

Abstract
Human cytomegalovirus (HCMV) has been suggested to contribute to the development of vascular diseases. Since matrix metalloproteinases (MMPs) have been implicated in atherosclerosis and plaque rupture, we investigated the effect of HCMV infection on MMP expression in human macrophages. We used quantitative real-time PCR, Western blotting, and gelatin zymography to study the expression and activity of MMP-2, -3, -7, -9, -12, -13, and -14 and of tissue inhibitor of metalloproteinase 1 (TIMP-1), -2, -3, and -4. HCMV infection reduced MMP-9 mRNA, protein, and activity levels but increased TIMP-1 mRNA and protein levels. Furthermore, a decrease in MMP-12, MMP-14, TIMP-2, and TIMP-3 mRNA levels could be detected. The MMP-9 and TIMP-1 mRNA alterations required viral replication. MMP-9 mRNA expression was affected by an immediate-early or early viral gene product, whereas TIMP-1 mRNA expression was affected by late viral gene products. We conclude that HCMV infection specifically alters the MMP-9/TIMP-1 balance in human macrophages, which in turn reduces MMP-9 activity in infected cells. Since MMP-9 prevents atherosclerotic plaque development in mice, these results suggest that HCMV may contribute to atherogenesis through specific effects on MMP-9 activity.
Human cytomegalovirus (HCMV), a member of the betaherpesvirus family, is a ubiquitous pathogen that causes life-threatening disease in immunologically immature or immunocompromised individuals such as transplant recipients, AIDS patients, and congenitally infected neonates (16). It rarely causes any symptoms in healthy individuals. After a primary infection, HCMV remains latent within its host but can be reactivated throughout life. Primary infection usually starts with replication of the viral genome in mucosal epithelial cells, but the virus can infect and replicate in many different cell types (12).
Atherosclerosis is a multifactorial, inflammatory disease that may be caused or exacerbated by various factors. Accumulating evidence suggests that HCMV is a triggering or contributing factor in the development of vascular diseases, including atherosclerosis, restenosis, and vascular sclerosis in transplanted organs. HCMV nucleic acids have been detected in the arterial wall and atherosclerotic lesions of atherosclerotic patients. Furthermore, HCMV antigenemia due to a primary infection or reactivation of latent infection has been reported in patients with acute myocardial infarction (reviewed in references 2, 19, and 22). Thus, HCMV DNA in an atherosclerotic lesion can elicit an immune response in the aorta, resulting in the production of cytokines that attract blood-borne myeloid cells to the site of infection.
Inflammation is central for the life cycle of CMV. CMV establishes latency in monocytes/premonocytic cells (20), and reactivation of latent virus in macrophages appears to be dependent on cytokine-mediated differentiation of monocytes into macrophages. Thus, latent virus in infiltrating monocytes might be reactivated when the cells differentiate into macrophages, stimulating further cytokine production and reactivation of latent virus in the arterial wall.
Macrophages in atheromata create an environment rich in inflammatory cytokines, chemokines, and matrix-degrading proteases (7). Matrix metalloproteinases (MMPs) are closely related proteinases belonging to a family of at least 25 proteases that can degrade all macromolecules in the connective tissue matrix. Thus, MMPs regulate the composition of the extracellular matrix. Because they also regulate connective tissue remodeling, thereby determining the expansion and stability of the atherosclerotic plaque and the ability of smooth muscle cells to proliferate, MMPs have been implicated in atherogenesis and acute coronary syndrome. Macrophages have been proposed to be the main source of MMP activity in atherosclerotic plaques (1, 11, 13), and we have shown that differentiation of monocytes into macrophages markedly increases their proteolytic capacity (27). HCMV infection downregulates MMP activity in endothelial cells and cytotrophoblasts, impairing endothelial cell migration and placental cytotrophoblast invasiveness (28). In contrast, HCMV infection upregulates MMP-2 protein levels and activity in human coronary artery smooth muscle cells (18).
In this study, we investigated the effects of HCMV infection on the expression and activity of MMPs and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), in human primary macrophages. We found that HCMV specifically decreased MMP-9 expression and activity but increased TIMP-1 mRNA and protein levels. These observations provide important information for the understanding of the role of HCMV in the development of vascular diseases.
MATERIALS AND METHODS
Cell culture conditions.
Peripheral blood mononuclear cells from healthy blood donors were isolated by density gradient centrifugation on Lymphoprep (Axis-Shield PoC AS, Oslo, Norway). Lymphocytes were washed and reconstituted repeatedly in sterile Hanks' balanced salt solution (Sigma-Aldrich, Steinheim, Germany) and finally reconstituted in Iscove's modified Dulbecco's medium (Gibco) supplemented with l-glutamine, 25 mM HEPES, and 1% penicillin-streptomycin, and 10% human AB serum. Cells were plated in six-well, flat-bottom Primaria plates or eight-well chamber glass slides (Falcon; Becton Dickinson Labware, Meylan, France) and incubated for 2 h. After removal of nonadherent cells, macrophage differentiation was induced with a supernatant containing cytokines produced by T cells and monocytes as described previously (21). After 24 h, the cells were washed repeatedly with phosphate-buffered saline (PBS) and “60/30/10” medium, consisting of 60% serum-free lymphocyte medium (Aim V; Gibco) and 30% Iscove's modified Dulbecco's medium (Gibco) supplemented with l-glutamine, 25 mM HEPES, 1% penicillin-streptomycin, and 10% human AB serum. The cells were cultured for 7 days at 37°C in a 5% CO2 atmosphere.
