtained and identified a conserved core region and mechanism of catalysis (Min et al., 2003; Sawada et al., 2004). Finally, hDOT1L has recently been directly implicated in leukemogenesis through misregulation of HOX genes (Okada et al., 2005).
One of the most striking phenotypes of dot1 mutant cells is the defect in telomeric silencing. The SIR complex, formed by Sir3, Sir4, and the Sir2 H4 K16 deacetylase, assembles and spreads from the end of chromosomes to form telomeric heterochromatin (Rusche et al., 2003; Shahbazian and Grunstein, 2007). Its spreading into neighboring euchromatin regions is thought to be blocked in part by specific histone modifications and histone H2A variant Htz1 (Meneghini et al., 2003; Shahbazian and Grunstein, 2007). Indeed, Sir3 binds chromatin through histone H4 tail, an interaction that is disrupted by H4 K16 acetylation (Carmen et al., 2002; Millar et al., 2004). Thus, a key determinant in the establishment of telomeric heterochromatin boundaries is the opposite actions of H4 K16 deacetylase (Sir2) and acetyltransferase (Sas2) (Kimura et al., 2002; Shia et al., 2006; Suka et al., 2002). Clearly, other modifications are also involved since histone H3 K79A mutant and Dot1 suppression/overexpression disrupt gene silencing by telomere position effect (TPE) (Ng et al., 2002; Singer et al., 1998; van Leeuwen et al., 2002). On the other hand, the underlying molecular events responsible for establishment of telomeric heterochromatin boundaries and their intricate relationships are far from being fully understood.
In this report, we present findings that shed light on the role of Dot1 and H3 K79 methylation in blocking the spreading of heterochromatin. We show that Dot1 needs to bind histone H4 tail in order to methylate histone H3 K79 residue in chromatin. Interestingly, Sir3 and Dot1 compete for the same short basic patch of H4 for binding to chromatin, but only Sir3 interaction is sensitive to H4 K16 acetylation. Thus, Sir3 binding to H4 tail blocks chromatin methylation by Dot1, an effect that can be suppressed by an H4 K16Q mutant or Sas2 HAT overexpression. Furthermore, we show that Sir3 also interacts with the region surrounding lysine 79 of H3, an interaction highly sensitive to Dot1-dependent methylation. Altogether, these results draw a detailed picture of the succession of events required to establish telomeric heterochromatin boundaries and underline the interplay of various modifications on distinct histones to differentially regulate binding to chromatin.
RESULTS
Histone H4 N-Terminal Tail Region Is Essential for Methylation of H3 at Lysine 79 In Vivo
Our initial interest was to determine putative crosstalk of histone H4 acetylation with other histone modifications. We measured different histone marks by western on whole-cell extracts from yeast strain harboring deletions of different histone tail regions (Figure 1A). The results clearly indicate that deletion of histone H4 N-terminal tail wipes out bulk methylation of histone H3 on lysine 79 in vivo, suggesting a putative crosstalk between these two histone regions. Other histone tail deletions do not affect levels of H3K79me2. To investigate if this crosstalk involved acetylation of the conserved lysine residues of H4 tail, we then looked at a mutant strain where all four lysines have been mutated. As shown in Figure 1B, the bulk methylation level of H3 K79 is not affected in this mutant. H4 point mutants for phosphorylation of serine 1 or methylation of arginine 3 were also tested and did not affect H3K79me3 either (see Figure S1A available online). Importantly, other methylated lysines of H3 are not affected by the H4 tail deletion (H3K4me3 and H3K36me3; Figure 1C and data not shown).
Figure 1. Histone H4 N-Terminal Tail Is Essential for Dot1-Dependent Methylation of Nucleosomal H3 at Lysine 79.

(A) Histone H4 tail is required for H3 K79 methylation in vivo. Western blot of RIPA extracts from wild-type, H4ΔN (Δ4–19), H3ΔN (Δ3–29), H2AΔN (Δ5–21), and H2AΔC (Δ121–132) yeast cells using anti-H3 diMeK79 antibody.
(B) Histone H4 lysine residues targeted for acetylation are not required for H3 K79 methylation in vivo. Western analysis of RIPA extracts was performed as in (A) including cells carrying K to Q substitutions for all four lysine residues of H4 tail domain. Anti-total H3 signals are included to monitor loading amounts.
(C) Histone H3 methylation at lysine 4 is not affected by H4 tail deletion. Western analysis of RIPA extracts was as in (A) using anti-H3 diMeK4 and anti-total H4 antibodies.
(D and E) Dot1 requires histone H4 tail to methylate H3 K79 on nucleosomes in vitro. HMT assays were performed using recombinant Dot1 and recombinant nucleosome core particles (rNCP) with or without H4 N-terminal tail (Δ2–20). Reactions were analyzed by gel/fluorography in (D) and filter binding/liquid scintillation counting in (E) (average of duplicate reactions with standard deviations).
Dot1 Requires Histone H4 Tail to Methylate Nucleosomal Histone H3 In Vitro
Since point mutants for all known modifications of histone H4 tail did not reproduce the effect of its deletion on H3K79 methylation in vivo, we investigated whether the region was directly required for Dot1 to methylate chromatin. We used recombinant nucleosome core particles with or without H4 tail region in HMT assays with purified recombinant Dot1. As shown in gel or liquid assays (Figures 1D and 1E), while Dot1 efficiently methylates recombinant nucleosomes, it is completely unable to modify the ones lacking the H4 tail region. These results suggest that Dot1 directly interacts with the H4 tail region during its association with nucleosomes and that this interaction is essential for H3K79 methylation by Dot1 in vivo and in vitro.
Dot1 Directly Binds Histone H4 Tail, an Interaction Not Required for Binding to Nucleosomes
To investigate if Dot1 does directly interact with the histone H4 tail, we performed GST pull-down assays with recombinant Dot1 and GST fusion proteins harboring the H4 tail region in its wild-type form or with its lysine residues mutated to glutamines. As shown in Figure 2A, Dot1 does efficiently bind H4 tail region in vitro. This interaction is unaffected by the lysine mutants, in agreement with the results in vivo (Figure 1B). Sir3, a known H4 tail-binding protein, is used for comparison (Figure 2B). In this case, mutation of the lysine residues greatly affects Sir3 binding, as previously reported (Hecht et al., 1995).
