

Abstract
Signaling proteins are localized and regulated by Src homology 2 domains which recognize phosphotyrosine-containing sequences. Recently, non-canonical ligands have been proposed for Src homology 2 domains including that of Abl and its breakpoint cluster region fusion, which causes chronic myelogenous leukemia. Here, the Abl Src homology 2 domain’s binding sites and affinities for phosphotyrosine- and phosphoserine-containing motifs, phosphoinositides as well as a pyridone-based peptidomimetic inhibitor were determined using nuclear magnetic resonance spectroscopy in order to define their roles. The cognate Crk peptide ligand was bound with an affinity of 69 μM and, like the higher affinity peptidomimetic, engages the phosphotyrosine and +3 hydrophobic pockets while putative phosphoserine-containing breakpoint cluster region ligands are ruled out. Surprisingly, phosphatidylinositol 4, 5 bisphosphate interacts with an overlapping site through an electrostatic mechanism that does not appear to involve hydrophobic insertion into micelles. The conserved Arg36 residue in the FLVRES motif is required for both phosphotyrosine binding and for localization to phosphatidylinositol 4, 5 bisphosphate-containing liposomes, while Arg59 in the βD strand is necessary for the phosphoinositide interaction. Thus the Src homology 2 domain of Abl, a myristoylated and membranelocalized protein, is able to interact directly with phosphoinositides through a multifunctional basic site that overlaps the phosphotyrosine pocket.
Keywords: NMR spectroscopy, phosphoinositide, phosphopeptide, pyridone, SH2 domain
Introduction
Tyrosine phosphorylation plays an essential physiological role by recruiting proteins that contain Src homology 2 (SH2) domains to signaling sites. However, Tyr phosphorylation represents only 0.05% of total protein phosphorylation in a human cell, and activated protein tyrosine kinase oncogene products increase this fraction to only about 0.5% (1). Alternative ligands for some SH2 domains have been proposed, and are more abundant than canonical phosphotyrosine (pTyr) ligands. However, there is little structural information about the recognition of alternative phosphorylated ligands, thus limiting the prediction of binding sites, targets and specificities.
Protein sequences that contain phosphoserine (pSer) have been suggested as possible SH2 ligands. For example, the Abl SH2 domain appears to recognize breakpoint cluster region (BCR) sequences in a pTyr-independent manner (2). BCR possesses a Ser/Thr kinase activity (3), and phosphorylates BCR residues including Ser354 and Ser356. Inhibition of the Bcr-Abl oncoprotein by Bcr requires its pSer 354-containing sequence, which reportedly interacts with the Abl SH2 domain (4). Moreover, the Raf-1 SH2 binds to Src family kinases in a pTyr-independent mechanism (5), the Lck SH2 domain binds pSer’s in Lck (6) and the p62 protein (7), and the SAP SH2 domain binds to an unphosphorylated tyrosine in SLAM (8,9), highlighting the functional diversity within the family.
Some SH2 domains including those of Src and phosphoinositide (PI) 3-kinase interact with PIs (10-13). Similarly, the membrane recruitment of PLC-γ1 involves the interaction of its C-terminal SH2 domain with PtdIns(3,4,5)P3 (11,12). Although no structures of lipid-bound SH2 domains have been reported, the interactions of PI’s and pTyr-peptides are competitive (13). Controversially, the PI interactions of the PI 3-kinase’s C-terminal SH2 domain have been reported as being relatively weak and non-specific (14) or specific for long chain PtdIns(3,4,5)P3 (10), emphasizing the need for comparative binding studies.
Despite the causal role of Abl in transformation and leukemia and its role in PI 3-kinase signaling (15), the identities of the ligands of its SH2 domain remain unclear: specific pSer and pTyr peptides and PIs have all been suggested, and its novel ligand interaction sites have not been experimentally mapped or disproven (4,13,16-18). Furthermore, Abl is recruited to various locations in the nucleus, cytosol and plasma membrane, suggesting a complex cellular targeting mechanism (19,20).
The Abl oncoprotein is the target of the first successful mechanism-based cancer drug, Glivec. This agent inhibits the kinase hyperactivity in BCR-Abl transformed cells (21). However in recent years, resistance to this drug has emerged (22). The BCR-Abl SH2 domain promotes transformation (23) and is required for PI3-kinase pathway activation (24). Moreover, BCR-Abl SH2 signaling specifically influences disease latency and phenotype in mice (25). Effective SH2 inhibitors have been developed, and focused on replacement of the phosphate group with non-hydrolyzable mimetics (26). Effective drug-like inhibitors of specific SH2 domains are still needed (27), such as the pyridone derivative investigated here that was designed to mimic pTyr’s interaction with SH2 domains. The pyridone moiety was designed to maintain ionic and hydrogen bond interactions with an Arg at the second position of the αA helix (denoted ArgαA2) and ArgβB5, while introducing a hydrogen bond with SerβB7, and being attached to a peptidomimetic developed to fit the P + 1 to P + 3 pockets (28). It therefore appeared to be suited for binding to the Abl SH2 domain, and hence was investigated here.