Infection of macrophages.
Macrophages were infected with HCMV strain VR1814, an endothelial cell-adapted clinical isolate (6) (kindly provided by Giuseppe Gerna, University of Pavia, Italy), at a multiplicity of infection (MOI) of 1 to 12 for 2 to 168 h. In some experiments, we used UV-irradiated virus, virus-cleared inocula, or supernatant from human umbilical vein endothelial cells; in others, cells were infected with VR1814 in the presence of 5 mM Foscavir (AstraZeneca AB, Södertälje, Sweden), which was added 2 h after infection to inhibit viral replication. After incubation, cells were washed in PBS, and fresh 60/30/10 medium was added. Infected cells and uninfected control cells were collected at 0, 2, 6, 12, 24, 72, 96, or 168 h after infection.
RNA isolation and cDNA synthesis.
Total cellular RNA was isolated from macrophages by adding 350 μl of RLT buffer (Qiagen, Hilden, Germany) containing 1% β-mercaptoethanol. RNA was further isolated with the RNeasy minikit and QIAshredder (Qiagen) and analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). cDNA was synthesized from 500 ng of RNA with the SuperScript III first-strand synthesis system for reverse transcription-PCR and oligo(dT20) primers (Invitrogen Life Technologies, Carlsbad, CA).
PCR and real-time PCR.
To confirm the presence or absence of major immediate-early (IE) gene transcripts, we carried out a nested PCR with a specific set of primers (outer primer set I 3′ [5′-GAG TTC TGC CAG GAC ATC TTT-3′] and I 5′ [5′-GAG TCC TCT GCC AAG AGA AA-3′]; inner primer set II 3′ [5′-CTC GGG GTT CTC GTT GCA AT-3′] and II 5′ [5′-GAG AAA GAT GGA CCC TGA TAA T-3′]). Glucose-6-phosphate dehydrogenase was analyzed as a positive control (primers I 3′ [5′-ATG ATC CCA AAT TCA TCG AAA TA-3′], I 5′ [5′-AAC CGC ATC ATC GTG GAG AA-3′], II 3′ [5′-AGG ATA ACG CAG GCG ATG TT-3′], and II 5′ [5′-GCG AAG GAG ATG GTG CAG AA-3′]).
Gene expression was analyzed with TaqMan assays for MMP-2 (Hs00234422_m1), MMP-3 (Hs00233962_m1), MMP-7 (Hs00159163_m1), MMP-9 (Hs00234579_m1), MMP-12 (Hs00159178_m1), MMP-13 Hs00233992_m1, MMP-14 (Hs00237119_m1), TIMP-1 (Hs00171558_m1), TIMP-2 (Hs00234278_m1), TIMP-3 Hs00165949_m1, TIMP-4 (Hs00162784_m1), and β2-microglobulin (Hs00187842_m1). The assay for HCMV late gene pp150 mRNAs was manufactured by the Custom TaqMan Gene Expression Assay Service (forward, GGCGCGGGAACCTCTT; reporter, 6-carboxyfluorescein-CAGCCGTCAGCCTCG; reverse, CCGTGGGCGACAAAACG). All assays were from Applied Biosystems (Foster City, CA).
Real-time PCR was performed with the ABI Prism 7000 or 7700 sequence detection system. Collected data were analyzed with the sequence detection system software. The standard curve method was used to calculate collected and analyzed data, as described in ABI Prism User Bulletin 2. All samples were normalized to the housekeeping β2-microglobulin gene.
Western blotting.
For Western blotting experiments, cytosolic protein fractions were collected from HCMV-infected or mock-infected human macrophages. The protein concentration was determined by the bicinchoninic acid method. Cytosolic proteins (30 μg) from each sample were mixed with 2× Tris-glycine-sodium dodecyl sulfate (SDS) sample buffer containing β-mercaptoethanol and heated at 95°C for 4 min. The samples were loaded on 1-mm, 4 to 20% Tris-glycine precast gels (PAGEr Gold; Cambrex BioScience, Rockland, ME) along with full-range Rainbow molecular weight markers (RPN800; Amersham Biosciences, Sweden) and electrophoresed for 1.5 h at 150 V. Proteins were transferred to polyvinylidene fluoride transfer membranes (Hybond-P; Amersham Biosciences, Little Chalfont, United Kingdom) in a blotting container (100-V setting) for 1 h at 4°C. The membranes were incubated overnight at 4°C in PBS with 1% milk together with the primary antibody (monoclonal anti-human MMP-9, MAB911, or anti-human TIMP-1 antibody [R&D Systems, Minneapolis, MN]) or for 1 h at room temperature with mouse anti-β-actin (ab6276 [Abcam]) after stripping of the membranes. After washing, the membranes were incubated with sheep anti-mouse immunoglobulin G-horseradish peroxidase-linked whole antibody (NA931V; Amersham Bioscience, United Kingdom) or horseradish peroxidase-linked rabbit anti-goat antibody (P0449; Dakocytomation, Denmark) for 2 h at 4°C, and proteins were detected with a peroxidase substrate kit (DAB SK-4100; Vector Laboratories, Burlingame, CA) or with the ECL system.
