

Abstract
Regulated expression of the recombinase RAG-1 and RAG-2 proteins is necessary for generating the vast repertoire of antigen receptors essential for adaptive immunity. Here, a retroviral cDNA library screen showed that the stress-regulated protein GADD45a activated transcription of the genes encoding RAG-1 and RAG-2 in transformed pro-B cells by a pathway requiring the transcription factor Foxo1. Foxo1 directly activated transcription of the Rag1-Rag2 locus throughout early B cell development, and a decrease in Foxo1 protein diminished the induction of Rag1 and Rag2 transcription in a model of receptor editing. We also found that transcription of Rag1 and Rag2 was repressed at the pro-B cell and immature B cell stages by the kinase Akt through its ‘antagonism’ of Foxo1 function. Thus, Foxo1 is a key regulator of Rag1 and Rag2 transcription in primary B cells.
Adaptive immunity depends on the concerted action of the lymphocyte-restricted products of recombination-activating gene 1 (RAG-1; A002009) and recombination-activating gene 2 (RAG-2; A002010), which catalyze the somatic DNA rearrangement of variable, diversity and joining gene segments forming the variable-domain exons of B cell antigen receptors (BCRs) and T cell antigen receptors (TCRs)1. In B cells, RAG activity occurs in two discrete waves: first at the common lymphoid progenitor and pro-B cell stages during immunoglobulin heavy-chain locus rearrangement and then again at the pre-B cell stage during immunoglobulin light-chain locus rearrangement2,3. Productive rearrangement of both heavy- and light-chain genes leads to BCR expression at the immature B cell stage. Basal signaling from a self-tolerant BCR limits RAG activity at this stage and ultimately leads to complete loss of expression of the genes encoding RAG-1 and RAG-2 (Rag1 and Rag2, respectively; collectively called ‘Rag’ here) as B cells mature further4. If, however, the BCR recognizes self antigen, development halts and Rag expression continues, resulting in further light-chain locus rearrangement (receptor editing) and altered BCR specificity until an ‘innocuous’ BCR is expressed or the potential for light-chain gene recombination is exhausted5,6. Regulated expression of RAG-1 and RAG-2 in B cells is thus necessary for both the nearly limitless repertoire of antigen receptors as well as the ‘pruning’ of this repertoire to maintain central tolerance.
Both the pre-BCR and BCR form signaling complexes that suppress Rag transcription at critical stages of B cell development4,7,8. This negative-feedback regulation of RAG activity by the products of recombination prevents genomic instability in large cycling pre-B cells, contributes to allelic exclusion of heavy- and light-chain expression and inactivates variable-(diversity)-joining recombination to stabilize genes encoding a self-tolerant receptor. The signaling pathways and transcription factors that mediate this regulation are ill defined. Given this lack of knowledge, we devised a functional screen for cDNA molecules able to induce Rag1 transcription in a transformed pro-B cell line. We found that the stress-regulated protein GADD45a (A001020) activated Rag transcription in these cells by a pathway involving mitogen-activated protein kinase signaling and the transcription factor Foxo1 (A000944). We also found that phosphatidylinositol-3-OH kinase (PI(3)K), the serinethreonine kinase Akt and Foxo1 were important in regulating Rag transcription in developing primary bone marrow B cells and during receptor editing.
RESULTS
Screen for regulators of Rag transcription identifies GADD45a
To screen for factors that regulate Rag transcription in B lymphocytes, we generated an indicator cell line using a published ‘knock-in’ mouse9 in which the endogenous Rag1 coding sequence is replaced with cDNA encoding green fluorescent protein (Rag1-GFP; Supplementary Fig. 1a online). We first confirmed previous work9 showing that the knock-in mutation did not affect normal regulation of Rag1 and that GFP expression was an accurate reflection of Rag1 promoter activity (data not shown). We then infected bone marrow from Rag1-GFP-heterozygous mice with Abelson murine leukemia virus (AMuLV). The viral oncogene v-abl selectively transforms cells and arrests their development at a stage that resembles that of large cycling pre-B cells10. Treatment of AMuLV-transformed pro-B cells with a small-molecule inhibitor of v-Abl, STI-571 (Gleevec), results in an increase in transcription of genes typical of pre-B cells, including Rag1 and Rag2 (ref. 11). As expected, treatment with STI-571 induced GFP expression in these AMuLV-transformed Rag1-GFP pro-B cells (Fig. 1a). In addition, we constructed a retroviral cDNA expression library with mRNA purified from mouse CD19+IgM− bone marrow B cell progenitors. The library contained approximately 5 × 106 inserts with an average size of 1.5 kilobases (data not shown). The retroviral library vector contained an internal ribosome entry site (IRES) followed by cDNA encoding Thy-1.1, which ‘marked’ infected cells with this surface protein.
Figure 1.

Identification of Gadd45a by a retroviral cDNA library screen for factors that induce Rag1 transcription in AMuLV-transformed B cells. (a) Flow cytometry of GFP expression in AMuLVtransformed Rag1-GFP B cells in normal culture conditions (filled histogram) or treated for 24 h with 5 μM STI-571 (solid line). Vertical axis (‘% of max’) indicates a scale of relative cell numbers with the median value set as 100%. (b) Flow cytometry of GFP expression in cells left uninfected (Uninf) or infected with retrovirus expressing Gadd45a (GADD45a), labeled with anti-Thy-1.1 (retroviral marker) and gated for infected Thy-1.1+ cells (solid line) or uninfected Thy-1.1− cells (filled histogram). Numbers above bracketed lines indicate percent GFP+ cells in the infected population (top number) and uninfected population (bottom number in parenthesis). (c) Quantitative RT-PCR analysis of Rag1 and Rag2 transcripts in sorted cells infected with empty vector retrovirus or retrovirus expressing GADD45a. Values are normalized to Hprt1 transcript abundance and are presented relative to expression in cells transduced with empty vector, set as 1. All data are representative of at least three independent experiments.
We infected AMuLV-transformed Rag1-GFP cells with our retroviral cDNA library and then selected for cells with increased GFP expression (Supplementary Fig. 1b). After two rounds of selection and cDNA recovery, the complexity of each ‘rescued’ cDNA pool was diminished to a few unique clones (Supplementary Fig. 1c). We isolated and sequenced each clone and retested their ability to induce GFP expression in the parental Rag1-GFP cell line. We chose one such cDNA clone encoding the stress-response molecule GADD45a for further study. Cells infected with GADD45a-expressing retrovirus consistently expressed eight- to tenfold more GFP than did uninfected cells (Fig. 1b) or cells infected with empty vector (parental retroviral vector with no insert cDNA). This result was also reflected in the basal amounts of Rag1 and Rag2 transcripts from the unaltered allelic Rag locus in sorted cells overexpressing GADD45a (Fig. 1c).
Characterization of the GADD45a pathway
Gadd45a was initially identified as a gene induced by DNA damage in Chinese hamster ovary cells12. The protein it encodes is one of three related proteins, GADD45a, GADD45b and GADD45g, that share over 50% amino acid identity. All three are induced by various cell stresses, including DNA damage, hypoxia and withdrawal of growth factor13. Among their other known functions, GADD45 proteins bind to and activate the mitogen-activated protein kinase MEKK4, which in turn leads to phosphorylation of the stress-associated protein kinases p38 and Jnk14. In T cells, GADD45 proteins are involved in propagating signals from the TCR or cytokine receptors to p38 and Jnk15-17. Additionally, Gadd45a and Gadd45b have been identified as transcripts induced in AMuLV-transformed pro-B cells treated with the Abl kinase inhibitor STI-571 (ref. 11).