Figure 2. Dot1 Directly Binds the Histone H4 Tail, but This Interaction Is Not Required for Binding to Nucleosomes.
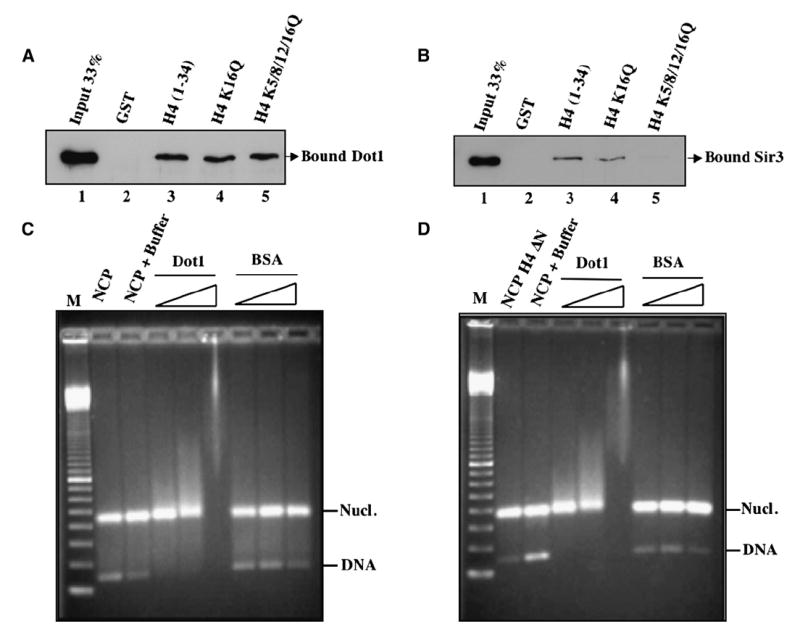
(A) Dot1 binds histone H4 tail region irrespective of acetylatable lysine residues. GST pull-down assays were performed using recombinant His-Dot1 and beads harboring GST-H4 fusion proteins (wild-type or K-to-Q substitution mutants at the indicated positions). GST is used as negative control. After washes, input (33%) and bound (100%) material were run on a 12% SDS-PAGE and revealed by western analysis using anti-His antibody.
(B) Sir3 binds histone H4 tail region in a lysine-dependent manner. GST pull-down assays were as in (A) but using recombinant His-Sir3 (620–978).
(C and D) Dot1 binds nucleosome core particles irrespective of histone H4 tail. Gel mobility shift assays were performed using recombinant nucleosome core particles (1.5 μg) with or without histone H4 tail (Δ2–20) and increasing amounts of Dot1 (0.1, 0.3, and 1 μg). Binding reactions were run on a 1.8% agarose gel followed by ethidium bromide staining. Mononucleosome and naked DNA bands are indicated. BSA was used as control. Marker is 100 bp ladder.
Since Dot1 interaction with H4 tail is essential for methylation to occur, a simple explanation would be that the interaction allows Dot1 to first bind nucleosomes, allowing subsequent modification of H3 K79. We performed mobility shift assays using Dot1 and recombinant mononucleosomes with or without H4 tail. As shown in Figures 2C and 2D, increasing amounts of Dot1 associate with nucleosome core particles and shift them up the gel while BSA has no effect. Interestingly, the lack of H4 tail has no effect on nucleosome binding by Dot1. This could be explained by the fact that Dot1 has intrinsic affinity for DNA and the domain responsible for it has been mapped (Sawada et al., 2004). Thus, Dot1 binds nucleosomes independently of H4 tails, but this binding is not sufficient for methylation of K79 of H3. This suggests that H4 tail binding by Dot1 on nucleosomes positions the enzyme in a proper orientation for its catalytic core to methylate K79 of H3.
Sir3 Inhibits Methylation of Chromatin by Dot1 In Vitro in an H4 Lysine 16-Dependent Manner
Based on the opposing relationship between the SIR complex and H3 methylation by Dot1 near telomeres (Ng et al., 2003; van Leeuwen et al., 2002), our finding of Dot1 interacting with H4 tail raised an interesting question about the impact of Sir3 binding to the same H4 tail. We performed HMT assays with Dot1 and recombinant mononucleosomes, adding increasing amounts of Sir3 protein to the reaction. We used as control another telomeric protein, Rap1, a DNA-binding factor that does not associate with H4 tails (Rusche et al., 2003). As presented in Figure 3A, Sir3 acts as a strong inhibitor to Dot1-dependent methylation, presumably by blocking Dot1 access to the H4 tail. Similar results were obtained with full-length Sir3 protein purified from yeast (data not shown). To test if this inhibitory effect could also occur on more physiological chromatin substrates, we repeated the experiment with purified native oligonucleosomes and obtained similar results (Figure 3B). Sir3 has been reported to also bind histone H3 tail, but it is binding to H4 tail that is sensitive to acetylation, primarily at H4K16 (Millar et al., 2004). Thus, an interesting mechanism would involve acetylation of H4K16 to disrupt Sir3 interaction, allowing Dot1 to bind and methylate H3K79. To test this hypothesis, we purified native yeast chromatin from wild-type, K16Q, and K5/8/12/16Q H4 strains and tested Sir3 inhibition of Dot1 activity. The results presented in Figure 3C indicate that mutation of H4 K16 to an acetylation mimicking glutamine is sufficient to suppress inhibition by Sir3. These data demonstrate that Sir3 and Dot1 compete for binding to histone H4 tail in chromatin and suggest that K16 acetylation near telomeres indirectly facilitates Dot1 binding by blocking Sir3 association.
Figure 3. Heterochromatin Protein Sir3 Specifically Blocks Nucleosomal H3 Methylation by Dot1 in an H4 Lysine 16-Dependent Manner.