We have analyzed the Abl SH2 domain’s affinities and binding sites for its proposed ligands. We found that the Crk pTyr but not BCR pSer peptide binds as expected in the basic pocket, while the pyridone-based pTyr analog binds with the highest affinity. The Abl SH2 domain also interacts with phosphatidylinositol 4, 5 bisphosphate (PtdIns(4,5)P2) in a site that overlaps the pTyr pocket, thus localizing the protein to membranes. Importantly, a discriminatory point mutation that specifically compromises the SH2 domain’s PI binding function is designed and demonstrated.
Methods and Materials
Protein purification
The murine c-Abl sequence encoding residues Asn120-Arg220 was cloned into pGEX-2T as a N-terminal tagged-glutathione S-transferase (GST) fusion protein and included a vector-derived DLVPRGSG sequence (29). The protein was expressed in Escherichia coli strain BL21 (DE3) pLysS. The protein was purified with glutathione sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ, USA), cleaved with thrombin and concentrated.
NMR spectroscopy
Samples contained 0.15-1.2 mM unlabeled or uniformly 15N-labeled protein in 25 mM perdeuterated Tris-HCl, pH 7.0, 1 mM NaN3 and 1 μM APMSF, as well as 135 mM KCl where indicated. All spectra were recorded at 25 °C on Varian Inova 500 and 600 MHz nuclear magnetic resonance (NMR) spectrometers. Resonance assignments in the presence of phospholipids were confirmed using 15N-edited heteronuclear single quantum coherence (HSQC)-NOESY spectra as previously described (29). The NMR data were analyzed using NMRPipe (30) and in-house software programs (http://biomol.uchsc.edu) on Sun Microsystems Ultra 10 and Silicon Graphics 02 workstations, and plotted on the Abl SH2 structure represented by PDB 2ABL.
Ligand-binding assays
Lipid binding was monitored from 15N-edited HSQC spectra of 150-250 μM Abl SH2 domain collected in concentrations of up to 1.6 mM of dibutanoyl (C4) PtdIns(3)P PtdIns(4)P, PtdIns(3,4)P2, PtdIns(3,5)P2, PtdIns(4,5)P2 and PtdIns(3,4,5)P3, as well as Ins(1,4,5)P3 and dioctanoyl (C8) PtdIns(4,5)P2 (Echelon Biosciences Inc., Salt Lake City, UT, USA). Similar spectra were collected in the presence of up to 3.2 mM pTyr or pSer amino acids (Sigma, St Louis, MO, USA), as well as synthetic peptides (Colorado State University) containing Crk residue pTyr221, and BCR pSer 354 and pSer354/p356. The peptides had sequences EPGP[pY]AQPSVNTP, GQSSRV[pS]PSPTTY and GQSSRV[pS]P[pS]PTTY, respectively (where pY and pS refer to pTyr and pSer, respectively). A pyridone-based pTyr analog CDS-009804 of chemical formula C27H46O8N5P (28) was also titrated into the Abl SH2 domain. As the HSQC spectra indicated that the ligand interactions occurred in the fast exchange regime of the NMR timescale, the progressive shifts in 15N,1H cross peaks were monitored to assign resonances and measure perturbations. The perturbations were normalized as the weighted average of the 1H and 15N chemical shift changes for each amino acid according to the formula (31). All the Kd values were calculated using the NMR titration data collected at up to millimolar ligand concentrations by a nonlinear least square analysis using the Xmgr program and the equation Y = (P × L)/(Kd + L), where L is the concentration of the lipid, Y is the observed difference in chemical shifts in the presence and absence of ligand, and P is the protein concentration.
Site-directed mutagenesis
Single point mutations were introduced into the wild type protein using PCR with the following primers: for Arg36Lys, 5′-GGCAGCTTCTTGGT-GAAGGAGAGTGAGAGCAGTCCTGGC-3′; Arg45Gln, 5′GAGAGCAGTCCTGGCC- AGCAGTCCATCTCGCTG-3′; and Arg59Gln, 5′-GGGAGGGTGTACCATTACCGA- TCAACACTGCTTCTGATGGC-3′. All the point mutations were confirmed by DNA sequencing.
Liposome binding assay
Lipid mixtures containing 48% phosphatidylethanolamine (PtdEth) (Avanti Polar Lipids, Alabaster, AL, USA), 12% phosphatidylcholine (PtdCho) (Avanti Polar Lipids), and 40% of either dipalmitoyl PtdIns(4,5)P2 or PtdIns (Echelon Biosciences Inc.) were solubilized into a solution containing 69% chloroform and 27% methanol to 1 mg/ml, and lyophilized overnight. The lipid was hydrated in NMR buffer and frozen in liquid nitrogen and thawed thrice. The liposomes were then subjected to the rapid extrusion technique (Avanti Polar Lipids) and incubated for 1 h with 10 μg of either the wild type or mutant Abl SH2 protein. The mixture was spun for 10 min at 44,000 rpm, and protein which partitioned to the pelleted liposome fraction and supernatant was analyzed by SDS-PAGE gel electrophoresis.