Zymography.
Cytosolic MMP-9 activity in HCMV-infected or mock-infected cells was assessed by gelatin zymography. For each sample, 30 μg of cytosolic proteins was mixed with 5 μl of Novex Tris-glycine-SDS sample buffer (2×) (Invitrogen Life Technologies). Samples and full-range Rainbow molecular weight markers were loaded onto a Novex 10% zymogram gel, placed in a Novex XCell SureLock electrophoresis minicell, covered with Novex Tris-glycine-SDS running buffer (Invitrogen Life Technologies), and electrophoresed at 125 V for 1.5 h. The gels were incubated for 30 min at room temperature in Novex zymogram renaturing buffer and later incubated in Novex zymogram developing buffer (Invitrogen Life Technologies) overnight at 37°C. The buffer was discarded, and the gels were incubated in Coomassie blue staining solution (400 ml of 40% ethanol, 1.25 g of Coomassie brilliant blue R-250, 500 ml of distilled water, and 100 ml of 10% acetic acid) for 2 h at room temperature and washed in water.
ELISA.
Supernatants collected from noninfected or HCMV-infected cell cultures were analyzed for secreted MMP-9 or TIMP-1 at 4 and 7 days after infection by using Quatikine enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN). The data obtained for each sample were normalized to the corresponding total protein concentration (bicinchoninic acid method).
Statistical analysis.
Data are expressed as means ± standard errors of the means. Statistical significances was tested using either an unpaired Mann-Whitney test or a paired Wilcoxon signed rank test; a P value of <0.05 was considered statistically significant. Analysis was performed using StatView software version 5.0 (Cary, NC).
RESULTS
HCMV infection downregulates MMP-9 mRNA and upregulates TIMP-1 mRNA in human macrophages.
To examine the effects of HCMV on expression of MMPs and TIMPs, we collected mRNA from uninfected and infected macrophages and analyzed the samples by real-time PCR at 4 days after infection, a time point when a full replication cycle of the viral genome is estimated to be completed. MMP-9, -12, and -14 were the only MMPs found to be significantly altered by HCMV infection compared to the control (Fig. 1A). MMP-3 and -13 mRNA levels were below the limit of detection (data not shown). TIMP-1, -2, and -3 mRNA levels were significantly altered in HCMV-infected cells (Fig. 1B). TIMP-1 mRNA levels were found to be increased, while TIMP-2 and -3 mRNA levels decreased after HCMV infection. However, the most profound effects after HCMV infection were seen in the alterations of MMP-9 and TIMP-1 mRNA levels.
FIG. 1.

Quantitative real-time PCR analysis of MMP-2, MMP-7, MMP-9, MMP-12, and MMP-14 (A) and TIMP-1, -2, -3, and -4 (B) mRNA levels in uninfected versus HCMV-infected macrophages (n = 8). The mRNA levels were normalized to β2-microglobulin (β2M) and are presented as percentages of control values (MOI = 1). *, P < 0.05. Experiments were performed 4 days after infection. Error bars indicate standard errors of the means.
MMP-9 and TIMP-1 expression appears to be regulated by different viral mechanisms.
To determine if viral replication is required to alter the expression of MMP-9 and TIMP-1 mRNAs, we performed experiments with UV-irradiated, replication-deficient HCMV inocula. MMP-9 and TIMP-1 mRNA levels were not significantly altered by infection with UV-irradiated HCMV or virus-cleared inocula. Thus, viral replication was required to significantly alter the expression of MMP-9 and TIMP-1 mRNAs, and soluble factors from infected or mock-infected cells were not responsible for any significant changes (Fig. 2A and B).
FIG. 2.