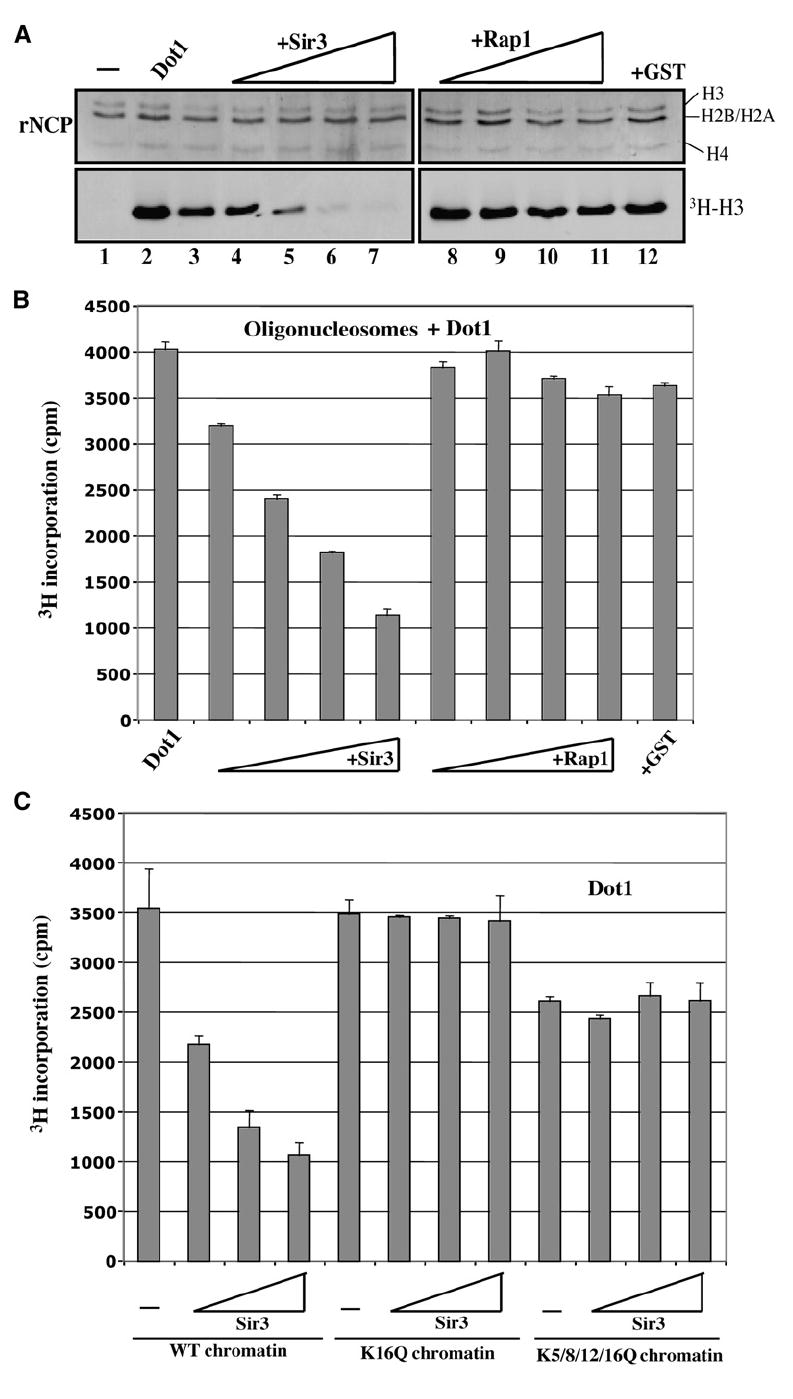
(A) Sir3 inhibits nucleosomal H3 methylation by Dot1. HMT assays are as in Figure 1D using recombinant nucleosome particles, Dot1, and increasing amounts of Sir3 or Rap1 proteins (0.5, 1.0, 1.5, and 2.0 μg).
(B) HMT assays were performed as in Figure 1E using purified native oligonucleosomes from human, Dot1, and increasing amounts of Sir3 or Rap1 (0.5–2.0 μg).
(C) Sir3 requires histone H4 lysine 16 to inhibit Dot1-dependent methylation in vitro. HMT assays as in (B) using purified native chromatin from yeast cells expressing wild-type histone H4 or substitution mutants for the indicated positions of lysine residues. Increasing amounts of Sir3 (1, 2, and 4 μg) were added to the reactions.
Histone H4 Basic Patch Is Essential for Dot1 to Methylate Chromatin
The region within the H4 tail that is essential for Sir3 binding has been mapped, and the short basic patch formed by residues 16–20 (KRHRK) is critical for Sir3 association (Hecht et al., 1995; Millar et al., 2004). Along with the H4 K16Q mutant, we also analyzed Dot1-dependent methylation with an H4 R17/19A mutant, expecting a similar effect on inhibition by Sir3. We were surprised to find a complete loss of bulk H3 K79 methylation in vivo in a R17/19A mutant, while H3 K4 methylation or H4 acetylation is unaffected (Figure 4A and Figure S1B). Since the abundant ISWI ATP-dependent chromatin remodeling complexes have also been shown to bind H4 basic patch (Clapier et al., 2002; Fazzio et al., 2005), it was important to test if the effect on H3 K79 methylation was indirect by affecting ISWI association throughout the yeast genome. This is not the case, since deletion of ISWI paralogs ISW1 and ISW2 has no effect on bulk levels of H3 K79 methylation (Figure 4B).
Figure 4. Histone H4 Basic Patch Residues Are Essential for Chromatin Methylation by Dot1 In Vivo and In Vitro.
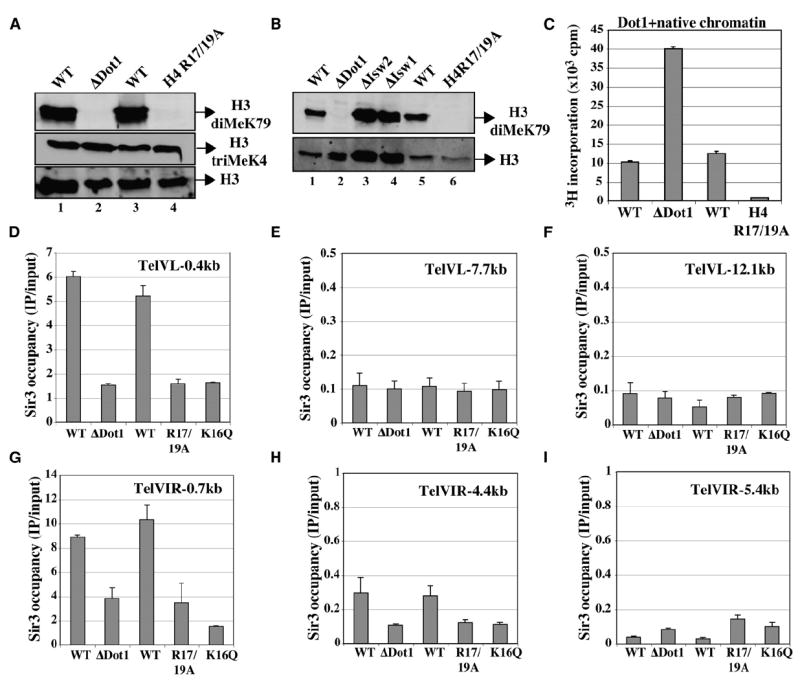
(A) Point mutants of histone H4 arginine residues at positions 17 and 19 lead to loss of Dot1-dependent methylation of H3 in vivo. Western analysis of RIPA extracts (as in Figures 1A–1C) from DOT1 deletion or histone H4 R17/19A substitution mutant cells and their isogenic wild-types was performed using anti-H3 diMeK79, anti-H3 triMeK4, and anti-H3 (total).