Results and Discussion
NMR screening identifies PtdIns(4,5)P2 ligand
The interactions of a series of candidate Abl SH2 domain ligands were tested by HSQC NMR experiments. When soluble C4-PtdIns(4,5)P2 was titrated into the 15N-labeled protein sample, significant and progressive chemical shift changes were detected in several amide cross peaks (Figure 1A). The largest perturbations upon PtdIns(4,5)P2 binding were observed in the βB residue Ser32 and Glu39 located on either side of the FLVRES motif. Substantial perturbations were also observed for the residues located in and around the α1 helix and βA and βC strands including Tyr12, Gly14, Val16, Ser17, Asn19, Ala21, Gly43 and Arg45 to Ser48 (Figure 1B). The perturbations greater than the average plus one standard deviation were mapped onto the structure (Figures 1C and D), indicating that the majority of the large perturbations occurred around the pTyr binding site. PtdIns(4,5)P2 also caused significant chemical shift changes in the Arg45, Arg59, and Arg104 residues, which are positioned nearby for interactions. The amide resonances of SH2 residues in the hydrophobic +3 pocket that normally accommodates an apolar residue three residues C-terminal to the pTyr remain largely unperturbed, except for Val70 in the EF loop and the proximal Tyr58. Thus, SH2’s +3 site is unlikely to be fully occupied by the hydrophobic acyl chains of C4 PtdIns(4,5)P2 or C8 PtdIns(4,5)P2, which yielded similar chemical shift changes (data not shown). Small perturbations occurred outside the canonical binding pockets in only a few residues such as His98 and Arg104, suggesting that the integrity of the fold is preserved, with only minor conformational changes accompanying the PtdIns(4,5)P2 interaction.
Figure 1. PtdIns(4,5)P2 caused significant chemical shift changes upon binding to the Abl SH2 domain.

15N-edited HSQC spectra of Abl SH2 domain were recorded in the presence of 135 mM KCl, and increasing concentrations of PtdIns(4,5)P2. (A) The spectra were overlaid to show the chemical shift perturbations of NH peaks due to C4-PtdIns(4,5)P2 binding; several including the G14 peak are folded. (B) The chemical shift changes in each residues’ signals at 0.8 mM ligand concentrations were plotted into histograms, with secondary structure taken from PDB 1OPK. The residues exhibiting significant PtdIns(4,5)P2-induced perturbations are shown on the ribbon (C) and molecular surface (D) in red, orange, and yellow indicating large (>10.0), medium (6.0-10.0) and small (2.0-6.0) normalized amide chemical shift changes, respectively.
Binding affinity for PtdIns(4,5)P2
The PtdIns(4,5)P2 molecule must interact with the Abl SH2 domain with significant relative affinity in order to compete with the canonical pTyr ligands in the cellular environment. In order to estimate the Abl SH2 domain’s affinity for C4-PtdIns(4,5)P2, the magnitude of the chemical shift changes were plotted as a function of the phospholipid concentration. Binding affinities of 130 and 85 μM were measured for C4-PtdIns(4,5)P2 and its IP3 head group, respectively, in the absence of KCl (Table 1). In the presence of 135 mM KCl the binding affinity for both compounds was around 500 μM, indicating the predominantly electrostatic nature of the PtdIns(4,5)P2 interaction. A small stabilizing hydrophobic effect was also indicated by the 1.6 fold stronger affinity of the C8 over C4 form of PtdIns(4,5)P2, although a precise estimate of the former Kd was precluded by micelle formation at higher PI concentrations.
Table 1.
Binding affinities of Abl SH2 domain ligands
| Ligands | SH2 sequence | Affinity in low salt (mM) | Affinity in high salt (mM) |
|---|---|---|---|
| diC4-PtdIns(3,4,5)P3 | Wild type | 0.2 ± 0.007 | 1.3 ± 0.18 |
| diC4-PtdIns(4,5)P2 | Wild type | 0.13 ± 0.001 | 0.8 ± 0.08 |
| diC8-PtdIns(4,5)P2 | Wild type | - | 0.5 ± 0.07 |
| diC4-PtdIns(3,5)P2 | Wild type | 0.09 ± 0.002 | 3.5 ± 0.45 |
| diC4-PtdIns(3,4)P2 | Wild type | 0.26 ± 0.022 | 1.5 ± 0.15 |
| diC4- PtdIns(4)P | Wild type | 0.7 ± 0.23 | >10 |
| diC4-PtdIns(3)P | Wild type | - | 3.1 ± 0.76 |
| Ins(1,4,5)P3 | Wild type | 0.085 ± 0.005 | 0.5 ± 0.03 |
| Phosphatidicacid | Wild type | - | 3.7 ± 0.38 |
| Phosphatidylserine | Wild type | - | Not detectable |
| BCR pSer354 peptide | Wild type | - | Not detectable |
| BCR pSer354/pSer356 peptide | Wild type | - | 26.1 ± 9.3 |
| Crk pTyr221 peptide | Wild type | - | 0.069 ± 0.0045 |
| Pyridone | Wild type | - | 0.030 ± 0.0013 |
| diC4-PtdIns(4,5)P2 | Arg45Gln | - | 0.9 ± 0.06 |
| diC4-PtdIns(4,5)P2 | Arg59Gln | - | >10 |
| diC4-PtdIns(4,5)P2 | Arg36Lys | - | Not detectable |
| pTyr | Arg45Gln | - | 0.39 ± 0.013 |
| pTyr | Arg59Gln | - | 2.5 ± 0.20 |
| pTyr | Arg36Lys | - | Not detectable |
The affinities of candidate phospholipid and phosphopeptide ligands of the Abl SH2 domain in the absence and presence of 135 mM KCl are listed, as well as related compounds. Ligands were tested for binding to the 15N-labeled SH2 domain by HSQC titration experiments, and the chemical shift perturbations were fitted to the binding curve to estimate the affinity. The standard errors are noted, the affinities that were very weak but detectable are indicated as being “>10 mM”, while “-” indicates not measured.