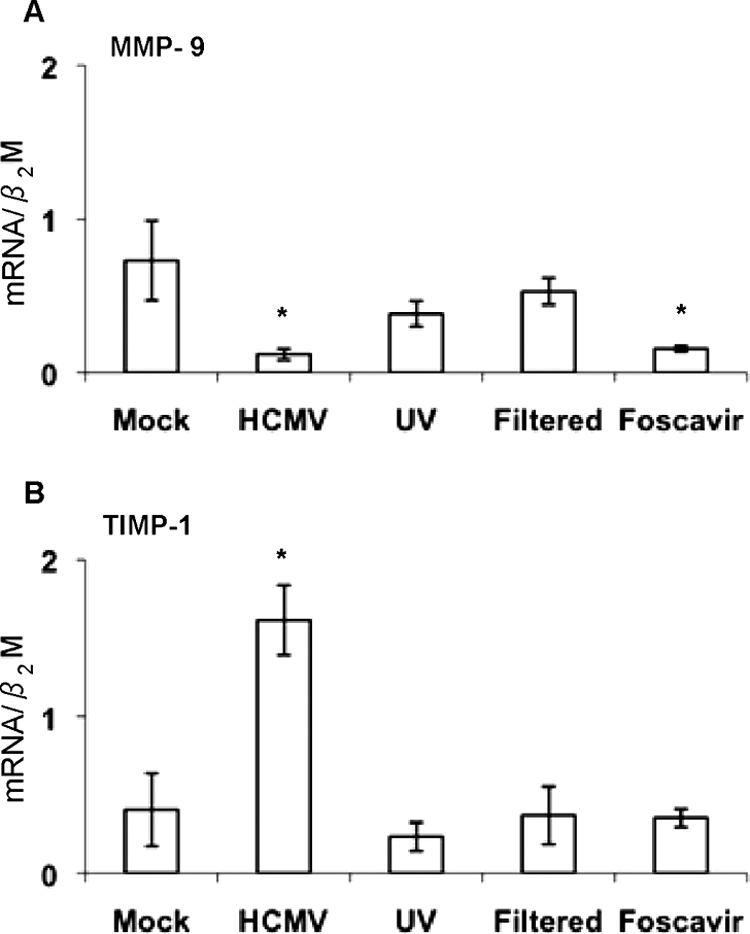
Quantitative real-time PCR analysis of MMP-9 (A) and TIMP-1 (B) mRNA levels in macrophages (n = 3). Macrophages were mock infected or challenged with HCMV, UV-irradiated HCMV inocula, filtered viral inocula from HCMV-infected cultures, or HCMV-infected macrophages treated with Foscavir 2 h after infection. The mRNA levels were normalized to β2-microglobulin (β2M) (MOI = 1). *, P < 0.05. Experiments were performed 4 days after infection. Error bars indicate standard errors of the means.
To further analyze whether IE/early or late viral genes were involved, we added Foscavir to the cell cultures at 2 h after infection to block late viral gene expression. MMP-9 mRNA levels were decreased (Fig. 2A), indicating that an IE or early HCMV gene product downregulated MMP-9 mRNA. However, the viral effect on TIMP-1 mRNA expression was blocked (Fig. 2B), indicating that late viral proteins are involved in HCMV's ability to affect TIMP-1 mRNA expression. UV treatment or filtration of viral inocula, as well as Foscavir treatment, inhibited late gene pp150 mRNA expression compared to that in mock-infected cells (Fig. 3A to C). The Foscavir-treated samples were all positive for IE mRNA in a nested PCR assay (Fig. 3D).
FIG. 3.
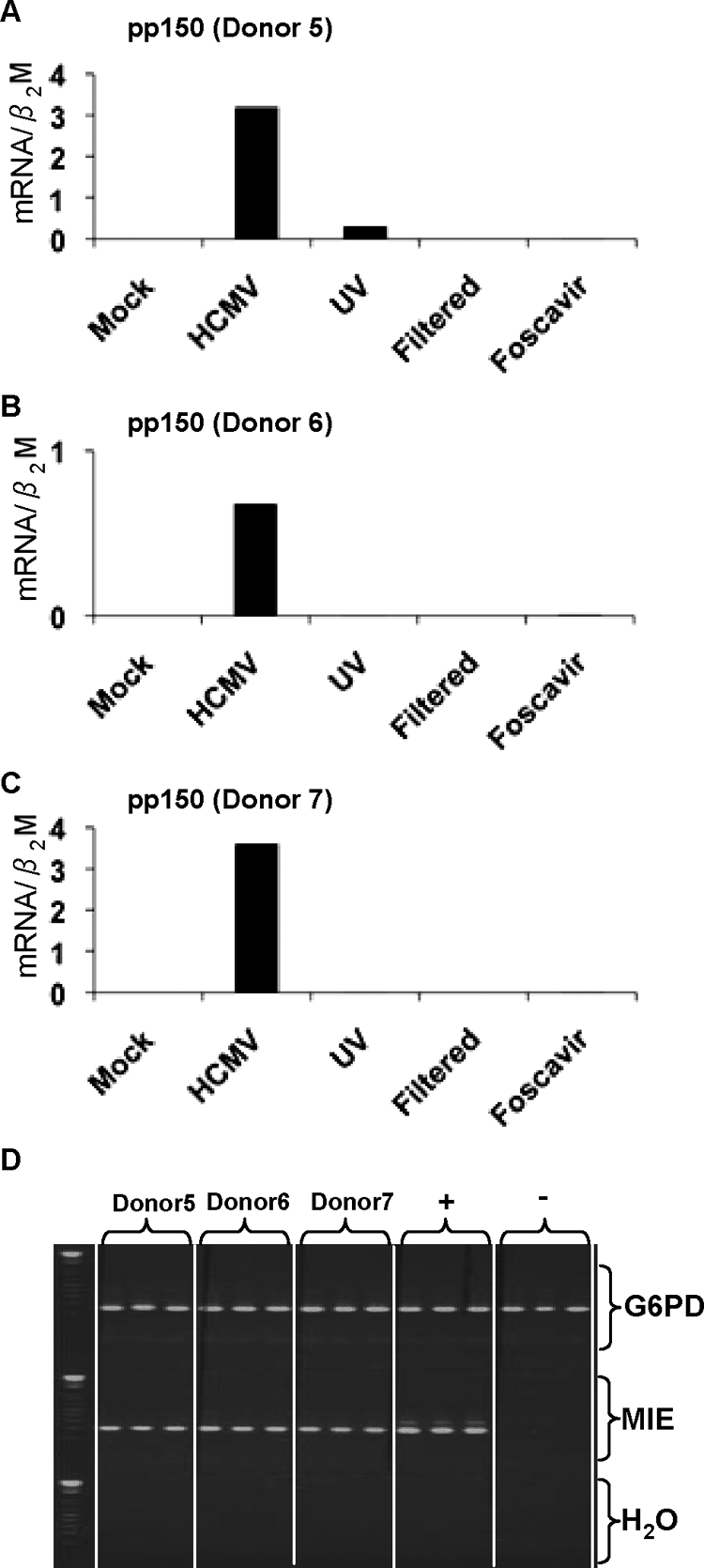
(A to C) Quantitative real-time PCR analysis of HCMV pp150 mRNA levels in macrophages from three donors. The macrophages were mock infected or challenged with HCMV, UV-irradiated HCMV inocula, filtered viral inocula from HCMV-infected cultures, or HCMV-infected macrophages treated with Foscavir 2 h after infection. The mRNA levels were normalized to β2-microglobulin (β2M). (D) Nested PCR for detection of the major IE (MIE) gene in the Foscavir-treated samples from three donors; a triplicate for each donor is displayed (MOI = 1). Experiments were performed 4 days after infection. G6PD, glucose-6-phosphate dehydrogenase.