To test whether the GADD45a overexpression phenotype was MEKK4 dependent, we created an inducible from of GADD45a by fusing the region encoding Gadd45a to a modified version of the estrogen receptor hormone-binding domain18(ER-GADD45a; Supplementary Fig. 2a online). In Rag1-GFP cells expressing ER-GADD45a, there was no change in GFP expression in the absence of ligand, which indicated tight regulation of GADD45a activity. The addition of tamoxifen (an estrogen analog) resulted in notably higher expression of GFP within 24 h (Fig. 2a). Tamoxifen had no such effect on cells not expressing ER-GADD45a. We next infected the cells expressing ER-GADD45a with retrovirus encoding short hairpin RNA (shRNA), targeting the gene encoding MEKK4 (Map3k4; called ‘Mekk4’ here)19. Two independent shRNA molecules decreased Mekk4 message abundance and blunted the increase in GFP expression caused by tamoxifen induction of ER-GADD45a activity (Fig. 2b and Supplementary Fig. 2b).
Figure 2.

GADD45a induces Rag1 transcription by a MEKK4- and p38-dependent pathway. (a) Flow cytometry of GFP expression in AMuLV-transformed Rag1-GFP cells infected with retrovirus expressing ER-GADD45a (Thy-1.1+; solid lines) and treated for 18 h with vehicle control (Veh) or tamoxifen (Tamox). Filled histograms, uninfected cells (Thy-1.1−) in the same culture. Numbers above bracketed lines indicate percent GFP+ cells in the infected population (top number) and uninfected population (bottom number in parenthesis). (b) GFP expression in ER-GADD45a-expressing cells infected with retrovirus encoded shRNA (above plots) and treated with tamoxifen for 24 h, then labeled with anti-hCD2 and gated for infected cells (expressing shRNA; solid lines) or uninfected cells (not expressing shRNA; filled histograms). Numbers above bracketed lines indicate percent GFP+ cells (top number) and mean fluorescence intensity (bottom number) in the shRNA-expressing population. Empty vector does not contain shRNA; GFP shRNA targets the transcript encoding GFP and serves as a positive control for GFP knockdown. (c) Flow cytometry of GFP expression in ER-GADD45a-expressing cells treated for 18 h with tamoxifen with or without a p38 or Jnk inhibitor, analyzed and gated as described in a. (d) Immunoblot of total and phosphorylated (p-) p38 and Jnk in ER-GADD45a-expressing cells treated for 6 h as described in c. In the Jnk blot, the lower band is Jnk1 (p46) and the upper band is Jnk2 and Jnk3 (p54). All data are representative of at least two independent experiments.
MEKK4 activates p38 and Jnk by phosphorylating ‘upstream’ mitogen-activated protein kinase kinases14,20. Because no other targets of MEKK4 activity are known, we used specific chemical inhibitors to test whether p38 or Jnk are ‘downstream’ of MEKK4 and GADD45a in the pathway leading to Rag1 transcription. In Rag1-GFP cells expressing ER-GADD45a, the addition of a chemical inhibitor of p38 (SB203580), along with tamoxifen, blocked the increase in GFP expression, whereas an inhibitor of Jnk had little effect (Fig. 2c). We confirmed the effectiveness of these inhibitors by immunoblot (Fig. 2d). We conclude that GADD45a increases Rag1 transcription by a MEKK4- and p38-dependent pathway in the AMuLV-transformed Rag1-GFP cell line.
GADD45a induces transcription of putative Foxo1 target genes
Initially, we hypothesized p38 activity might interfere with v-Abl activity and thus act in a way similar to STI-571. However, except for Rag1 and Rag2, the transcript abundance of a large set of genes known to be regulated by v-Abl was unchanged in cells overexpressing GADD45a (Supplementary Fig. 3a online). GADD45a thus seemed to act by a mechanism distinct from v-Abl inhibition. We then considered targets ‘downstream’ of p38 activity that might lead to Rag transcription. As p38 has many phosphorylation targets, we decided to assess global gene expression by microarray analysis in an attempt to narrow the number of candidates. We reasoned that Rag1 and Rag2 might belong to a related ‘cohort’ of genes induced by GADD45a overexpression and coregulated by a common transcription factor. Comparison of uninfected cells with AMuLV-transformed Rag1-GFP cells overexpressing GADD45a identified a small number of transcripts whose abundance varied by more than threefold (Supplementary Fig. 3b,c).
Because analysis of the microarrary data did not identify obvious functional commonality among the transcripts with altered expression, we evaluated whether the group of genes with the largest increase might be targets of a common transcription factor. We used a whole-genome rVista program to evaluate conserved sequences for potential transcription factor-binding sites21. For any given set of genes, the program assesses whether there is a statistical over-representation of conserved transcription factor-binding sites among the set relative to the whole genome. In the 5 kilobases upstream of the start sites of Rag1, Cd36, Slc7a3, Mmp13, Igj, Atf and Cth, there were 11 transcription factors with over-represented conserved binding sites shared by mice and humans (Supplementary Table 1 online). Of these, only a few are known to be expressed in B cells and T cells. We focused our attention on those factors whose activity might be controlled by p38 or PI(3)K signaling (as PI(3)K has been shown to be necessary for suppressing Rag transcription in immature B cells4) and whose genetic ablation has been shown to affect lymphocyte function; thus, we studied the Foxo family of transcription factors.
Foxo1 regulates Rag transcription
There are four closely related genes encoding Foxo proteins in mammals: Foxo1, Foxo3a, Foxo4 and Foxo6 (refs. 22,23). These genes encode molecules that are part of a much larger family of forkhead box (Fox) transcription factors characterized by a ‘winged helix’ DNA-binding motif24. The group is further divided into subgroups (such as subgroup O) on the basis of homology. The Foxo proteins are direct targets of the Akt-SGK kinase family25. Each contains three consensus Akt-phosphorylation sites responsible for the regulation of Foxo cellular localization and activity. After being phosphorylated by Akt, Foxo proteins are actively shuttled out of the nucleus and are retained in the cytoplasm or degraded by targeted proteolysis. In the absence of Akt activity, Foxo proteins are relocalized to the nucleus. Foxo1 and Foxo3a are the predominantly expressed family members in B cells (data not shown).
To evaluate whether Foxo transcription factors are required for GADD45a to activate transcription of the Rag locus, we expressed shRNA molecules targeting Foxo1, Foxo3a and Foxo4 individually in AMuLV-transformed Rag1-GFP cells also expressing ER-GADD45a. These shRNA molecules decreased Foxo1 and Foxo3a abundance by 46% and 41%, respectively (Fig. 3a). After the addition of tamoxifen for 24 h, only the shRNA targeting Foxo1 substantially diminished GADD45a-induced GFP expression, whereas ‘knockdown’ of Foxo3a or Foxo4 had little effect (Fig. 3b). To confirm our interpretation of the knockdown results, we overexpressed individual Foxo proteins in conjunction with ER-GADD45a (Fig. 3a). As expected on the basis of the shRNA results, overexpression of Foxo1 augmented ER-GADD45a-induced GFP expression (Fig. 3c). Expression of Foxo3a or Foxo4 had little effect on the mean fluorescence intensity of GFP. As further confirmation that Foxo1 is the ‘downstream’ target of GADD45a activity, we found that GADD45a overexpression increased Foxo1 protein abundance by 50% in AMuLV-transformed B cells (Foxo1 transcript amounts were unaltered; Fig. 3d and data not shown).
Figure 3.

Foxo1 functions ‘downstream’ of GADD45a to increase Rag1 transcription in AMuLV-transformed pro-B cells. (a) Immunoblot analysis of Foxo1, Foxo3A and Foxo4 in AMuLV-transformed B cells infected with retroviruses expressing shRNA (top) or mRNA (bottom). For shRNA knockdown, numbers below lanes indicate the amount of Foxo1 or Foxo3a, after normalization to β-actin (loading control), relative to that in cells infected with empty vector (set as 100). Foxo4 protein is undetectable in the absence of overexpression. For protein overexpression, cells infected with retrovirus-expressing Foxo protein are compared with cells infected with empty vector retrovirus, with β-actin as a loading control. (b,c) GFP expression in ER-GADD45a-expressing cells infected with retrovirus encoding shRNA (b) or mRNA (c) and treated with tamoxifen for 24 h, then labeled with anti-hCD2 or anti-Thy-1.1 and gated for infected cells (solid lines) or uninfected cells (filled histograms). Numbers above bracketed lines indicate percent GFP+ cells (top number) and the mean fluorescence intensity (bottom number). (d) Immunoblot analysis of Foxo1 in AMuLV-transformed B cells infected with empty virus retrovirus or retrovirus expressing GADD45a. Numbers below lanes indicate the ratio of Foxo1 to β-actin. All data are representative of three independent experiments.