Micromolar affinities for soluble PI molecules are typical for PI binding domains. For example, the Vam7p PX domain binds PtdIns(3)P with 300 μM affinity (32), while the FYVE domain of the EEA1 protein binds this ligand at affinities between 71 μM and 1.4 mM depending on the pH (33). The affinities of the PtdIns(3,4,5)P3 interaction of the p85α C-terminal SH2 domain ranges from >500, 93 and 23 μM for the C4, C8 and C16 forms, respectively (10). Thus the PtdIns(4,5)P2 affinity of the Abl SH2 domain is comparable to those of other domains for less abundant PIs.
Micelle insertion
Other protein modules that recognize PIs such as FYVE and PX domains insert hydrophobic clusters of residues into the bilayer’s hydrocarbon interior, thus enhancing membrane affinity (32,33). Consequently we tested whether the Abl SH2 domain also inserts into micelles. However, addition of dodecylphosphocholine or 1,2-diheptanoyl-phosphocholine up to and over the respective critical micelle concentrations caused only minimal chemical shift changes and did not significantly perturb any hydrophobic or aromatic residues near the PI interacting surface of the Abl SH2 structure, either in the presence or absence of PtdIns(4,5)P2 (data not shown). Thus interaction of the Abl SH2 domain with PtdIns(4,5)P2 does not involve detectable hydrophobic insertion into PC-based micelles.
PI specificity
Interactions with other soluble PIs were investigated in order to evaluate the intrinsic specificity of the Abl SH2 domain. Each lipid was titrated stepwise into a 15N-labeled Abl SH2 domain sample and HSQC spectra were collected. The profile of the chemical shift changes was generally similar to that seen with the titration of PtdIns(4,5)P2, indicating that a common set of residues can accommodate different PI headgroups (Table 1). However, the affinities of the other phospholipids were substantially lower, ranging from 1.3 mM for PtdIns(3,4,5)P3 to more than 10 mM for PtdIns(4)P in the presence of 135 mM KCl. The multiply phosphorylated PI’s generally bound more tightly and induced more extensive chemical shift perturbations than the monophosphorylated species, supporting an electrostatic mechanism. The interactions with phosphatidylserine (PtdSer) and phosphatidic acid (PtdOH) were very weak, with affinities in the millimolar range. Considering that PtdIns(4,5)P2 represents about 99% of the doubly phosphorylated PI and 5% of total PI in the cell (34), and is concentrated in membranes (35) where c-Abl is localized (17), it is reasonable to infer that PtdIns(4,5)P2 is the most likely PI to interact with the Abl SH2 domain, particularly in the absence of tyrosine phosphorylation of its protein ligands.
Phosphopeptide binding by the Abl SH2
The most established physiological ligand of the Abl SH2 domain is the Crk sequence spanning pTyr221, providing a basis for comparison with the PI interactions. The Crk pTyr221 peptide was titrated to a 16-fold molar excess into the Abl SH2 domain, and induced significant chemical shift changes (Figure 2A). Perturbations of the chemical shifts of residues on either side of Argα2A and Arg36, Glu37 and Ser38 in the FLVRES motif indicated a canonical interaction with the bound pTyr. These changes in chemical shifts were mapped onto the Abl SH2 domain, showing the exposed residues which are involved in phosphopeptide recognition (Figure 2C). Several residues C-terminal to Crk’s pTyr221 are also engaged based on the substantial chemical shift changes in several EF and BG loop residues. The affinity of the Abl SH2 domain for the Crk pTyr221 peptide was 69 μM at 135 mM KCl (Figure 2B). Thus the Crk ligand binds the Abl SH2 domain with a higher affinity than C4-PtdIns(4,5)P2 and comparable to C16-PtdIns(4,5)P2 (data not shown). There are some residues that overlap with those involved in PtdIns(4,5)P2 binding, such as Gly14, ValSer(16,17), Asn19, Ala21, Gly43, ArgSer-Ile (45-47) and GluSer (37,38), suggesting that the interactions would be mutually exclusive.
Figure 2. The Crk pTyr221 binds tightly to the Abl SH2 domain.