MMP-9 mRNA levels were found to be slightly decreased already at 2 h after infection, and major effects were observed at 72 to 168 h after infection (Fig. 4A). In contrast, TIMP-1 mRNA levels increased first at 72 h after infection (Fig. 4B). These observations support the notion that MMP-9 mRNA expression is affected by an IE or early viral gene product and TIMP-1 by a late gene product. MMP-9 and TIMP-1 mRNA levels were altered in a dose-dependent manner with increasing viral doses of HCMV (Fig. 4C and D).
FIG. 4.

(A and B) Quantitative real-time PCR analysis of the viral kinetic effect on the expression of MMP-9 (A) and TIMP-1 (B) mRNA levels in HCMV-infected macrophages (MOI = 1). (C and D) Quantitative real-time PCR analysis of MMP-9 (C) and TIMP-1 (D) mRNA levels in macrophages that were uninfected or infected with HCMV at different MOIs. Dose-dependent effects on the expression of MMP-9 and TIMP-1 mRNA levels were observed 4 days after infection. The mRNA levels were normalized to β2-microglobulin (β2M) and presented as percentages of control values. *, P < 0.05. Error bars indicate standard errors of the means.
HCMV downregulates MMP-9 protein expression and activity but upregulates TIMP-1 protein expression.
To determine if MMP-9 and TIMP-1 protein levels and MMP-9 activity were altered by HCMV, we performed Western blot experiments, gelatin zymography, and ELISA. Cytosolic levels of MMP-9 protein were clearly reduced by HCMV infection in human macrophages at 4 days after infection (Fig. 5A). In contrast, cytosolic TIMP-1 protein levels increased after HCMV infection (Fig. 5B). Further, MMP-9 activity was consistently reduced in macrophages infected with HCMV, in contrast to the case for the mock-infected cells (Fig. 5C). We further analyzed supernatants from noninfected or HCMV-infected macrophages, collected at 4 and 7 days after infection, by ELISA. At 4 days after infection we observed a trend for decreased MMP-9 secretions in HCMV-infected cells compared to control cells (Fig. 6A). At 7 days after infection we observed a significant decrease in secreted MMP-9 in HCMV-infected cells compared to control cells (Fig. 6B). TIMP-1 secretion was significantly increased in HCMV-infected cells at 4 days after infection (Fig. 6C) but not at 7 days after infection (Fig. 6D).
FIG. 5.