Foxo1 regulates Rag transcription in developing B cells
Although we identified roles for GADD45a and Foxo1 in Rag expression using AMuLV-transformed pro-B cells, our ultimate goal was to link those findings to normal development of B cells. To determine whether GADD45a or Foxo1 regulates the expression of Rag1 and Rag2 in developing B cells, we used retroviral vectors to modulate the expression of these genes in primary bone marrow cultures from mice heterozygous for the Rag1-GFP knock-in. In bone marrow infected with retrovirus expressing GADD45a or GADD45b, neither protein increased GFP fluorescence to any substantial degree in any B cell subpopulation relative to that of cells infected with an empty vector control virus (Fig. 4a and data not shown). GFP fluorescence was also unchanged when we used ER-GADD45a virus and short-term tamoxifen treatment or shRNA targeting Mekk4 (data not shown). Culture of Rag1-GFP mouse bone marrow for 2 d in the presence of the p38 inhibitor did, however, decrease the mean fluorescence intensity of GFP in B220+IgM− pro-B cells or pre-B cells relative to treatment with dimethyl sulfoxide (DMSO) vehicle control (Fig. 4b). These results suggest that although the ‘upstream’ components may not act or be limiting in primary B cells, p38 activity does influence Rag1 transcription.
Figure 4.

Overexpression of Foxo1 increases Rag1 transcription in primary B lymphocytes, whereas constitutively active Akt suppresses it. (a) GFP expression in cultured B220+IgM− bone marrow B cells from Rag1-GFP-heterozygous mice, infected with various retroviral vectors (above plots; gated on retrovirus-infected cells; solid lines) or empty vector (filled histograms; repeated in each plot for reference), collected 3-4 d later and labeled with antibodies to delineate B cell developmental subsets and ‘mark’ retrovirus-infected cells; cells infected with retrovirus expressing Foxo1-ER were additionally treated with tamoxifen for 9 h before analysis. Numbers at top right indicate GFP mean fluorescence intensity (all cells are GFP+ compared with non-B cells in the same culture). Myr-Akt, myristilated Akt; IκBΔN (DN), dominant negative form of IκBα. (b) GFP expression in B220+IgM- bone marrow B cells from Rag1-GFP-heterozygous mice, cultured for 2 d in the presence of IL-7 (2 ng/ml) and 1 μM SB203580 (p38 inhibitor; solid line) or DMSO vehicle alone (filled histogram). Numbers at top left indicate GFP mean fluorescence intensity for cells treated with SB203580 (top number) or DMSO (bottom number in parenthesis). All data are representative of at least two independent experiments.
To assess whether Foxo1 influences Rag expression in primary B cells, we infected bone marrow from mice heterozygous for Rag1-GFP with retrovirus expressing Foxo1-ER (an inducible estrogen receptor fused to Foxo1). Treatment with tamoxifen for 9 h induced a substantial increase in GFP expression (as measured by mean fluorescence intensity) in B220+IgM− cells expressing Foxo1-ER relative to that in cells infected with an empty vector control (Fig. 4a and Supplementary Fig. 4a online). Retroviral expression of unmodified wild-type Foxo1 protein gave a result similar to that of Foxo1-ER, whereas Foxo3a protein expression, as in the cell line, had no effect on GFP expression (data not shown). Active Akt phosphorylates Foxo1, leading to its cytoplasmic sequestration and opposing its function25. Retroviral infection and expression of a constitutively active mutant Akt (myristilated Akt26) substantially suppressed GFP expression in B220+IgM− pro-B cells and pre-B cells (Fig. 4a and Supplementary Fig. 4b). That set of results were reproducible in many similar experiments. We compared them with results obtained with other transcription factors reported to be involved in Rag expression: Foxp1 and NF-κB27,28. Expression of a dominant negative form of IκBα (which suppresses NF-κB activity) in B cells had little effect on GFP expression (Fig. 4a and Supplementary Fig. 4c). Foxp1 expression, however, did induce a small reproducible increase in GFP fluorescence (Fig. 4a).
To examine the function of Foxo1 during B cell development more specifically, we decreased Foxo1 protein expression in the bone marrow of Rag1-GFP-knock-in mice with retrovirus expressing an shRNA targeting Foxo1. This shRNA (similar to the control shRNA targeting GFP) decreased the mean fluorescence intensity of GFP by 40-50% at all stages of early B cell development relative to that of cells infected with empty vector virus or virus expressing shRNA targeting Foxo3a (Fig. 5a,b). This decrease was reproducible in many independent experiments and emphasizes the specific importance of Foxo1 (relative to that of Foxo3a) for Rag expression in B cells. Flow cytometry showed that shRNA expression did not alter the survival of specific subsets of B cells during the course of this experiment (Supplementary Fig. 5 online). Pro-B cells expressing the Foxo1-specific shRNA did not grow well in longer-term culture and were eventually lost, which prevented us from transplanting such cells into mice (data not shown).
Figure 5.
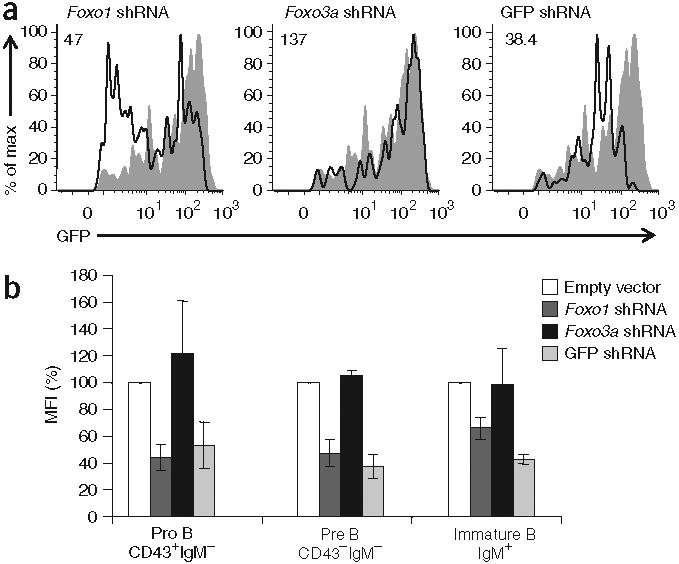
A decrease in Foxo1 protein in primary B lymphocytes decreases Rag1 transcription at all stages of B cell development. (a) GFP expression in cultured hCD2+B220+CD43−IgM− bone marrow pre-B cells from Rag1-GFP-heterozygous mice, infected with retrovirus expressing shRNA specific for Foxo1, Foxo3a or GFP (solid lines) or empty vector (filled histograms; repeated in each plot for reference), collected 3-4 d later and labeled with antibodies to delineate B cell developmental subsets and ‘mark’ retrovirus-infected cells. Numbers at top left indicate GFP mean fluorescence intensity; the value for the population infected with empty vector retrovirus is 127. (b) GFP mean fluorescence intensity (MFI) for shRNA retrovirus infection experiments done as described in a, presented as a percent of the GFP mean fluorescence intensity for the population infected with empty vector retrovirus (set as 100%), analyzing pro-B cell, pre-B cell and immature B cell populations gated on live cells and B220. Data are representative of three experiments (a) or are the average of three independent experiments (±s.d.; b).