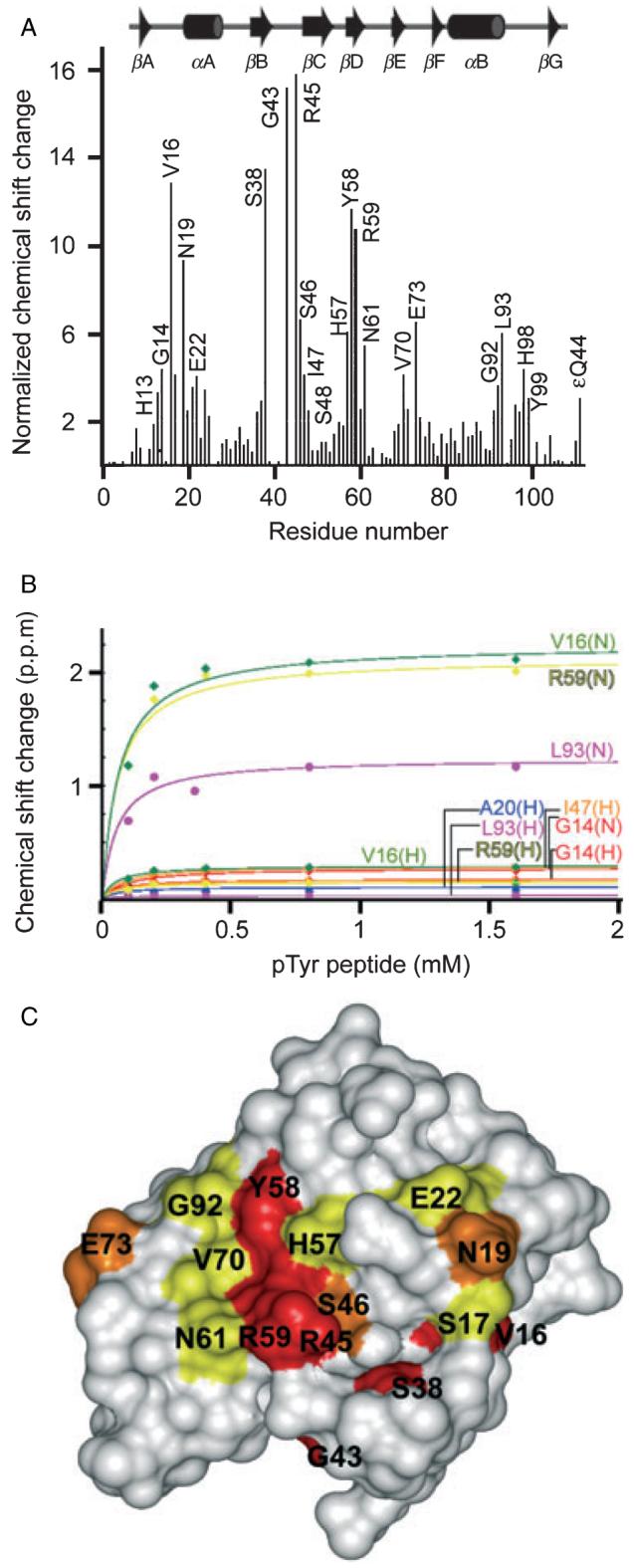
The Crk pTyr221 peptide was tested for binding to the Abl SH2 domain by HSQC NMR titration experiments. (A) Plotting of the chemical shifts into a histogram shows several residues affected upon pTyr-binding. (B) The binding affinity of 69 μM was calculated from the binding curve. (C) Structure with surface residues colored in red, orange, and yellow indicating large (>10.0), medium (6.0-10.0) and small (2.0-6.0) normalized chemical shift changes, respectively.
In addition, we investigated the interactions of the Abl SH2 domain with pSer-containing BCR sequences that had been observed by GST-pull down assays (4,16,36). The singly phosphorylated (pSer354) and the doubly phosphorylated (pSer354/pSer356) BCR peptides were titrated into the 15N labeled Abl SH2 domain sample to a 16 fold excess concentration. However neither peptide caused any significant chemical shift changes in the HSQC spectra (data not shown), indicating that the BCR pSer354 or pSer356 peptide interactions are nonexistent, extremely weak or indirect.
Mutations that selectively alter ligand binding
Based on the interaction sites mapped by NMR, we designed conservative point mutations in order to manipulate specific ligand affinities while preserving structural integrity. When the absolutely conserved Arg36 in the FLVRES motif was mutated to Lys, not only binding to pTyr but also to PtdIns(4,5)P2 was abolished (Figures 3A and D), consistent with its crucial role. The amide resonances of Arg45 and Arg59 are also perturbed by pTyr and PI binding, and are well-positioned for electrostatic interactions with the additional phosphates presented by the PtdIns(4,5)P2 ring. Each Arg was mutated to a Gln residue, and the ligand-induced chemical shift changes were compared to those observed with the wild type Abl SH2 domain. The Arg45Gln mutant had a similar affinity for C4-PtdIns(4,5)P2 compared to the wild type protein (Table 1), and the ValSer (16-17), Ala21, Gly43, and Val70 resonances were less involved based on their minimal chemical shift changes, while Gly31, Gln45 and Thr88 were more perturbed (Figure 3B and Table 1). On the other hand, the same Arg45Gln mutant showed a 0.39 mM affinity for pTyr (Table 1 and Figure 3E), indicating that this residue is not required for pTyr binding. Importantly, the Arg59Gln mutation almost completely disrupted PtdIns(4,5)P2 interaction based on the lack of significant chemical shift changes, while the pTyr affinity was reduced to millimolar levels (Table 1), suggesting that Arg59 is absolutely required for the PtdIns(4,5)P2 interaction and also contributes to pTyr binding (Figures 3C and F). Interestingly, the Arg59 residue is conserved in the PLCγ SH2 domain which binds PIs with high affinity (11,12), suggesting that it is a predictor of dual PI/pTyr specificity.
Figure 3. Effects of Arg mutations in the Abl SH2 domain.