(B) Abundant Isw1/2 ATP-dependent chromatin remodelers that interact with histone H4 basic patch do not regulate bulk levels of Dot1-dependent H3 methylation in vivo. Western analysis was performed as in (A), including RIPA extracts from isw1 and isw2 mutant cells.
(C) Dot1 requires arginine residues of histone H4 tail basic patch to methylate H3 K79 in chromatin in vitro. HMT assays were performed as in Figure 1E using Dot1 and yeast native chromatin purified from DOT1 deletion or histone H4 R17/19A substitution mutant cells and their isogenic wild-types. Since the majority of H3 K79 is methylated in wild-type cells in vivo, ΔDot1 mutant chromatin was used as control for the H4 mutant chromatin (neither contain MeK79, as shown in [A]).
(D–I) In vivo association of Sir3 protein with telomeric regions is greatly decreased in dot1, H4 K16Q, and H4 R17/19A mutant cells. Sir3 binding to the telomeres of chromosome V left arm (D–F) and VI right arm (G–I) was analyzed by ChIP in the indicated strains. Occupancy represents IP/input signal (%) using a strain carrying chromosomal SIR3 tagged with 13 Myc epitopes. Signals are determined by real-time PCR with primers corresponding to the indicated positions relative to the end of the chromosomes. These values are based on two independent experiments with standard errors.
These data suggest that Dot1 binds the same short basic patch of histone H4 tail as Sir3. To test this, we purified native yeast chromatin from the R17/19A mutant strain and used it in HMT assay with Dot1. Since the majority of H3 K79 is already methylated in wild-type native chromatin (van Leeuwen et al., 2002), but not in the R17/19A mutant, we had to use a control chromatin purified from a DOT1-deleted strain to compare methylation levels (both having no H3K79me to start with). As expected, Dot1 is much more active in vitro on ΔDot1 chromatin compared to wild-type, reflecting the higher ratio of free K79 available (Figure 4C). In contrast, Dot1 is unable to methylate chromatin harboring the H4 R17/19A mutation (Figure 4C). This effect is not general to all proteins/enzymes binding the H4 tail, since in vitro chromatin acetylation by the NuA4 HAT complex is not affected by the R17/19A mutation (Figure S1C). These results confirm that Dot1 needs to bind H4 basic patch region in order to methylate chromatin and that this interaction is surprisingly unaffected by the adjacent K16, in contrast to Sir3. This differential effect of K16 on Sir3 and Dot1 binding to the basic patch is clearly meaningful in blocking the spreading of telomeric heterochromatin. Chromatin immunoprecipitation (ChIP) analysis of Sir3 occupancy at telomeres on chromosomes V and VI confirmed that ΔDot1, H4 R17/19A, and H4 K16Q mutants all greatly diminished Sir3 association to chromosomes ends in vivo (Figures 4D–4I). These signals truly reflect SIR complex association, since Sir2 occupancy is similarly affected in the mutant strains (Figures S2A–S2E).
Sir3 Binds H3 Lysine 79 Peptide Region In Vitro, an Interaction Disrupted by Methylation
While the H4 basic patch mutants directly block Sir3 from binding to telomeres in vivo (Hecht et al., 1996), the loss of Dot1 decreases this local Sir3 binding by allowing it to invade euchromatin (Ng et al., 2003; van Leeuwen et al., 2002). Overexpression of Dot1 in vivo should favor it to compete with Sir3 for binding to the H4 tail basic patch in chromatin. Indeed, ChIP analysis shows that Dot1 over-expression increases methylation of H3 K79 at telomeres on chromosomes V and VI while Sir3 occupancy is concomitantly decreased (Figure 5A, left and middle). Importantly, levels of histone H4 K16 acetylation are not affected by Dot1 overexpression, arguing that Dot1 action is directly responsible for Sir3 displacement (Figure 5A, right). Also in agreement with Sir3-Dot1 competition on the H4 tail, overexpression of Sir3 instead was previously shown to decrease H3 K79 methylation near telomeres (Ng et al., 2003).
Figure 5. Dot1 and H3 K79 Methylation Directly Affect the Ability of Sir3 to Bind Chromatin.

(A) Overexpression of Dot1 increases methylation of H3 K79 near telomeres with a corresponding decrease in Sir3 occupancy while not affecting acetylation of H4 K16. ChIP analysis as in Figure 4E was performed in a strain carrying a galactose-inducible DOT1 gene on a high-copy plasmid (over a wild-type DOT1 background). IPs were done on chromatin prepared from mid-log phase cells grown from 2 hr in glucose or in galactose. IPs were done with anti-H3 triMeK79 and anti-total H3 (left), anti-Myc (middle), or anti-H4 AcK16 and anti-total H3 (right). Data are presented as the change of IP/input signals when Dot1 is overexpressed (standard errors are based on two independent experiments). Total H3 signal is used to correct for potential changes/variations in nucleosome occupancy (for MeK79 and AcK16).
(B) Sir3 binds to peptides corresponding to histone H3 K79 region in a methylation-sensitive manner. Sir3 pull-down assays were performed with the indicated biotinylated peptides. Mock reaction is Sir3 incubated without peptides.
(C) Even monomethylation of K79 disrupts Sir3 interaction with H3 peptides. Pull downs were done as in (B) with Sir3 and the indicated peptides. The membrane was also probed with anti-biotin to monitor relative peptide amounts bound to the beads.