The PI(3)K-Akt signaling axis controls Rag expression
We assessed whether Akt might function ‘upstream’ of Foxo1 in developing B cells to modulate Rag expression. Incubation of IgM+IgD− immature B cells from Rag1-GFP-knock-in mice with a specific Akt inhibitor resulted in a notable increase in GFP fluorescence and a decrease in phosphorylated Akt and phosphorylated Foxo1 relative to that of cells treated with a DMSO vehicle control (Fig. 6a and Supplementary Fig. 6a online). Because PI(3)K activity is often ‘upstream’ of Akt activation, we also assessed its function29. Similar to published results (and similar to Akt inhibition), GFP fluorescence in immature B cells increased when the cells were incubated with a PI(3)K inhibitor (Fig. 6a and Supplementary Fig. 6a). We concluded that the basal BCR signals that repress Rag transcription in immature B cells proceed in part through a PI(3)K and Akt-dependent pathway.
Figure 6.
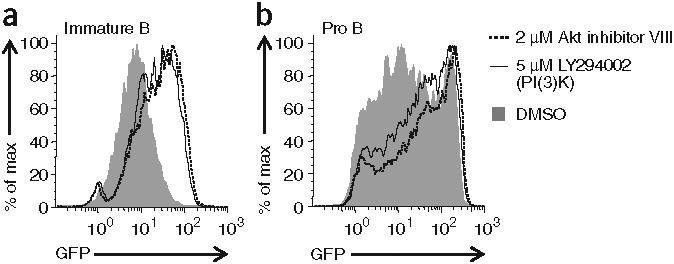
IL-7R and BCR signaling repress Rag transcription through the PI(3)K-Akt signaling axis. (a) GFP expression in B220+IgM+IgD− primary immature B cells from Rag1-GFP-heterozygous mice, cultured for 24 h with Akt or PI(3)K inhibitors or DMSO vehicle alone. (b) GFP expression in large B220+CD43+IgM− bone marrow B cells from Rag1-GFP-heterozygous mice, cultured for 4 d in the presence of IL-7 (2 ng/ml), then treated for an additional 2 d with Akt or PI(3)K inhibitors or DMSO vehicle alone (while still in the presence of IL-7) and then collected and labeled with antibodies to distinguish developing B cell subsets. Data are representative of two independent experiments.
Earlier in B cell development, pro-B cells are sensitive to the mitogenic cytokine interleukin 7 (IL-7), which is necessary for their growth and survival. Reports suggest that IL-7 might partially repress Rag expression in cycling pro-B cells and pre-B cells30,31. To confirm that observation, we incubated IL-7-dependent pro-B cells from Rag1-GFP-knock-in mice for 2 d in various concentrations of IL-7 (Supplementary Fig. 6b). As expected, decreasing the concentration of IL-7 in culture increased the GFP fluorescence of pro-B cells in a concentration-dependent way. Given that IL-7 receptor (IL-7R) signaling has been shown to activate PI(3)K and, in turn, Akt, we tested if repression of Rag expression by IL-7 required these kinases32,33. We cultured bone marrow from Rag1-GFP-knock-in mice for several days in the presence of IL-7 and then treated the cells with an inhibitor of either PI(3)K or Akt (Fig. 6b and Supplementary Fig. 6a). Inhibition of either kinase increased GFP fluorescence in large CD43+B220+pro-B cells. IL-7R signaling thus represses Rag transcription in pro- B cells via PI(3)K and Akt in a way similar to BCR signaling in immature B cells.
Receptor editing is sensitive to Foxo1 expression
Studies of receptor editing suggest that downregulation of BCR signaling might be crucial to the regulation of Rag transcription4,34. As basal BCR signaling activates PI(3)K and Akt, we reasoned that loss of such signaling after BCR ligation and internalization (as occurs during editing) might result in dephosphorylation and nuclear accumulation of Foxo1 and this in turn would drive Rag expression (Supplementary Fig. 7 online). To test that, we induced receptor editing by crosslinking immunoglobulin M (IgM) on cultured primary immature B cells in which Foxo1 expression was diminished by shRNA. In immature B cells expressing a control shRNA targeting GFP (which has no endogenous target in wild-type mice), the addition of antibody to IgM (anti-IgM) increased the abundance of Rag1 and Rag2 transcripts approximately 5- and 50-fold, respectively (Fig. 7). However, the same treatment increased Rag1 by 1.5-fold and Rag2 by 8-fold in immature B cells expressing shRNA targeting Foxo1, which thus linked Foxo1 to the regulation of this critical tolerance mechanism. Control experiments with immature B cells expressing a Foxo1 yellow fluorescent protein fusion protein confirmed the supposition that crosslinking with anti-IgM as well as pharmacologic inhibition of either PI(3)K or Akt resulted in relocalization of Foxo1 from the cytoplasm to the nucleus (Supplementary Fig. 8 online).
Figure 7.
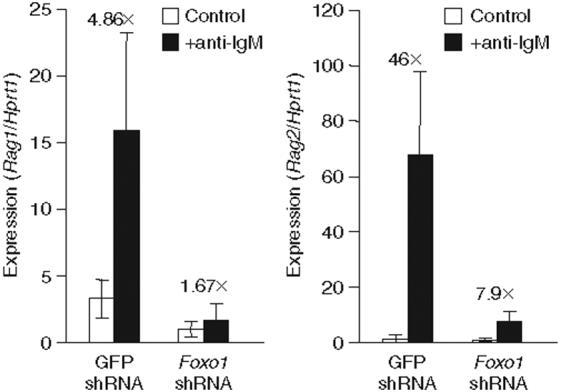
A decrease in Foxo1 protein in primary immature B cells interferes with Rag1 and Rag2 transcript induction in response to BCR crosslinking. Quantitative RT-PCR analysis of Rag1 or Rag2 transcripts in cultured immature B cells from Rag1-GFP-heterozygous mice, infected with retrovirus encoding shRNA targeting either GFP or Foxo1, then collected 4 dlater, labeled with the appropriate antibodies and sorted by flow cytometry to isolate B220loIgM+ immature B cells bearing the retroviral marker protein. Sorted immature B cells were then left untreated or incubated for 12-15 h with anti-IgM before being collected for RNA isolation. Rag1 and Rag2 transcripts quantified by real-time PCR were normalized to Hprt1 transcripts, with the lowest value set as 1. Numbers above bars indicate the ‘fold difference’ (×) in transcript abundance between control cells and cells treated with anti-IgM. Data are representative of two independent experiments.
Foxo1 acts directly on the Rag locus
To determine whether Foxo1 acts directly on the Rag locus, we assessed the kinetics of GFP induction in AMuLV-transformed Rag1-GFP pro-B cells expressing Foxo1-ER protein (Fig. 8a). After the addition of tamoxifen, GFP fluorescence began to increase within 4 h, with most cells becoming GFP+ by 8 h. That result contrasted with those of cells expressing ER-GADD45a, in which no change in GFP fluorescence was evident until 12-22 h after the addition of tamoxifen (data not shown). To extend those results, we did a similar experiment in the presence of cyclohexamide to prevent new protein synthesis. The addition of cyclohexamide did not affect the ability of Foxo1-ER to induce Rag transcription in response to tamoxifen (Fig. 8b). Thus, new protein synthesis is not required for Foxo1 to induce Rag1-GFP transcription. To test whether DNA binding by Foxo1 is required for it to induce Rag transcription, we expressed a DNA binding-defective point mutant of Foxo1 (ref. 35) in AMuLV-transformed pro-B cells as well as primary bone marrow B cells. The mutant Foxo1 failed to increase Rag expression in either context relative to wild-type Foxo1 (Supplementary Fig. 9a online). These results collectively indicate that Foxo1 acts by binding directly to the Rag locus through its DNA-binding domain.
Figure 8.