Basic residues near the Abl SH2 domain binding site were mutated conservatively. The interactions of C4-PtdIns(4,5)P2 and pTyr amino acid at final concentrations of 0.8 mM and 3.2-4 mM, respectively, were analyzed by HSQC NMR titrations and compared to the wild type (see Figures 1 and 2) for each of three different mutants Arg36Lys (A, D), Arg45Gln (B, E) and Arg59Gln (C, F). HSQC spectra were recorded and overlaid to visualize the chemical shift changes which were plotted by residue position in the histograms.
PtdIns(4,5)P2-containing bilayers recruit the Abl SH2 domain
We investigated the membrane localization of the Abl protein through PtdIns(4,5)P2 binding by its SH2 domain. Liposomes were prepared using a mixture of PtdEth, PtdCho and PI lipids to mimic the membrane composition. The wild type Abl SH2 domain interacted specifically with PtdIns(4,5)P2 containing liposomes while the Arg36Lys mutant failed to associate with any liposome (Figure 4). This parallels the NMR binding data, and underscores the critical role of the FLVRES motif in recognition of PI-containing membranes. Since Abl is myristoylated on its N-terminal Gly residue (37) and is localized to the cell membrane (38) and apparent cytoplasmic vesicles (20,39), we propose that Abl docking to the membrane also involves the interaction of the SH2 domain’s basic site with PtdIns(4,5)P2, particularly in the absence of tyrosine hyperphosphorylation following mitogenic stimulation or cellular transformation.
Figure 4. PtdIns(4,5)P2-containing liposomes interact with the Abl SH2 domain.

The wild type Abl SH2 and Arg36Lys mutant (10 μg final protein) were incubated with liposomes containing 40% of PtdIns(4,5)P2 or PtdIns, 48% PtdEth and 12% PtdCho, centrifuged to separate the supernatant and pelleted liposome fractions, and identified by SDS PAGE.
Pyridone peptidomimetic interaction
We sought to define the binding mode and affinity of the pyridone-containing compound CDS-009804 by HSQC NMR experiments. The peptidomimetic bound more tightly to the Abl SH2 domain than any other ligand, with an estimated Kd of 30 μM in physiological salt levels (Figure 5 and Table 1). The CDS-009804 interaction induced significant chemical shift changes comparable to the Crk pTyr221 peptide, indicating engagement of both the basic pTyr and the hydrophobic pTyr +3 pockets (Figure 5A). Interaction of the FLVRES motif with the pyridone is indicated by the Ser38 perturbation. However, the amide resonances in the Ser17Arg18Asn19 sequence are less affected, suggesting a compromised interaction with αA residue Arg18. The chemical shift changes were plotted into histogram (Figure 5B), and the largest perturbation was observed for Arg59, a residue shown here to be particularly important for PtdIns(4,5)P2 binding. Mapping chemical shift perturbations onto the backbone ribbon and surface of the SH2 domain (Figures 5C and D) reveals that the pyridone, PI and pTyr binding sites overlap extensively, and suggests mutually exclusive interactions. Thus the CDS-009804 compound is a promising lead as a relatively high affinity competitive inhibitor of the Abl SH2 domain’s physiological interactions.
Figure 5. Pyridone-based pTyr analog binds to the Abl SH2 domain.
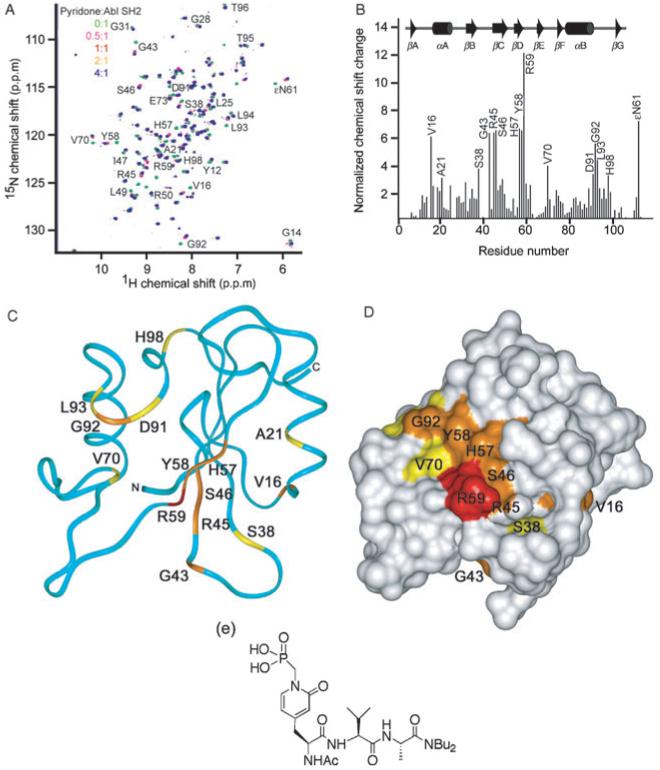
The CDS-009804 compound was tested for binding to the Abl SH2 domain by HSQC NMR titration experiments. Plotting of the chemical shifts into a histogram shows several residues affected upon binding (A, B), and a binding affinity of 31 μM was estimated. The residues involved in binding the compound are depicted in the ribbon (C) and molecular surface (D) colored in red, orange, and yellow to indicate those with large (>10.0), medium (6.0-10.0) and small (3.3-6.0) normalized chemical shift changes, respectively. The Structure of the compound is also shown (E).