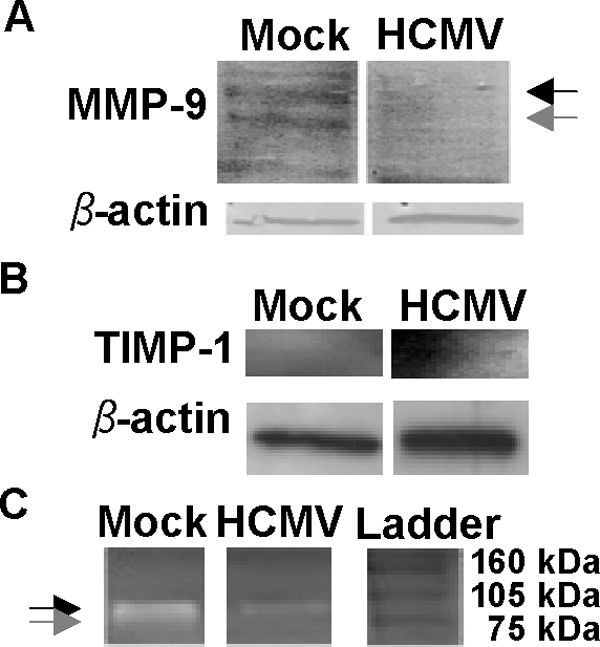
(A) Representative Western blot analysis for pro-MMP-9 (black arrow) and active-MMP-9 (gray arrow) protein levels in mock-infected macrophages and macrophages infected with HCMV; β-actin was used as a loading control. (B) Representative Western blot analysis for TIMP-1 protein levels in mock-infected macrophages and macrophages infected with HCMV; β-actin was used as a loading control. (C) A representative zymograph image of mock-infected and HCMV-infected macrophages assayed by gelatin zymography. The black arrow indicates pro-MMP-9, and the gray arrow indicates active-MMP-9. Experiments were performed 4 days after infection at an MOI of 1.
FIG. 6.
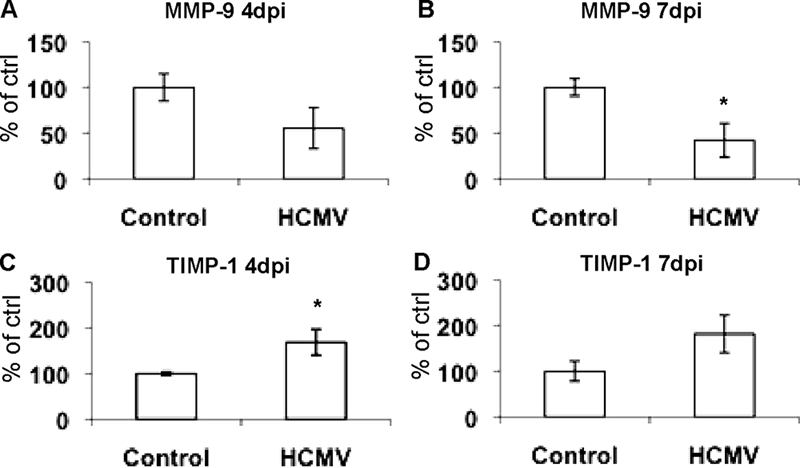
ELISA measurements of secreted MMP-9 and TIMP-1 in supernatants from noninfected or HCMV-infected macrophages at 4 and 7 days after infection. (A) MMP-9 levels in supernatants 4 days after infection. (B) MMP-9 levels in supernatants 7 days after infection. (C) TIMP-1 levels in supernatants 4 days after infection. (D) TIMP-1 levels in supernatants 7 days after infection. Data are presented as percentages of control values (n = 3). *, P < 0.05. Error bars indicate standard errors of the means.
DISCUSSION
Through their ability to control extracellular remodeling, MMPs figure prominently in inflammatory diseases, such as cardiovascular diseases, pulmonary diseases, cancer, and autoimmune diseases (5, 13, 17, 26). Here, we show that HCMV infection of human macrophages reduces MMP-9 mRNA expression and activity and increases the expression of its inhibitor, TIMP-1. The decrease in MMP-9 mRNA levels was mediated by HCMV IE or early gene products, whereas the increase in TIMP-1 expression was mediated by late viral gene products.
We also found significant effects on MMP-12, MMP-14, TIMP-2, and TIMP-3 mRNA expression after HCMV infection. However, since the most profound effects were on MMP-9 and TIMP-1 mRNA levels and because MMP-9 has been described as one of the key proteolytic enzymes in vascular diseases, we focused our further analysis on MMP-9 and TIMP-1.
How the HCMV-mediated downregulation of macrophage MMP-9 activity would affect atherosclerosis is difficult to predict. Until recently, all MMPs were thought to contribute to vascular disease progression by weakening the atherosclerotic plaque and increasing the risk of plaque rupture. However, a more complicated picture has emerged. Johnson et al. recently examined the effect of deficiency in various MMPs and apolipoprotein E (apoE) on atherosclerosis development in mice (9). apoE-knockout mice (29) develop spontaneous hypercholesterolemia and arterial lesions and are widely used for experimental atherosclerosis studies. In apoE/MMP-9 double-knockout mice, the plaque area was larger than in strain-matched apoE single-knockout controls (9). These mice also had more buried fibrous layers per plaque (suggesting previous plaque rupture), fewer smooth muscle cells, and more macrophages in the plaques than the single-knockout mice. These data suggest that MMP-9 plays a protective role in atherosclerosis by promoting plaque stability. Thus, the HCMV-mediated decrease in MMP-9 activity may lead to larger, unstable plaques. This finding suggests yet another molecular link between HCMV and vascular diseases.