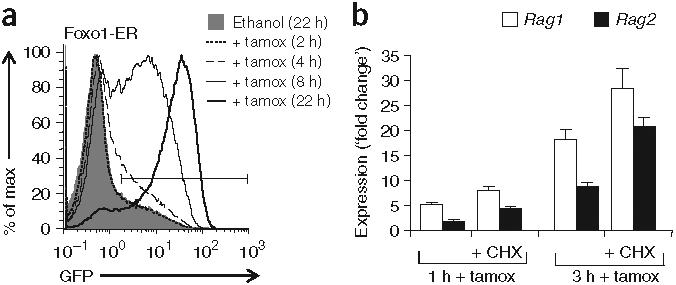
Foxo1 acts directly on the Rag locus. (a) Flow cytometry of GFP expression in AMuLV-transformed Rag1-GFP pro-B cells expressing Foxo1-ER protein and treated for 2-22 h (key) with tamoxifen or ethanol (vehicle control). (b) Quantitative RT-PCR analysis of Rag1 and Rag2 transcripts in the cells in a, treated for 1 or 3 h with tamoxifen or cyclohexamide (CHX) or both. Data are presented as the ratio of Rag mRNA in tamoxifen-treated cells to that in ethanol vehicle-treated control cells; Rag transcript abundance was normalized to that of Hprt1 transcripts. Data are representative of two independent experiments.
An enhancer has been identified that regulates Rag expression during B cell development (Erag)36. Multispecies genomic alignment of this region has identified several conserved forkhead-binding sites. Therefore, we considered whether Foxo1 might exert its influence on the Rag locus by binding to Erag. To test that, we infected cultures of primary C57BL/6 or Erag-deficient mouse B cells with retrovirus expressing Foxo1-ER and then treated the cells for 15 h with tamoxifen or vehicle control. After sorting for infected pro-B cells and pre-B cells, we measured the abundance of Rag1 and Rag2 transcripts. Although Rag transcription was 80-90% lower in Erag-deficient B cells, the addition of tamoxifen increased Rag1 expression by a similar amount (threefold) in both Erag-deficient and wild-type B cells (Supplementary Fig. 9b). We conclude that whereas Foxo1 might act in part through sites in Erag, it must also influence Rag expression through elements outside Erag.
DISCUSSION
Several transcription factors are necessary for expression of Rag1 and Rag2 early in lymphoid development. Deficiency in either E47 or Foxp1 prevents RAG expression beginning at the common lymphoid progenitor-pro-B cell stage27,37,38. These factors may be responsible for establishing or maintaining chromatin modifications at the Rag locus that permit transcription without actually promoting transcription. Indeed, in established pro-B cell lines, E47 can be ablated with no discernible affect on Rag expression39. Also, overexpression of E47 failed to increase Rag transcript abundance in B cells, which makes it unlikely to account for the changes in Rag expression in early B cell development (data not shown). Studies have suggested that NF-κB proteins are the relevant transcription factors driving Rag expression during receptor editing28, yet early B cell development seems completely unaffected in mice deficient in the NF-κB subunits p65 or c-Rel or classical NF-κB signaling40-42. In our experiments, overexpression of a dominant negative IκBα mutant (that prevented NF-κB nuclear translocation) did not affect GFP expression in primary pro-B cells or pre-B cells. However, we used a different method for our experiments than those described by other groups, and further work needs to be done to clarify the function of NF-κB proteins in this system.
Unlike previous studies identifying regulators of Rag transcription, our work here has shown that Foxo1 can activate abundant transcription from the endogenous Rag locus. Also, a decrease in Foxo1 protein in primary B cells diminished Rag transcription at all developmental stages. Many studies have shown that Foxo1 activity is blocked by Akt-dependent phosphorylation and cytoplasmic sequestration23,25. We found that pharmacological inhibition of either PI(3)K or Akt in B cells increased Rag expression in both pro-B cells and immature B cells. In pro-B cells, PI(3)K and Akt are activated by IL-7R signaling32,33. In our bone marrow cultures, high concentrations of IL-7 effectively decreased Rag expression in pro-B cells. IL-7 had no such effect on small pre-B cells or immature B cells, which do not express IL-7R (data not shown). In vivo, IL-7R signaling may act in synergy with the pre-BCR on large cycling pre-B cells to limit Rag expression and prevent the introduction of double-strand DNA breaks in these proliferating cells. Later in development, activation of PI(3)K and Akt ‘downstream’ of tonic BCR signaling is important for the repression of Foxo1 activity and Rag transcription as immature B cells transition from the T1 to the T2 developmental stage4. In our model, the presence of autoantigen would cause the ‘offending’ BCR to be removed from the cell surface, resulting in a loss of PI(3)K and Akt signaling, increased nuclear accumulation of active Foxo1 and consequent Rag transcription. Accordingly, we found that BCR cross linking (a model for receptor editing43) in immature B cells induced dephosphorylation and nuclear accumulation of Foxo1.
In agreement with our results, ablation of the PI(3)K subunits p110δ or p85α results in a partial block in B cell development and more Rag transcription in immature B cells8,44. There are three partially redundant Akt proteins in mammals45. Although B cell specific triple-Akt-knockout mice have yet to be reported, we predict that such mice will also have higher Rag expression. Our results contrast with a published report that did not find involvement of Akt in Rag expression in immature B cells4. The critical difference between those experiments and ours is the Akt chemical inhibitor used. We used a newer Akt inhibitor that is noncompetitive in terms of ATP and depends on the pleckstrin homology domain of the protein46. This inhibitor shows much greater selectivity for Akt than for SGK kinases. Given that overexpression of a constitutively active Akt mutant substantially repressed Rag expression in primary B cells, we believe our results are accurate.
Our results have shown that Foxo1 bound directly to the Rag locus to activate transcription. Although we found conserved forkhead-binding sites in Erag, deletion of this enhancer had no discernible effect on Foxo1-mediated Rag induction in B cells. This result indicates that Foxo1 influences the Rag locus through binding sites outside Erag (though it does not rule out the possibility of a Foxo1-Erag interaction). Neither the minimal Rag1 promoter nor the minimal Rag2 promoter has conserved canonical forkhead-binding sites. Accordingly, Foxo1 expression did not increase Rag1 or Rag2 promoter activity in cell lines containing integrated Rag1 or Rag2 promoter reporter constructs (data not shown). As the rVista analysis demonstrated, there are conserved Foxo1-binding sites throughout the Rag locus.
We have done experiments to assess the function of this pathway in thymocytes. In the several thymocyte cell lines tested, Foxo1 expression did not increase Rag1 or Rag2 transcript abundance (data not shown). These experiments should be repeated in primary thymocytes. However, given that T cell-specific knockout of Foxo1, Foxo3a and Foxo4 in mice fail to show a defect in Rag transcription, we believe that Foxo1 function in this context is specific to the B lineage47.
We originally identified Foxo1 as a target of GADD45a activity in our AMuLV-transformed progenitor B cell line. In primary cells, overexpression of either GADD45a or GADD45b had a negligible effect on GFP expression. It is possible that GADD45 is not limiting in primary cells, so expressing more GADD45 by means of a retrovirus does nothing to modulate Rag expression. However, we found that p38 inhibition did decrease Rag expression in primary pro-B cells and pre-B cells. Both GADD45a- and MEKK4-knockout mice have been generated, and neither is reported to have an early B cell defect17,48,49. Those data suggest that in primary B cells, p38 activation may lead to increased Rag expression, but that the ‘upstream’ pathway may not be GADD45 and MEKK4 dependent. Because there are many pathways that can activate p38, this is a likely scenario. Data from our cell line experiments have shown that p38 activity increases Foxo1 protein abundance and may also stimulate its transcriptional activity. The connection between these proteins should be explored more thoroughly.