(D) Sir3 interaction with H3 N-terminal peptides is also regulated by site-specific methylation but seems less sensitive. Pull-down assays were done as in (C) with Sir3 and the indicated peptides (reactions done at the same time but run on two different gels).
Previous results suggest that Dot1 binding to the H4 tail could be sufficient to block the spreading of Sir3 into euchromatin regions next to telomeres. While the H3 lysine 79 to alanine point mutation or Dot1 overexpression disrupts telomeric silencing/Sir3 binding, the H3Lys79Arg mutation, which keeps the positive charge but disallows methylation, has no effect or even increased silencing (Fry et al., 2006; Park et al., 2002; van Leeuwen et al., 2002). The absence of a phenotype for the H3 K79R mutation suggests that even though Dot1 cannot methylate the residue, its binding to the H4 tail is sufficient to block Sir3 spreading and to maintain normal telomeric heterochromatin boundaries. On the other hand, mutation of residues surrounding H3 K79 also creates silencing defects and affects binding of the SIR complex in vivo (Fry et al., 2006; Park et al., 2002; Thompson et al., 2003). Closer examination of that region in the nucleosome core structure indicates that the close proximity of H3 L1 and H4 L2 loops forms a cluster of basic residues, all of which affect telomeric silencing and SIR binding when mutated (Fry et al., 2006; Luger et al., 1997; Park et al., 2002; Thompson et al., 2003). Since this cluster of basic residues is reminiscent of Sir3/Dot1 target on the H4 tail, it raises the hypothesis that Sir3 could also interact with that region at the surface of the nucleosome and that methylation of K79 would disrupt this interaction. This would explain the silencing defects of point mutants of residues in this region that remove their positive charge. To test that hypothesis, peptide pull-down assays were performed with Sir3 and biotinylated peptides corresponding to the H4 tail and H3 K79 region, in unmodified or methylated forms (Figure 5B). The results obtained indicate that Sir3 does interact with the H3 K79 region in vitro, similar to its binding to the H4 tail. Strikingly, trimethylation of K79 completely disrupts this binding, whereas trimethylation of K20 on the H4 tail peptide has no effect on Sir3 association. We then verified if the level of methylation has a differential effect on Sir3 binding (Figure 5C). Surprisingly, even monomethylated K79 is sufficient to disrupt Sir3 association. Finally, we wanted to compare these results with Sir3 binding to the H3 tail region, since this was reported to be sensitive to Set1-dependent methylation of K4 (Santos-Rosa et al., 2004). In our binding assays, we also detected Sir3 binding to the H3 tail region, and we observed that this binding is sensitive to K4 methylation (Figure 5D). In contrast to methylated H3 K79 peptide, we could still detect some binding to the methylated H3 K4 peptide. Together, these results indicate that Sir3 and Dot1 target the same two regions within nucleosomes, i.e., the basic patches on the H4 tail and the globular region of H3. In addition, two distinct modifications disrupt binding of Sir3 to each region, i.e., K16Ac in H4 tail and K79Me in H3 globular domain.
Increase of H4 Lysine 16 Acetylation at Telomeres Stimulates Local H3 Lysine 79 Methylation
Based on the absence of telomere silencing defect in H3 K79R mutant strains (Park et al., 2002) we suggested that Dot1 binding to the H4 tail, not H3 K79 methylation per se, was sufficient to establish normal heterochromatin boundaries/block Sir3 spreading. In addition, this implies that Sir3 binding to the H4 tail is critical and forms the foundation for SIR complex binding to chromatin (binding to H3 tail and/or K79 basic patch is not sufficient) (Millar et al., 2004). Based on this model, disrupting binding of Sir3 by increasing acetylation of H4 K16 should favor association of Dot1 and H3 K79 methylation at telomeres. We tested this model by overexpressing the Sas2 H4 K16 acetyltransferase in vivo. In this experiment, we detect a large increase of H4 K16 acetylation near telomeres VL and VIR (Figure 6A). H3 K79 methylation is also greatly increased while there is a loss of SIR complex from the same regions (Figures 6B and 6C). Since the level of H3K79me is much higher than obtained by directly over-expressing Dot1 and H4 K16 acetylation was not affected by Dot1 (Figure 5A), these data support the upstream and primary role of H4 K16 acetylation in defining telomeric heterochromatin boundaries. In agreement with such a primary role, Sas2 overexpression also stimulated incorporation of histone variant Htz1 (H2A.Z) at telomeres (Figure 6D), confirming a recent report that linked acetylation of K16 to Htz1 incorporation near chromosome ends (Shia et al., 2006).
Figure 6. Increased Sas2-Dependent Acetylation of H4 K16 Greatly Enhances H3 K79 Methylation and Htz1 Incorporation near Telomeres.

Overexpression of Sas2 H4 K16 acetyltransferase greatly stimulates methylation of H3 K79 near a telomere in vivo. ChIP analysis as in Figure 5A was performed in a strain carrying a galactose-inducible SAS2 gene on a high-copy plasmid (over a wild-type SAS2 background). IPs were done on chromatin prepared from mid-log phase cells grown from 2 hr in glucose or in galactose. IPs were done with anti-H4 AcK16 (A), anti-H3 triMeK79 (B), Anti-Sir2 (C), anti-Htz1 (D), and anti-total H3. Data are presented as the change occupancy when Sas2 is overexpressed. Histone marks/variants are corrected for nucleosome occupancy using total H3 signal.
DISCUSSION
Since the identification of Dot1 as a HMT, chromatin modification by this enzyme has been implicated in many nuclear processes, namely establishment of telomeric chromatin boundaries, transcription elongation, and DNA damage response (Shilatifard, 2006; Wysocki et al., 2005). Methylation of H3 lysine 79 is prevalent in budding yeast chromatin and is found in coding regions of transcribed genes and near telomeres (Millar and Grunstein, 2006). It is therefore seen as a euchromatin-specific modification. A functional role of methylated K79 in DNA damage response is to allow binding of ScRad9 to chromatin near DSBs through the protein tudor domain (Wysocki et al., 2005). However, the precise mechanistic roles of Dot1/K79 methylation during transcription elongation and establishment telomeric heterochromatin boundaries are not understood. Our findings showing that Dot1 interacts with a short region of histone H4 tail in chromatin and that this interaction is essential for methylation of lysine 79 on H3 provide critical new information that shed light on its function in blocking the spreading of heterochromatin.