Studies with apoE-knockout mice also suggest that CMV contributes to atherogenesis. As in MMP-9/apoE double-knockout mice (9), lesion size was increased in apoE-knockout mice infected with murine CMV (3). In another study of apoE-knockout mice, murine CMV infection increased the number and size of early atherosclerotic lesions at 2 weeks after infection and the number of advanced lesions at 20 weeks (24). In addition, there was evidence that murine CMV contributes to atherosclerosis through both direct effects (viral DNA and increased production of proinflammatory cytokines in the aortic arch) and an indirect effect (increased systemic levels of proinflammatory cytokines) (24).
The molecular mechanisms of the CMV-mediated decrease in MMP-9 expression and activity are not understood. In apoE-knockout mice infected with murine CMV, interleukin-18 (IL-18) and gamma interferon expression increased in infected macrophages (25). Gamma interferon-mediated STAT-1α activation of the transcriptional target class II major histocompatibility complex transactivator (CIITA or MHC2TA) reportedly suppresses MMP-9 expression by competitive binding of the CREB-binding protein (CBP) (15), a histone acetyltransferase that regulates the expression of many genes. The HCMV-encoded IE2-p86 protein interacts with the acetyltransferase domains of p300 and CBP (8). Hence, the competitive binding of CBP that is necessary for the activation of the MMP-9 promoter by IE2-p86 may influence the expression of MMP-9 in HCMV-infected cells. Interestingly, a polymorphism in CIITA was recently associated with increased risk of myocardial infarction (23).
Consistent with our results, HCMV downregulated MMP activity in endothelial cells and cytotrophoblasts, impairing the migration of endothelial cells and the invasiveness of placental cytotrophoblasts (28). After HCMV infection, levels of human IL-10 were elevated, and infected cells secreted a functional HCMV-encoded IL-10 homologue (UL111.5A or cmvIL-10) (28). IL-10 inhibits metalloproteinase production and enhances TIMP-1 expression (10). Since cmvIL-10 encoded by the UL111.5A gene is expressed as a gene product in the viral replication cycle (4) and because cells infected by HCMV induce human IL-10 mRNA at 24 h postinfection (14), it is possible that human IL-10 or cmvIL-10 could be responsible for the downregulation of MMP-9 mRNA levels. The upregulation of TIMP-1 mRNA at later time points might also be influenced by elevated levels of IL-10 produced by HCMV-infected cells.
In summary, HCMV infection reduced MMP-9 activity in macrophages and altered the MMP-9/TIMP-1 balance. This finding suggests that HCMV may contribute to the growth and destabilization of atherosclerotic plaques and provides another possible molecular link between the virus and vascular disease development.
Acknowledgments
This work was supported by grants from the Swedish Heart-Lung Foundation (20070446 and 20060425), the VR Swedish Research Council (12660 and K2007-56X-12615-10-3), and the Swedish Cancer Foundation (060253). C.S.-N. is a Royal Swedish Academy of Science Research Fellow supported by a grant from the Knut and Alice Wallenberg Foundation.
Footnotes
Published ahead of print on 22 October 2008.
REFERENCES
- 1.Brauer, P. R. 2006. MMPs—role in cardiovascular development and disease. Front. Biosci. 11447-478. [DOI] [PubMed] [Google Scholar]
- 2.Bruggeman, C. A., H. J. Marjorie, and G. Nelissen-Vrancken. 1999. Cytomegalovirus and atherogenesis. Antiviral Res. 43135-144. [DOI] [PubMed] [Google Scholar]
- 3.Burnett, M. S., C. A. Gaydos, G. E. Madico, S. M. Glad, B. Paigen, T. C. Quinn, and S. E. Epstein. 2001. Atherosclerosis in apoE knockout mice infected with multiple pathogens. J. Infect. Dis. 183226-231. [DOI] [PubMed] [Google Scholar]
- 4.Chambers, J., A. Angulo, D. Amaratunga, H. Guo, Y. Jiang, J. S. Wan, A. Bittner, K. Frueh, M. R. Jackson, P. A. Peterson, M. G. Erlander, and P. Ghazal. 1999. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J. Virol. 735757-5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galis, Z. S., and J. J. Khatri. 2002. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 90251-262. [PubMed] [Google Scholar]
- 6.Grazia Revello, M., F. Baldanti, E. Percivalle, A. Sarasini, L. De-Giuli, E. Genini, D. Lilleri, N. Labo, and G. Gerna. 2001. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J. Gen. Virol. 821429-1438. [DOI] [PubMed] [Google Scholar]
- 7.Hansson, G. K. 2005. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 3521685-1695. [DOI] [PubMed] [Google Scholar]