We began these experiments with the aim of identifying pathway components or transcription factor(s) responsible for transmitting signals from the pre-BCR or BCR to the Rag locus. On the basis of our data presented here, we propose that IL-7R and the pre-BCR or BCR repress Rag expression through a PI(3)K- and Akt-dependent pathway leading to phosphorylation and cytoplasmic sequestration of Foxo1. In the absence of those receptors, Foxo1 accumulates in the nucleus and is the main transcription factor driving Rag expression in developing B cells. Because Foxo1−/− mice die early in embryogenesis, this is the first report, to our knowledge, of a specific nonredundant function for Foxo1 in B cells50. Furthermore, because pro-B cells with lower Foxo1 protein expression do not grow well in culture, we suspect that Foxo1 may have other functions in B cell development.
METHODS
Mice
Rag1-GFP-knock-in mice9and Erag-deficient mice36have been described. Rag1-GFP-knock-in mice (a gift from N. Sakaguchi) were back-crossed to the C57BL/6 background for at least six generations. C57BL/6 mice were from Jackson Laboratories. Use of all mice was with the approval of the Animal Care and Use Committee of the University of California at Berkeley.
Cell culture, retrovirus production and infection
The AMuLV-transformed Rag1-GFP-knock-in cell line was generated by infection of bone marrow from a mouse heterozygous for Rag1-GFP knock-in with AMuLV10. Cytokine-independent, transformed B cells were cloned by limiting dilution and were then screened for low basal GFP expression and responsiveness to treatment with STI-571 (Novartis). A single clone was chosen for all experiments. Cells were maintained in RPMI 1640 medium supplemented with 10% (vol/vol) FCS, L-glutamine (2 mM), penicillin (100 μg/ml), streptomycin (100 μg/ml) and 2-mercaptoethanol (50 μM) and were grown at 37 °C in 5% CO2.
Retrovirus was produced with the EcoPack2 packaging cell line (Clontech), maintained according to the manufacturer's protocol. EcoPack2 cells were transfected with retroviral plasmid with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and viral supernatant was collected 48 h later and filtered before use.
AMuLV-transformed Rag1-GFP pro-B cells were infected by resuspension of the cells directly in viral supernatant containing polybrene (4 μg/ml; Sigma) at a final cell density of 1 × 105 to 2 × 105 cells per 2 ml virus in one well of a six-well dish. Cells were left in viral supernatant overnight and their populations were expanded the next day by tenfold dilution of the cells directly into complete media.
For primary cell infection, bone marrow was collected from 1- to 2-month old mice and was depleted of red blood cells by density centrifugation with Histopaque-1083 (Sigma). The resulting leukocytes were then cultured together with EcoPack2 cells (transfected with retroviral plasmid 6 h before) in standard RPMI media described above and additionally supplemented with polybrene (4 μg/ml) and recombinant IL-7 (2 ng/ml; R&D Systems). After 2 d of coculture, bone marrow cells were removed and washed and were plated onto irradiated S17 mouse stromal cells in RPMI medium with IL-7 (2 ng/ml). In some experiments, bone marrow was infected by direct incubation of cells at a density of 10 × 106 cells per well in a six-well plate in 2-3 ml of fresh high-titer retroviral supernatant supplemented with polybrene. Retroviral supernatant was replaced after 24 h with fresh viral supernatant and the cells were incubated for an additional 24 h. Cells were then collected, washed and plated onto S17 cells as described above. Both methods gave equivalent results. Primary cells were labeled with anti-IgM (II/41), anti-IgD (11-26c), anti-B220 (RA2-6B2) and anti-CD43 (S7), plus anti-Thy-1.1 (OX-7), anti-human CD4 (RPA-T4) or anti-human CD2 (RPA-2.10) and were analyzed or sorted 3-4 d after retroviral infection. For treatment with tamoxifen or other pharmacological inhibitors (Calbiochem), the drug was added to cultures 8-24 h before analysis.
For induction of editing in immature B cells, bone marrow was retrovirally infected and, after 4 d, culture cells were collected, were labeled with anti-IgM, anti-IgD, anti-B220 and anti-human CD2 and were sorted by flow cytometry for retrovirus-infected immature B cells (B220loIgM+hCD2+ cells). All sorting and cell handling was done on ice to minimize BCR signaling at this point. Half the sorted cells were then immediately lysed in TRIzol reagent (Invitrogen) and were frozen at −80 °C (control cells). The other half were plated into wells of a 96-well plate in RPMI medium containing F(ab′)2 anti-mouse IgM (10 μg/ml; Jackson ImmunoResearch) and were cultured for 12-15 h before being collected and lysed in TRIzol.
Retroviral cDNA library production
Bone marrow was collected from 30 C57BL/6 mice and red blood cells were removed by incubation in ammonium chloride-potassium bicarbonate lysis buffer (0.15 M NH4Cl, 1 mM KHCO3 and 0.1 mM EDTA, pH 7.3). Bone marrow was depleted of IgM+ cells with anti-IgM paramagnetic beads (Miltenyi) on an AutoMACS cell separator (Miltenyi). The IgM− fraction was then labeled with biotinylated anti-CD19 (1D3), followed by incubation with streptavidin paramagnetic beads and selection of CD19+ cells on magnetic MS columns (Miltenyi). The purity of each cell-fractionation step was monitored by flow cytometry. The final CD19+IgM− fraction was over 95% pure in three separate purifications.
An Oligotex Direct mRNA mini kit (Qiagen) was used to isolate mRNA from 1.1 × 108 CD19+IgM− cells according to the manufacturer's protocol. Purified mRNA (7 μg total) was determined to be intact and undegraded by denaturing gel electrophoresis and was converted into cDNA with the Super-Script Choice System for cDNA Synthesis (Invitrogen) according to the manufacturer's protocol with the following modifications. No radioactive dNTPs were used at any point during synthesis and, where noted in the manufacturer's protocol, were replaced with water. For first-strand synthesis, an oligo(dT) primer containing a SfiI-A restriction site was used instead of the primer provided (no random hexamers were used). After second-strand synthesis, a ‘custom’ double-stranded DNA adaptor containing a SfiI-B restriction site was ligated to the cDNA in place of the manufacturer's adaptor. Adaptor-ligated cDNA was digested with SfiI (NEB) and was fractionated by size with a Sephacryl S-500 HR column for removal of all DNA below 500 base pairs. Digested cDNA was ligated overnight at 16 °C to SfiI-digested CMSCV-IRES-Thy-1.1 retroviral vector with T4 DNA ligase (NEB).
Competent ElectroMAX DH10B bacteria (Invitrogen) were transformed with the ligated cDNA according to the manufacturer's protocol. A total of eight transformations were done, and each was then plated and maintained separately in individual pools for screening. Each transformation was plated onto 25 15-cm agar plates with carbenicillin (100 μg/ml) and was grown for 30 h at 30 °C. Dilutions of each transformation were also plated to determine transformation efficiency. Bacteria were scraped off the plates and were pooled before plasmid purification. A random sample of 40 bacterial colonies was picked and the plasmids were analyzed for the presence and size of the cDNA insert; 95% of the plasmids had cDNA inserts with an average size of 1.5 kilobases. On the basis of bacterial colony counts from diluted transformations, each pool is estimated to contain about 6 × 105 cDNA clones and the total cDNA library is estimated to have a complexity of about 4.8 × 106 clones.
Retroviral cDNA library screen
AMuLV-transformed Rag1-GFP pro-B cells were infected with retrovirus prepared from the library in five separate pools of 1.2 × 106 cells each. After 1 week, cells were sorted by flow cytometry for high GFP expression (highest, 1%) and the presence of the Thy-1.1 retroviral marker. Sorted cells were cultured for an additional week before being sorted once again for high GFP expression. DNA was extracted from a subset of the selected cells (2 × 106to 3 × 106 cells) and the retroviral cDNA inserts were amplified by PCR. The Advantage 2 PCR Enzyme System (BD Biosciences) was used for PCR according to the manufacturer's protocol with primers specific for the retroviral vector and 100 ng genomic DNA as a template. PCR cycling conditions were as follows: 94 °C for 2 min, followed by 30 cycles of 94 °C for 15 s and 68 °C for 6 min, with a final extension step of 68 °C for an additional 6 min. PCR products were purified by phenol-chloroform extraction or Qiaquick PCR cleanup kit (Qiagen) and then were digested overnight with SfiI (NEB). Digested inserts were purified by agarose gel purification and were ligated into SfiI-digested CMSCV-IRES-Thy-1.1 retroviral vector and were then used to transform competent bacteria. Transformed bacteria were grown in bulk culture with ampicillin selection and plasmids were purified with an endotoxin-free plasmid purification kit (Qiagen). Retrovirus made with this plasmid was used to infect parental AMuLV-transformed Rag1-GFP pro-B cells. The selection and ‘rescue’ scheme was then repeated until the cDNA complexity in each pool was decreased to a few clones. Each PCR-amplified cDNA was then individually isolated, recloned and sequenced and was tested for the ability to induce GFP expression in the parental cell line.