We showed that Sir3, the chromatin-binding component of the SIR complex, which spreads from chromosome ends to form heterochromatin and silence gene expression, is a potent inhibitor of Dot1-dependent methylation. This is explained by the fact that Sir3 and Dot1 compete for the same binding site on H4 tail, a short basic patch between residues 16 and 20. It was known that Sir3 binding to H4 tail is disrupted by acetylation lysine 16, but this is not the case for Dot1. This supports the concept that the regulation of H4 K16 acetylation by Sas2 acetyltransferase and Sir2 deacetylase is a key primary step controlling the establishment of heterochromatin boundaries (Figure 7) (Millar et al., 2004). Our results indicate that a pivotal function of H4 K16 acetylation is to displace Sir3, which in turn allows Dot1 to bind and methylate H3 K79. The role of AcK16 in Dot1-dependent methylation is restricted to the region near Sir3-containing chromatin since K16 mutation has no effect on bulk levels of MeK79 or in vitro HMT activity (Figures 1B and 3C).
Figure 7. Model of Succession of Events during the Establishment of Telomeric Heterochromatin Boundaries in Yeast.

Histone H4 tail and its K16 acetylation site is depicted as coming out the nucleosome cores. Methylation of H3 K79 is depicted by the “M”-marked red circle in the cores, and “Z”-marked ovals represent histone variant Htz1.
Our results also reconcile contradictory data present in the literature. While it was known that deletion of DOT1, its overexpression, or mutation of H3 lysine 79 to an alanine had similar effect on telomeric silencing (van Leeuwen et al., 2002), it was puzzling to see that a K79R mutation, which cannot be methylated, had no phenotype or even increased silencing (Park et al., 2002). These data suggest that Dot1 binding to the H4 tail, even without K79 methylation, is sufficient to block SIR complex spreading into euchromatin regions. It is clearly not that simple, since expression of catalytically dead mutants of Dot1 only slightly suppresses the telomeric silencing defects of DOT1 deletion/overexpression (van Leeuwen et al., 2002). Since Dot1 does not appear to methylate other histone residues in vitro (van Leeuwen et al., 2002), these results could reflect a lower affinity of the Dot1 mutants for chromatin. Maybe AdoMet binding by Dot1 induces a conformational change allowing coordinated binding to both H4 tail and H3 K79 region at the surface of the nucleosome. As for the H3 K79A mutation, it suggests that losing the positive charge has a similar effect as methylation (which does not remove the charge). Since mutation of other basic residues neighboring H3 K79 in the nucleosome also creates defect in silencing (Fry et al., 2006; Park et al., 2002; Thompson et al., 2003), we tested if in fact Sir3 was also interacting with a second basic patch in the nucleosomes, centered on H3 K79. We showed that it was the case and that the interaction is extremely sensitive to K79 methylation (Figure 5). Altogether, our results indicate that Sir3 and Dot1 target the same two positively charged patches on nucleosomes. Binding of Sir3 to both targets is apparently necessary for its function, while Dot1 binding to the H4 tail/ nucleosome without methylation could be sufficient to block Sir3 spreading (based on K79A/R phenotypes) (Figure 7).
The data and concepts presented in this report could also impact our understanding of DNA DSB repair. While Dot1-dependent methylation has been implicated for binding of ScRad9 and checkpoint activation (Wysocki et al., 2005), the SIR complex is also recruited near DSBs but at later time points (Martin et al., 1999). In this situation, one would expect that H3 K79 would have to be demethylated in order to allow local SIR (Sir3) binding.
It will be interesting to investigate the role of Dot1 in the establishment of chromatin boundaries in higher eukaryotes, since the mechanisms may significantly differ. The H4 K16 acetyltransferase and deacetylase have been identified in hMOF and Sirtuins (Michan and Sinclair, 2007; Smith et al., 2005), but no homolog of Sir3 or SIR complex has been characterized. Artificial targeting of human H4 K16 deacetylase Sirt1 to a gene in cultured cells results in local loss of H3 K79 methylation, heterochromatinization, and gene silencing (Vaquero et al., 2004). Furthermore, in higher eukaryotes, H4 is methylated on K20 in heterochromatin (Kouzarides, 2007). The impact of this modification on mammalian Dot1-binding activity needs to be studied. It is tempting to speculate that H4 MeK20 could regulate Dot1 binding to the basic patch as Sir3 does in yeast. Accordingly, single H4 molecules do not carry simultaneous AcK16 and MeK20 residues, and H4 K16 acetylation inhibits K20 methylation in vitro (and vice versa) (Garcia et al., 2007; Nishioka et al., 2002). Finally, a recent report identified two homologs of Dot1 in trypanosoma, each one being selective for di- or trimethylation of H3. Such selectivity in the level of methylation argues for distinct functional roles (Janzen et al., 2006).
The data presented here underline the critical role played by a short basic patch of histone H4 tail in chromatin function. This small region is now known to physically interact with Sir3, Sas2/MOF HAT, Sir2 HDAC, Dot1 HMT, and the ISWI family of ATP-dependent chromatin remodelers. Coordinated interaction of Sir3 or Dot1 with both the H4 tail and H3 K79 region is compatible with the previously established close proximity of these two regions in the nucleosome structure (Luger et al., 1997; Thompson et al., 2003). An independent study also identified Dot1 interaction with H4 tail and mapped the region necessary for binding to a short acidic patch in Dot1 (Fingerman et al., 2007).