Conclusions and Future Directions
The normal function of the Abl SH2 domain is thought to be the recognition of pTyr-containing protein sequence (40,41), and its activity is required in order to malignantly transform cells (23-25). However, Abl is activated by a variety of stimuli, and many models of its regulation have been proposed. It has been suggested for example that PtdIns(4,5)P2 and possibly its effectors are involved in maintaining c-Abl in an inactive conformation before PDGF stimulation, and that activation occurs only after PtdIns(4,5)P2 hydrolysis by PLC-γ or dephosphorylation (17). Here we found that PtdIns(4,5)P2 binds to the Abl SH2 domain, implying that it may directly localize and orient the Abl kinase domain on membranes. However the affinity of PtdIns(4,5)P2 for the Abl SH2 domain is lower than pTyr peptide ligands, suggesting that PtdIns(4,5)P2 binding is more likely to happen in resting cells with little tyrosine phosphorylation of its protein ligands. This novel link between c-Abl and phosphoinositide turnover could contribute to the regulation of c-Abl activity by lipid signaling pathways (17). Further studies of the PI interactions of SH2 domains are needed to define the biological consequences, and can now be designed based on identification of Arg59 as an essential determinant of PI recognition. Finally, the demonstration that the pyridone peptidomimetic binds the Abl SH2 domain competitively and with relatively high affinity bodes well for the design of novel experimental and therapeutic agents that manipulate SH2 activities in signaling pathways.
Acknowledgments
We thank B. J. Mayer and A. L. Castelhano for generously providing the Abl DNA and pyridone compound, respectively, C. Finkielstein and R. Eyeson, R. Gatewood and P. Kohli for discussions. This work was supported by the National Institute of Health (T. K. and M.O.) American Cancer Society (D.G.S.C.) and Cancer Research UK (M.O.), and the University of Colorado Health Sciences Center’s DNA sequencing, Biophysical and NMR Facilities.
References
- 1.Machida K, Mayer BJ, Nollau P. Profiling the global tyrosine phosphorylation state. Mol Cell Proteomics. 2003;2:215–233. doi: 10.1074/mcp.R300002-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON. BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell. 1991;66:161–171. doi: 10.1016/0092-8674(91)90148-r. [DOI] [PubMed] [Google Scholar]
- 3.Maru Y, Witte ON. The BCR gene encodes a novel serine/threonine kinase activity within a single exon. Cell. 1991;67:459–468. doi: 10.1016/0092-8674(91)90521-y. [DOI] [PubMed] [Google Scholar]
- 4.Hawk N, Sun T, Xie S, Wang Y, Wu Y, Liu J, Arlinghaus RB. Inhibition of the Bcr-Abl oncoprotein by Bcr requires phosphoserine 354. Cancer Res. 2002;62:386–390. [PubMed] [Google Scholar]
- 5.Cleghon V, Morrison DK. Raf-1 interacts with Fyn and Src in a non-phosphotyrosine-dependent manner. J Biol Chem. 1994;269:17749–17755. [PubMed] [Google Scholar]
- 6.Park I, Chung J, Walsh CT, Yun Y, Strominger JL, Shin J. Phosphotyrosine-independent binding of a 62-kDa protein to the src homology 2 (SH2) domain of p56lck and its regulation by phosphorylation of Ser-59 in the lck unique N-terminal region. Proc Natl Acad Sci USA. 1995;92:12338–12342. doi: 10.1073/pnas.92.26.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joung I, Strominger JL, Shin J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci USA. 1996;93:5991–5995. doi: 10.1073/pnas.93.12.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poy F, Yaffe MB, Sayos J, Saxena K, Morra M, Sumegi J, Cantley LC, Terhorst C, Eck MJ. Crystal structures of the XLP protein SAP reveal a class of SH2 domains with extended, phosphotyrosine-independent sequence recognition. Mol Cell. 1999;4:555–561. doi: 10.1016/s1097-2765(00)80206-3. [DOI] [PubMed] [Google Scholar]
- 9.Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, van Schaik S, Notarangelo L, Geha R, Roncarolo MG, Oettgen H, De Vries JE, Aversa G, Terhorst C. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- 10.Ching TT, Lin HP, Yang CC, Oliveira M, Lu PJ, Chen CS. Specific binding of the C-terminal Src homology 2 domain of the p85α subunit of phosphoinositide 3-kinase to phosphatidylinositol 3,4,5-trisphosphate. Localization and engineering of the phosphoinositide-binding motif. J Biol Chem. 2001;276:43932–43938. doi: 10.1074/jbc.M105159200. [DOI] [PubMed] [Google Scholar]
- 11.Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC, Cantley LG. Phosphoinositide 3-kinase regulates phospholipase Cγ-mediated calcium signaling. J Biol Chem. 1998;273:23750–23757. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- 12.Bae YS, Cantley LG, Chen CS, Kim SR, Kwon KS, Rhee SG. Activation of phospholipase Cγ by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:4465–4469. doi: 10.1074/jbc.273.8.4465. [DOI] [PubMed] [Google Scholar]