Gene-expression analysis
For gene-expression analysis by RT-PCR, total RNA was isolated from cells with TRIzol reagent (Invitrogen) and was reverse-transcribed with random hexamers and SuperScriptII Reverse Transcriptase (Invitrogen; both steps according to the manufacturer's protocols). Jumpstart Taq (Sigma) and the following cycling conditions were used for both standard RT-PCR and quantitative RT-PCR assays: 95 °C for 3 min, followed by 25-45 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min, with a final extension step of 72 °C for 2 min. TaqMan fluorescent primer probes (MWG Biotech) were used for real-time PCR assay of transcripts of Rag1, Rag2 and Hprt1 (encoding hypoxanthine guanine phosphoribosyl transferase); transcript abundance was normalized to that of Hprt1 transcript. Products of standard RT-PCR reactions were separated by 2% agarose gel electrophoresis and were visualized by ethidium bromide staining and ultraviolet illumination. The sequences of gene-specific primers and Taqman probes are in Supplementary Table 2 online.
Expression plasmids
All retroviral plasmids were based on the MSCV retroviral vector and were modified to contain an IRES in-frame with a surface marker protein (Thy-1.1, human CD4 or human CD2) to ‘mark’ retrovirus-infected cells. The cDNA was cloned upstream of the IRES sequence in a multiple cloning site by standard techniques (SfiI restriction sites in the library vector).
The ER-GADD45a fusion construct was created by PCR amplification of the estrogen receptor domain from Foxo1-ER and the Gadd45a coding region from the cDNA library clone. Pfu TurboUltra (Stratagene) was used for PCR according to the manufacturer's protocol with 5 ng linearized plasmid template and 18 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 2 min. These two PCR fragments were digested with the appropriate restriction enzymes and were ligated together with CMSCV-IRES-Thy-1.1 in a trimolecular reaction.
Mekk4 cDNA (provided by K. Murphy) was cloned into MSCV-IRES-Thy-1.2. Retroviral plasmids encoding human Foxo1, Foxo1-ER, DNA binding- defective Foxo1 (Foxo1(H215R)-ER) and Foxo3a (provided by D. Fruman) have been described35. Mouse Foxo4 cDNA (provided by E. Olson) was cloned into CMSCV-IRES-hCD4. IκBαΔN-IRES-Thy-1.1 was provided by W. Sha (University of California at Berkeley). Foxp1a cDNA (provided by A. Rao) was cloned into CMSCV-IRES-Thy-1.1. Myr-Akt (provided by N. Rosenberg) was cloned into MSCV-IRES-Thy-1.1.
The shRNA was expressed by a retrovirus containing human CD2 cDNA followed by a modified human miR-30 microRNA precursor19. The mir-30 microRNA context was cloned by PCR amplification from the MSCV/ LTRmiR30-PIG vector (OpenBiosystems). In conjunction, human CD2 surface protein was also cloned by PCR from the MSCV-IRES-hCD2 vector (a gift from G. Barton). Both fragments were amplified with primers containing compatible restriction sites. Pfu TurboUltra was used for PCR by a procedure identical to that described for the ER-GADD45a construct. Fragments were digested with the appropriate restriction enzymes and were ligated together with a similarly digested MSCV retroviral vector in a trimolecular reaction. The sequence of shRNA targeting the various transcripts was obtained from the RNAi Codex and was cloned into the miR-30 context as described19. Specific shRNA sequences are in Supplementary Table 3 online.
Inhibition of Akt or PI(3)K in primary cells
For immature B cells, bone marrow was collected from several mice heterozygous for knock-in of Rag1-GFP approximately 1 month in age and was depleted of red blood cells by Histopaque density centrifugation. The resulting leukocytes were plated at a density of 2 × 106 cells per well in 24-well plates in standard RPMI media in the presence of DMSO vehicle control, 5 μM LY294002 (PI(3)K inhibitor) or 2 μM Akt inhibitor VIII (Calbiochem). After 24 h in culture, cells were collected, were labeled with appropriate antibodies to distinguish immature B cells and were analyzed by flow cytometry. For pro-B cells, bone marrow was collected as described above and the resulting white blood cells were plated onto irradiated S17 stromal cells at a density of 10 × 106 cells per six-well plate in RPMI medium supplemented with recombinant IL-7 (2 ng/ml); IL-7 was ‘refreshed’ after 2 d. After 4 d, cells were collected and washed and were plated onto S17 cells at a density of 2 × 106 cells per 24-well plate in the presence of IL-7 (2 ng/ml) and DMSO or a PI(3)K or Akt inhibitor as described above. After 2 d, cells were collected, labeled with anti-B220, anti-CD43, anti-IgM and anti-IgD and analyzed by flow cytometry.
Flow cytometry
Single-cell suspensions depleted of red blood cells were prepared from mice or from cultured cells and were incubated for at least 10 min with Fc receptor-blocking antibody (2.42G; purified from a hybridoma) and then were labeled with fluorochrome- or biotin-conjugated antibodies by standard techniques. A FC500 or an Elite XL flow cytometer (Beckman Coulter) was used for flow cytometry; a MoFlo high-speed cell sorter (Dako-Cytomation) was used for sorting (sorting parameters provided in Results and figure legends). Flow cytometry plots represent at least 1 × 105 live cells (by forward and side scatter) for cell-line experiments or at least five live cells for primary cell experiments. Data were analyzed with FlowJo software (Tree Star). All antibodies were from eBiosciences, except anti-B220 (Caltag Laboratories) and anti-CD43 and anti-Thy-1.1 (both from BD Pharmingen).
Immunoblot analysis
AMuLV-transformed Rag1-GFP pro-B cells were lysed directly in 2× sample buffer (1 × 105 cells/μl) and were boiled for 5 min. After centrifugation to clear insoluble material, 5-15 μl of each sample was separated by 8% SDS-PAGE and then transferred to nitrocellulose membranes. Membranes were blocked and then were labeled with primary and secondary antibodies according to the manufacturer's instructions. Membranes were analyzed with the Odyssey Infrared Imaging System (LI-COR Biosciences). Anti-Foxo1 (9462), anti-Foxo3a (9467), anti-p38 (9212), antibody to phosphorylated p38 (9211), anti-Jnk (9252) and antibody to phosphorylated Jnk (9251) were from Cell Signaling Technologies, and anti-actin (sc-1615) was from Santa Cruz Biotechnology. Infrared dye-conjugated secondary antibodies were from Molecular Probes-Invitrogen.
Additional methods
Information on gene-expression analysis by microarray, immunoblot analysis, imaging analysis of Foxo1 cellular localization and bioinformatics is available in the Supplementary Methods online.