Altogether, our results explain through a histone H4-H3 crosstalk why Dot1 methyltransferase activity can only modify chromatin substrates, not free histones/peptides. Even more importantly, they suggest a precise succession of molecular events leading to the establishment of telomeric heterochromatin boundaries, local control of histone H4 K16 acetylation specifying the direct binding of Sir3 or Dot1. Both proteins compete for the same molecular targets at the surface of the nucleosome. If Dot1 is allowed to bind, then methylation of H3 K79 directly blocks further Sir3 association/spreading from telomeres. The reported role of H2A variant Htz1 (H2AZ) in blocking heterochromatin spreading is also dependent on the H4 tail, since Htz1 incorporation near telomeres requires K16 acetylation (Figure 7) (Shia et al., 2006). On the other hand, the impact of Htz1 on telomeric silencing seems less dramatic than Dot1, suggesting that the incorporation and the role of Htz1 could be subsequent and accessory to the H4 acetylation/H3 methylation steps (Dhillon and Kamakaka, 2000). It will be very interesting to investigate the role of Dot1 in chromatin boundaries in higher eukaryotes. Distinct histone modifications could functionally interact with Dot1/H3 K79 methylation, but the primary role of H4 basic patch as regulatory target certainly remains. These findings also impact research efforts at dissecting Dot1 function in DNA damage response, transcription elongation, and leukemogenesis.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Yeast strains carrying different histone mutations have been described (Bird et al., 2002; Fazzio et al., 2005; Hirschhorn et al., 1995; Utley et al., 2005; Zhang et al., 1998) (O. Jobin-Robitaille and J.C., unpublished data). pRS425-GAL-FLAG-DOT1 yeast overexpression vector (2 μm) was derived from pRS416-ADH-FLAG-DOT1 (Fingerman et al., 2007), while GAL-SAS2 yeast overexpression strain (from a 2 μm vector), isw1, isw2, and dot1 deletion strains were obtained from Open Biosystems. The Sir3-13Myc-expressing yeast strain was obtained following standard PCR-mediated procedure to introduce 13 Myc epitopes at the C terminus of SIR3 (Longtine et al., 1998). Bacterial vectors for expression of recombinant Dot1, GST-H4, and GST-Rap1 have been described (Hecht et al., 1995; Lacoste et al., 2002; Nourani et al., 2004). PCR-mediated cloning of SIR3 (corresponding to amino acids 620–978; functional chromatin-binding/dimerization [Hecht et al., 1995]) into pET15b vector was performed following standard procedures.
Purification of Proteins, Nucleosome Core Particle, and Native Chromatin
Recombinant His6-tagged Dot1 (aa 1–582) and Sir3 (aa 620–978), GST-Rap1 (aa 629–827), -H4 (aa 1–34), -H4-K16Q (aa 1–34), and -H4-K5,8,12,16Q (aa 1–34) were bacterially expressed and purified with Ni2+-nitrilotriacetic acid-agarose (QIAGEN) or glutathione-Sepharose (GE) following the manufacturer’s protocol (Figure S3A). Tandem affinity-purified yeast NuA4 complex has been described (Lacoste et al., 2002).
Preparation of nucleosome core particles with recombinant histones from Xenopus was done as before (Selleck et al., 2005). Native H1-depleted oligonucleosomes were purified from HeLa cells as described (Utley et al., 1996). Yeast native chromatin was purified following a related procedure described previously (Lacoste et al., 2002) (see the Supplemental Data). After microccocal nuclease digestion, the chromatin samples were concentrated and fractionated by gel filtration (Superose 6, Amersham Biosciences) in high-salt buffer. Fractions were analyzed on DNA and protein gels (Figure S3B), and those containing dinucleosomes and longer chromatin fragments (>300 base pairs) were pooled and concentrated.
GST/Peptide Pull Down and Gel Shift Assays
GST-H4 fusion proteins (3 μg) coupled to glutathione beads were incubated with recombinant Dot1 or Sir3 (0.3 μg) in 25 mM HEPES (pH 7.5), 100 mM NaCl, 10% glycerol, 100 μg/ml BSA, 0.5 mM DTT, 0.1% Tween 20, and 1 μg/ml Leupeptin, Pepstatin, and Aprotinin for 1 hr at RT on a rotary shaker. Beads were then washed three times with binding buffer, and bound proteins were analyzed by western using anti-His antibody. Peptide pull-down assays were performed by incubating 1 μg of biotinylated histone peptides with Sir3 (0.2 μg) under similar conditions as above except that 150 mM NaCl was used. Peptides were then isolated with magnetic streptavidin beads and analyzed by western with anti-His and anti-biotin antibodies.
For gel shift assays, different amounts of recombinant Dot1 protein (0.1–1 μg) were incubated with 1.5 μg of nucleosome core particles in 10 μl binding buffer containing 20 mM Tris-HCl (pH 8), 4 mM EDTA, 1 mM PMSF, 0.5 mM DTT, and 3% glycerol for 1 hr at 30°C. Binding reactions were resolved on a 1.8% agarose gel and visualized by ethidium bromide staining.
Western Blotting and Histone Methyltransferase Assay
For analysis of in vivo bulk histone modifications in different mutant backgrounds, yeast cells were lysed in RIPA buffer (50 mM HEPES [pH 7.9], 2 mM EDTA, 0.25 M NaCl, 0.1% SDS, 0.1% DOC, 1% Triton X-100) with glass beads. Extracts were separated on 18% SDS-PAGE, transferred on nitrocellulose membranes, and probed with anti-di- or anti-trimethyl lysine 79 of H3 (gift of Y. Zhang/Abcam), anti-di- or anti-trimethyl lysine 4 of H3 (Upstate/Abcam), anti-acetyl lysine 8 or 12 of H4 (Upstate), and anti-H3 or anti-H4 (gift of A. Ruiz-Carrillo).
HMT assays were performed in 30 μl total volume using 0.5 μg of nucleosomes/chromatin and 1 μCi of [3H]AdoMet (S-adenosyl-L-[methyl-3H]methionine, 80 Ci/mmol; SAM) in HMT buffer (20 mM Tris/HCl [pH 8], 50 mM KCl, 0.1 mM EDTA, 5% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride) for 45 min at 30°C. For Sir3 inhibition assays, (oligo)nucleosomes were first incubated with Sir3 for 45 min at 30°C followed by addition of Dot1 and SAM. To visualize radiolabeled histones, the reactions were resolved on 18% SDS-PAGE, Coomassie stained, and subjected to fluorography/autoradiography. In parallel, methylation was quantified by spotting duplicate reactions on P81 filters, followed by washes and liquid scintillation counting (values are presented with standard deviations).
Chromatin Immunoprecipitation Assays
ChIPs were performed similarly to what has been described (Wysocki et al., 2005). Crosslinked chromatin was sonicated (Diagenode Bioruptor) to yield DNA fragments of an average size of 500 base pairs, and 100 mg was used for immunoprecipitations with antibodies against Myc (Babco), Sir2 (Santa Cruz), H3 trimethyl-K79 (Abcam), H4 acetyl-K16 (Serotec), Htz1 (Abcam), and H3 (C terminus, Abcam). Primers used in the PCR reactions were analyzed for linearity range and efficiency with a LightCycler (Roche) in order to accurately evaluate occupancy (percent of IP/input). All PCR reactions were at least duplicates, and variation was less than 15%. IP and chromatin samples were also repeated with similar results. The numbers presented with standard errors are based on two independent experiments. For the experiments with overexpressed Dot1 and Sas2, cells were grown in media containing raffinose (A600 = 0.6) followed by galactose induction for 2 hr.