- 13.Rameh LE, Chen CS, Cantley LC. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell. 1995;83:821–830. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- 14.Surdo PL, Bottomley MJ, Arcaro A, Siegal G, Panayotou G, Sankar A, Gaffney PR, Riley AM, Potter BV, Waterfield MD, Driscoll PC. Structural and biochemical evaluation of the interaction of the phosphatidylinositol 3-kinase p85α Src homology 2 domains with phosphoinositides and inositol polyphosphates. J Biol Chem. 1999;274:15678–15685. doi: 10.1074/jbc.274.22.15678. [DOI] [PubMed] [Google Scholar]
- 15.Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65:2047–2053. doi: 10.1158/0008-5472.CAN-04-3888. [DOI] [PubMed] [Google Scholar]
- 16.Arlinghaus RB. Bcr: a negative regulator of the Bcr-Abl oncoprotein in leukemia. Oncogene. 2002;21:8560–8567. doi: 10.1038/sj.onc.1206083. [DOI] [PubMed] [Google Scholar]
- 17.Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-γ1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- 18.Barila D, Mangano R, Gonfloni S, Kretzschmar J, Moro M, Bohmann D, Superti-Furga G. A nuclear tyrosine phosphorylation circuit: c-Jun as an activator and substrate of c-Abl and JNK. Embo J. 2000;19:273–281. doi: 10.1093/emboj/19.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith JM, Mayer BJ. Abl: mechanisms of regulation and activation. Front Biosci. 2002;7:d31–d42. doi: 10.2741/a767. [DOI] [PubMed] [Google Scholar]
- 20.Hantschel O, Wiesner S, Guttler T, Mackereth CD, Rix LL, Mikes Z, Dehne J, Gorlich D, Sattler M, Superti-Furga G. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell. 2005;19:461–473. doi: 10.1016/j.molcel.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 21.Redondo P, Lloret P, Andreu EJ, Inoges S. Imatinib mesylate in cutaneous melanoma. J Invest Dermatol. 2004;123:1208–1209. doi: 10.1111/j.0022-202X.2004.23496.x. [DOI] [PubMed] [Google Scholar]
- 22.Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- 23.Afar DE, Goga A, McLaughlin J, Witte ON, Sawyers CL. Differential complementation of Bcr-Abl point mutants with c-Myc. Science. 1994;264:424–426. doi: 10.1126/science.8153630. [DOI] [PubMed] [Google Scholar]
- 24.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarski P, Perrotti D, Chan TO, Wasik MA, Tsichlis PN, Calabretta B. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Wong R, Hao SX, Pear WS, Ren R. The SH2 domain of bcr-Abl is not required to induce a murine myeloproliferative disease; however, SH2 signaling influences disease latency and phenotype. Blood. 2001;97:277–287. doi: 10.1182/blood.v97.1.277. [DOI] [PubMed] [Google Scholar]
- 26.Sawyer TK, Bohacek RS, Dalgarno DC, Eyermann CJ, Kawahata N, Metcalf CA, III, Shakespeare WC, Sundaramoorthi R, Wang Y, Yang MG. SRC homology-2 inhibitors: peptidomimetic and nonpeptide. Mini Rev Med Chem. 2002;2:475–488. doi: 10.2174/1389557023405765. [DOI] [PubMed] [Google Scholar]
- 27.Shakespeare WC. SH2 domain inhibition: a problem solved? Curr Opin Chem Biol. 2001;5:409–415. doi: 10.1016/s1367-5931(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 28.Fu JM, Castelhano AL. Design and synthesis of a pyridone-based phosphotyrosine mimetic. Bioorg Med Chem Lett. 1998;8:2813–2816. doi: 10.1016/s0960-894x(98)00503-4. [DOI] [PubMed] [Google Scholar]
- 29.Overduin M, Mayer B, Rios CB, Baltimore D, Cowburn D. Secondary structure of Src homology 2 domain of c-Abl by heteronuclear NMR spectroscopy in solution. Proc Natl Acad Sci USA. 1992;89:11673–11677. doi: 10.1073/pnas.89.24.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol. 1995;NMR6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- 32.Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- 33.Lee SA, Eyeson R, Cheever ML, Geng J, Verkhusha VV, Burd C, Overduin M, Kutateladze TG. Targeting of the FYVE domain to endosomal membranes is regulated by a histidine switch. Proc Natl Acad Sci USA. 2005;102:13052–13057. doi: 10.1073/pnas.0503900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irvine R. Proliferation. J Cell Sci. 2000;113(Pt 21):3683–3684. [Google Scholar]
- 35.Cockcroft S. Biology of Phosphoinositides. Oxford University Press; Oxford; New York: 2000. [Google Scholar]
- 36.Sun T, Campbell M, Gordon W, Arlinghaus RB. Preparation and application of antibodies to phosphoamino acid sequences. Biopolymers. 2001;60:61–75. doi: 10.1002/1097-0282(2001)60:1<61::AID-BIP1004>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, Superti-Furga G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 38.Jackson P, Baltimore D. N-terminal mutations activate the leukemogenic potential of the myristoylated form of c-abl. Embo J. 1989;8:449–456. doi: 10.1002/j.1460-2075.1989.tb03397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, Hope TJ. Nuclear-cytoplasmic shuttling of c-ABL tyrosine kinase. Proc Natl Acad Sci USA. 1998;95:7457–7462. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol. 1994;14:2883–2894. doi: 10.1128/mcb.14.5.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]