Accession codes
UCSD-Nature Signaling Gateway (http://www.signaling-gateway.org): A002009, A002010, A001020 and A000944
ACKNOWLEDGMENTS
We thank all who provided mice (N. Sakaguchi; Kumamoto University) and plasmid constructs (K. Murphy (University of Washington at St. Louis), A. Rao (Harvard Medical School), E. Olson (University of Texas Southwestern Medical Center), D. Fruman (University of California at Irvine), W. Sha (University of California at Berkeley), N. Rosenberg (Tufts Medical School) and G. Barton (University of California at Berkeley)); H. Nolla (Cancer Research Laboratories, University of California at Berkeley) for help with flow cytometry cell sorting; and P. Herzmark (University of California at Berkeley) for help and advice with live microscopy. Supported by the National Institutes of Health (RO1 HL48702 and RO1 AI57487 to M.S.S.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
Supplementary Material
References
- 1.Schatz DG. V(D)J recombination. Immunol. Rev. 2004;200:5–11. doi: 10.1111/j.0105-2896.2004.00173.x. [DOI] [PubMed] [Google Scholar]
- 2.Borghesi L, et al. B lineage-specific regulation of V(D)J recombinase activity is established in common lymphoid progenitors. J. Exp. Med. 2004;199:491–502. doi: 10.1084/jem.20031800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grawunder U, et al. Down-regulation of RAG1 and RAG2 gene expression in PreB cells after functional immunoglobulin heavy chain rearrangement. Immunity. 1995;3:601–608. doi: 10.1016/1074-7613(95)90131-0. [DOI] [PubMed] [Google Scholar]
- 4.Verkoczy L, et al. Basal B cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes B cell-positive selection. J. Immunol. 2007;178:6332–6341. doi: 10.4049/jimmunol.178.10.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat. Immunol. 2004;5:645–650. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 6.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geier JK, Schlissel MS. Pre-BCR signals and the control of Ig gene rearrangements. Semin. Immunol. 2006;18:31–39. doi: 10.1016/j.smim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Llorian M, Stamataki Z, Hill S, Turner M, Martensson IL. The PI3K p110δ is required for down-regulation of RAG expression in immature B cells. J. Immunol. 2007;178:1981–1985. doi: 10.4049/jimmunol.178.4.1981. [DOI] [PubMed] [Google Scholar]
- 9.Kuwata N, Igarashi H, Ohmura T, Aizawa S, Sakaguchi N. Cutting edge: absence of expression of RAG1 in peritoneal B-1 cells detected by knocking into RAG1 locus with green fluorescent protein gene. J. Immunol. 1999;163:6355–6359. [PubMed] [Google Scholar]
- 10.Rosenberg N, Baltimore D, Scher CD. In vitro transformation of lymphoid cells by Abelson murine leukemia virus. Proc. Natl. Acad. Sci. USA. 1975;72:1932–1936. doi: 10.1073/pnas.72.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muljo SA, Schlissel MS. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat. Immunol. 2003;4:31–37. doi: 10.1038/ni870. [DOI] [PubMed] [Google Scholar]
- 12.Fornace AJ, Jr, Alamo I, Jr, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proc. Natl. Acad. Sci. USA. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res. 2005;569:133–143. doi: 10.1016/j.mrfmmm.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 14.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 15.Lu B, et al. GADD45γ mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45β required in cytokine-induced, but not TCR-induced, IFN-γ production. Nat. Immunol. 2001;2:157–164. doi: 10.1038/84264. [DOI] [PubMed] [Google Scholar]
- 17.Chi H, Lu B, Takekawa M, Davis RJ, Flavell RA. GADD45β/GADD45γ and MEKK4 comprise a genetic pathway mediating STAT4-independent IFNγ production in T cells. EMBO J. 2004;23:1576–1586. doi: 10.1038/sj.emboj.7600173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattioni T, Louvion JF, Picard D. Regulation of protein activities by fusion to steroid binding domains. Methods Cell Biol. 1994;43:335–352. doi: 10.1016/s0091-679x(08)60611-1. [DOI] [PubMed] [Google Scholar]
- 19.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral micro-RNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerwins P, Blank JL, Johnson GL. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J. Biol. Chem. 1997;272:8288–8295. doi: 10.1074/jbc.272.13.8288. [DOI] [PubMed] [Google Scholar]
- 21.Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217–221. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biggs WH, III, Cavenee WK, Arden KC. Identification and characterization of members of the FKHR (FOX O) subclass of winged-helix transcription factors in the mouse. Mamm. Genome. 2001;12:416–425. doi: 10.1007/s003350020002. [DOI] [PubMed] [Google Scholar]
- 23.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 24.Coffer PJ, Burgering BM. Forkhead-box transcription factors and their role in the immune system. Nat. Rev. Immunol. 2004;4:889–899. doi: 10.1038/nri1488. [DOI] [PubMed] [Google Scholar]
- 25.Biggs WH, III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellacosa A, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 27.Hu H, et al. Foxp1 is an essential transcriptional regulator of B cell development. Nat. Immunol. 2006;7:819–826. doi: 10.1038/ni1358. [DOI] [PubMed] [Google Scholar]
- 28.Verkoczy L, et al. A role for nuclear factor κB/rel transcription factors in the regulation of the recombinase activator genes. Immunity. 2005;22:519–531. doi: 10.1016/j.immuni.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 30.Billips LG, et al. Immunoglobulin recombinase gene activity is modulated reciprocally by interleukin 7 and CD19 in B cell progenitors. J. Exp. Med. 1995;182:973–982. doi: 10.1084/jem.182.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson K, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Corcoran AE, et al. The interleukin-7 receptor α chain transmits distinct signals for proliferation and differentiation during B lymphopoiesis. EMBO J. 1996;15:1924–1932. [PMC free article] [PubMed] [Google Scholar]
- 33.Venkitaraman AR, Cowling RJ. Interleukin-7 induces the association of phosphatidylinositol 3-kinase with the α chain of the interleukin-7 receptor. Eur. J. Immunol. 1994;24:2168–2174. doi: 10.1002/eji.1830240935. [DOI] [PubMed] [Google Scholar]
- 34.Tze LE, et al. Basal immunoglobulin signaling actively maintains developmental stage in immature B cells. PLoS Biol. 2005;3:e82. doi: 10.1371/journal.pbio.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Yusuf I, Andersen HM, Fruman DA. FOXO transcription factors cooperate with δEF1 to activate growth suppressive genes in B lymphocytes. J. Immunol. 2006;176:2711–2721. doi: 10.4049/jimmunol.176.5.2711. [DOI] [PubMed] [Google Scholar]
- 36.Hsu LY, et al. A conserved transcriptional enhancer regulates RAG gene expression in developing B cells. Immunity. 2003;19:105–117. doi: 10.1016/s1074-7613(03)00181-x. [DOI] [PubMed] [Google Scholar]
- 37.Bain G, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 38.Choi JK, Shen CP, Radomska HS, Eckhardt LA, Kadesch T. E47 activates the Ig-heavy chain and TdT loci in non-B cells. EMBO J. 1996;15:5014–5021. [PMC free article] [PubMed] [Google Scholar]
- 39.Lazorchak AS, Schlissel MS, Zhuang Y. E2A and IRF-4/Pip promote chromatin modification and transcription of the immunoglobulin κ locus in pre-B cells. Mol. Cell. Biol. 2006;26:810–821. doi: 10.1128/MCB.26.3.810-821.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alcamo E, et al. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J. Immunol. 2001;167:1592–1600. doi: 10.4049/jimmunol.167.3.1592. [DOI] [PubMed] [Google Scholar]
- 41.Kontgen F, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki Y, et al. Canonical NF-κB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Melamed D, Nemazee D. Self-antigen does not accelerate immature B cell apoptosis, but stimulates receptor editing as a consequence of developmental arrest. Proc. Natl. Acad. Sci. USA. 1997;94:9267–9272. doi: 10.1073/pnas.94.17.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fruman DA, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- 45.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindsley CW, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 47.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Hollander MC, et al. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 1999;23:176–184. doi: 10.1038/13802. [DOI] [PubMed] [Google Scholar]
- 49.Salvador JM, et al. Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity. 2002;16:499–508. doi: 10.1016/s1074-7613(02)00302-3. [DOI] [PubMed] [Google Scholar]
- 50.Furuyama T, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J. Biol. Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.