Supplementary Material
Supplemental Data include Supplemental Experimental Procedures and three figures and can be found with this article online at http://www.molecule.org/cgi/content/full/28/6/■■■/DC1/.
Acknowledgments
We are grateful to B. Hnatkovich for the preparation of recombinant nucleosome core particles; I. Fingerman for Dot1 overexpression vector; M. Grunstein for the GST-H4 tail fusion vectors; O. Gozani and E. Smith for the biotinylated histone peptides; Y. Zhang and A. Ruiz-Carrillo for antibodies; and O. Jobin-Robitaille, F. Winston, S. Dent, T. Tsukiyama, and M. Smith for histone mutant yeast strains. This work was supported by grants from the National Institutes of Health (GM74183) (to S.D.B.) and the Canadian Institutes of Health Research (CIHR) (to J.C.). J.C. is a CIHR Investigator.
References
- Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- Carmen AA, Milne L, Grunstein M. Acetylation of the yeast histone H4 N terminus regulates its binding to heterochromatin protein SIR3. J Biol Chem. 2002;277:4778–4781. doi: 10.1074/jbc.M110532200. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Nightingale KP, Becker PB. A critical epitope for substrate recognition by the nucleosome remodeling ATPase ISWI. Nucleic Acids Res. 2002;30:649–655. doi: 10.1093/nar/30.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon N, Kamakaka RT. A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Mol Cell. 2000;6:769–780. doi: 10.1016/s1097-2765(00)00076-9. [DOI] [PubMed] [Google Scholar]
- Fazzio TG, Gelbart ME, Tsukiyama T. Two distinct mechanisms of chromatin interaction by the Isw2 chromatin remodeling complex in vivo. Mol Cell Biol. 2005;25:9165–9174. doi: 10.1128/MCB.25.21.9165-9174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- Fingerman IM, Li HC, Briggs SD. A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Dev. 2007;21:2018–2029. doi: 10.1101/gad.1560607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry CJ, Norris A, Cosgrove M, Boeke JD, Peterson CL. The LRS and SIN domains: two structurally equivalent but functionally distinct nucleosomal surfaces required for transcriptional silencing. Mol Cell Biol. 2006;26:9045–9059. doi: 10.1128/MCB.00248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CD, Hunt DF. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grun-stein M. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature. 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Bortvin AL, Ricupero-Hovasse SL, Winston F. A new class of histone H2A mutations in Saccharomyces cerevisiae causes specific transcriptional defects in vivo. Mol Cell Biol. 1995;15:1999–2009. doi: 10.1128/mcb.15.4.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazo-netis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- Janzen CJ, Hake SB, Lowell JE, Cross GA. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol Cell. 2006;23:497–507. doi: 10.1016/j.molcel.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet. 2002;32:370–377. doi: 10.1038/ng993. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lacoste N, Utley RT, Hunter JM, Poirier GG, Cote J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002;277:30421–30424. doi: 10.1074/jbc.C200366200. [DOI] [PubMed] [Google Scholar]
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112:725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar CB, Grunstein M. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol. 2006;7:657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- Millar CB, Kurdistani SK, Grunstein M. Acetylation of yeast histone H4 lysine 16: a switch for protein interactions in heterochromatin and euchromatin. Cold Spring Harb Symp Quant Biol. 2004;69:193–200. doi: 10.1101/sqb.2004.69.193. [DOI] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci USA. 2003;100:1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- Nourani A, Utley RT, Allard S, Cote J. Recruitment of the NuA4 complex poises the PHO5 promoter for chromatin remodeling and activation. EMBO J. 2004;23:2597–2607. doi: 10.1038/sj.emboj.7600230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Park JH, Cosgrove MS, Youngman E, Wolberger C, Boeke JD. A core nucleosome surface crucial for transcriptional silencing. Nat Genet. 2002;32:273–279. doi: 10.1038/ng982. [DOI] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- San-Segundo PA, Roeder GS. Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell. 2000;11:3601–3615. doi: 10.1091/mbc.11.10.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Bannister AJ, Dehe PM, Geli V, Kouzarides T. Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J Biol Chem. 2004;279:47506–47512. doi: 10.1074/jbc.M407949200. [DOI] [PubMed] [Google Scholar]
- Sawada K, Yang Z, Horton JR, Collins RE, Zhang X, Cheng X. Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J Biol Chem. 2004;279:43296–43306. doi: 10.1074/jbc.M405902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleck W, Fortin I, Sermwittayawong D, Cote J, Tan S. The Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase complex requires the Enhancer of Polycomb A domain and chromodomain to acetylate nucleosomes. Mol Cell Biol. 2005;25:5535–5542. doi: 10.1128/MCB.25.13.5535-5542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shia WJ, Li B, Workman JL. SAS-mediated acetylation of histone H4 Lys 16 is required for H2A.Z incorporation at subtelomeric regions in Saccharomyces cerevisiae. Genes Dev. 2006;20:2507–2512. doi: 10.1101/gad.1439206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol. 2005;25:9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–383. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- Thompson JS, Snow ML, Giles S, McPherson LE, Grunstein M. Identification of a functional domain within the essential core of histone H3 that is required for telomeric and HM silencing in Saccharomyces cerevisiae. Genetics. 2003;163:447–452. doi: 10.1093/genetics/163.1.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley RT, Owen-Hughes TA, Juan LJ, Cote J, Adams CC, Workman JL. In vitro analysis of transcription factor binding to nucleosomes and nucleosome disruption/displacement. Methods Enzymol. 1996;274:276–291. doi: 10.1016/s0076-6879(96)74024-7. [DOI] [PubMed] [Google Scholar]
- Utley RT, Lacoste N, Jobin-Robitaille O, Allard S, Cote J. Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol Cell Biol. 2005;25:8179–8190. doi: 10.1128/MCB.25.18.8179-8190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 1998;17:3155–3167. doi: 10.1093/emboj/17.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Supplemental Experimental Procedures and three figures and can be found with this article online at http://www.molecule.org/cgi/content/full/28/6/■■■/DC1/.