- 8.Hsu, C. H., M. D. Chang, K. Y. Tai, Y. T. Yang, P. S. Wang, C. J. Chen, Y. H. Wang, S. C. Lee, C. W. Wu, and L. J. Juan. 2004. HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J. 232269-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson, J. L., S. J. George, A. C. Newby, and C. L. Jackson. 2005. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc. Natl. Acad. Sci. USA 10215575-15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacraz, S., L. P. Nicod, R. Chicheportiche, H. G. Welgus, and J. M. Dayer. 1995. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J. Clin. Investig. 962304-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libby, P., P. M. Ridker, and A. Maseri. 2002. Inflammation and atherosclerosis. Circulation 1051135-1143. [DOI] [PubMed] [Google Scholar]
- 12.Mocarski, E. S., and C. T. Courcelle. 2001. Cytomegaloviruses and their replication, p. 2629-2673. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 13.Newby, A. C. 2005. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol. Rev. 851-31. [DOI] [PubMed] [Google Scholar]
- 14.Nordoy, I., H. Rollag, E. Lien, H. Sindre, M. Degre, P. Aukrust, S. S. Froland, and F. Muller. 2003. Cytomegalovirus infection induces production of human interleukin-10 in macrophages. Eur. J. Clin. Microbiol. Infect. Dis. 22737-741. [DOI] [PubMed] [Google Scholar]
- 15.Nozell, S., Z. Ma, C. Wilson, R. Shah, and E. N. Benveniste. 2004. Class II major histocompatibility complex transactivator (CIITA) inhibits matrix metalloproteinase-9 gene expression. J. Biol. Chem. 27938577-38589. [DOI] [PubMed] [Google Scholar]
- 16.Pass, R. F. 2001. Cytomegalovirus, p. 2675-2705. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Wiliams & Wilkins, Philadelphia, PA. [Google Scholar]
- 17.Ram, M., Y. Sherer, and Y. Shoenfeld. 2006. Matrix metalloproteinase-9 and autoimmune diseases. J. Clin. Immunol. 26299-307. [DOI] [PubMed] [Google Scholar]
- 18.Reinhardt, B., M. Winkler, P. Schaarschmidt, R. Pretsch, S. Zhou, B. Vaida, A. Schmid-Kotsas, D. Michel, P. Walther, M. Bachem, and T. Mertens. 2006. Human cytomegalovirus-induced reduction of extracellular matrix proteins in vascular smooth muscle cell cultures: a pathomechanism in vasculopathies? J. Gen. Virol. 872849-2858. [DOI] [PubMed] [Google Scholar]
- 19.Soderberg-Naucler, C. 2006. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J. Intern. Med. 259219-246. [DOI] [PubMed] [Google Scholar]
- 20.Soderberg-Naucler, C., K. N. Fish, and J. A. Nelson. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91119-126. [DOI] [PubMed] [Google Scholar]
- 21.Soderberg-Naucler, C., D. N. Streblow, K. N. Fish, J. Allan-Yorke, P. P. Smith, and J. A. Nelson. 2001. Reactivation of latent human cytomegalovirus in CD14+ monocytes is differentiation dependent. J. Virol. 757543-7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Streblow, D. N., S. L. Orloff, and J. A. Nelson. 2001. Do pathogens accelerate atherosclerosis? J. Nutr. 1312798S-2804S. [DOI] [PubMed] [Google Scholar]
- 23.Swanberg, M., O. Lidman, L. Padyukov, P. Eriksson, E. Akesson, M. Jagodic, A. Lobell, M. Khademi, O. Borjesson, C. M. Lindgren, P. Lundman, A. J. Brookes, J. Kere, H. Luthman, L. Alfredsson, J. Hillert, L. Klareskog, A. Hamsten, F. Piehl, and T. Olsson. 2005. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat. Genet. 37486-494. [DOI] [PubMed] [Google Scholar]
- 24.Vliegen, I., A. Duijvestijn, G. Grauls, S. Herngreen, C. Bruggeman, and F. Stassen. 2004. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect. 617-24. [DOI] [PubMed] [Google Scholar]
- 25.Vliegen, I., A. Duijvestijn, F. Stassen, and C. Bruggeman. 2004. Murine cytomegalovirus infection directs macrophage differentiation into a pro-inflammatory immune phenotype: implications for atherogenesis. Microbes Infect. 61056-1062. [DOI] [PubMed] [Google Scholar]
- 26.Vu, T. H., and Z. Werb. 2000. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 142123-2133. [DOI] [PubMed] [Google Scholar]
- 27.Whatling, C., H. Bjork, S. Gredmark, A. Hamsten, and P. Eriksson. 2004. Effect of macrophage differentiation and exposure to mildly oxidized LDL on the proteolytic repertoire of THP-1 monocytes. J. Lipid Res. 451768-1776. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto-Tabata, T., S. McDonagh, H. T. Chang, S. Fisher, and L. Pereira. 2004. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J. Virol. 782831-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang, S. H., R. L. Reddick, J. A. Piedrahita, and N. Maeda. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258468-471. [DOI] [PubMed] [Google Scholar]