

Abstract
Pancreatic endocrine tumors (PETs) have long fascinated clinicians and investigators despite their relative rarity. Their clinical presentation varies depending upon whether the tumor is functional or not and also according to the specific hormonal syndrome produced. Tumors may be sporadic or inherited but little is known about their molecular pathology, especially the sporadic forms. Chromogranin A appears to be the most useful serum marker for diagnosis, staging and monitoring. Initially, therapy should be directed at the hormonal syndrome as this has the major initial impact on the patient's health. Most PETs are relatively indolent but ultimately malignant, except for insulinomas which are predominantly benign. Surgery is the only modality that offers the possibility of cure although it is generally noncurative in patients with Zollinger-Ellison syndrome or nonfunctional PETs with MEN1. Preoperative staging of disease extent is necessary to determine the likelihood of complete resection though debulking surgery is often felt to be useful in unresectable patients. Once metastatic, biotherapy is usually the first modality employed because it is generally well tolerated. Systemic or regional therapies are generally reserved until symptoms occur or tumor growth is rapid. Recently a number of newer agents, as well as receptor-directed radiotherapy, are being evalulated for patients with advanced disease. This review addresses a number of recent advances regarding the molecular pathology, diagnosis, localization and management of PETs including discussion of peptide receptor radionuclide therapy and other novel antitumor approaches. We conclude with a discussion of future directions and unsettled problems in the field.
Introduction
Pancreatic endocrine tumors (PETs) have long fascinated clinicians and investigators because of their unusual and florid symptoms as well as the insights they provide into the actions of gastrointestinal (GI) hormones. PETs share many pathological and biological features with GI carcinoids, but they have important differences which affect treatment as well as having a different pathogenesis1, 2, and thus the two groups of gastrointestinal neuroendocrine tumors(NETs) are best considered separately. There have been a number of recent advances in various aspects of PETs including diagnosis, management, insights into molecular changes, tumor localization, and the treatment of advanced disease. This paper will briefly review a number of these advances as well as their current management. All aspects of PETs will not be covered, because many features are recently covered in reviews or consensus conferences3-7.
Epidemiology
PETs occur in 0.5-1.5% of autopsies but are functional or symptomatic in <1/1000, resulting in a clinical detection rate of 1:100,000 population, which comprise 1-2% of pancreatic neoplasms8. In older studies nonfunctional PETs (NF-PETs), insulinomas and gastrinomas had equal frequency9, however in recent studies NF-PETs are twice as frequent10, 11. The relative frequency of PETs varies in surgical or medical series, but most studies suggest a relative order of: NF-PET >insulinoma>gastrinoma >glucagonoma >VIPomas >somatostatinomas>others9, 11. Four inherited disorders have an increased incidence of PETs: Multiple Endocrine Neoplasia-type 1 (MEN1), von Hippel-Lindau disease (VHL), von Recklinghausen's disease (VRD or neurofibromatosis 1[NF-1]) and tuberous sclerosis12, 13. The most important is MEN1 because 80-100% of these patients develop NF-PETs, 50-60% gastrinomas, 20% insulinomas and 3-5% VIPomas or glucagonomas with the result that 20-25% of all gastrinomas and 4% of insulinomas are due to this syndrome12, 13. PETs (primarily NF-PETs) develop in 10-17% of VHL patients, 0-10% of NF-1 patients (primarily duodenal somatostatinomas) and <1% of tuberous sclerosis patients (primarily NF-PETs)12, 13.
Classification/Pathology
PETs are divided clinically into two groups: functional and nonfunctional (NF-PETs). Functional PETs secrete biologically active peptides causing one of nine well-established syndromes (Table 1). NF-PETs are not associated with a specific hormonal syndrome either because no peptide is secreted or the substance secreted does not cause specific symptoms. Most (>70%) NF-PETs are not truly nonfunctional because they secrete substances such as pancreatic polypeptide (PPoma), other peptides (neurotensin, ghrelin, etc), neuron-specific enolase (NSE), chromogranins or human chorionic-gonadotropin subunits, each of which does not cause specific symptoms9, 14(Table 1). In addition to the well-established PET syndromes (Table 1), small numbers of patients are described with PETs producing other biologically active substances and new syndromes have been proposed, although in most cases too few patients have been described to clearly establish this point or its spectrum. GI tumors have been described secreting luteinizing-hormone causing masculinization15, secreting renin causing erythrocytosis16 and secreting PYY causing constipation (primarily ovarian tumors)17.
Table 1.
Pancreatic endocrine tumor syndromes
| Name of Tumor [syndrome] | Hormone Causing Syndrome | Signs or Symptoms | Primary Location | Malignant (%) |
|---|---|---|---|---|
| Gastrinoma | Gastrin | Abdominal Pain | Pancreas-60% | 60-90 |
| [Zollinger-Ellison syndrome] | Diarrhea | Duodenum-30% | ||
| Esophageal symptoms | Other-10% | |||
| Insulinoma | Insulin | Hypoglycemic symptoms | Pancreas- 99-100% | 5-15 |
| Glucagonoma | Glucagon | Rash, anemia | Pancreas- 99-100% | 60 |
| Diabetes/glucose intolerance | ||||
| Weight loss | ||||
| Thromboembolic disease | ||||
| VIPoma | Vasoactive intestinal peptide (VIP) | Severe watery diarrhea | Pancreas-90% | 80 |
| [Verner-Morrison, Pancreatic cholera WDHA] | Hypokalemia | Other-10% (neural, adrenal, periganglionic tissue) | ||
| Somatostatinoma | Somatostatin | Diabetes mellitus | Pancreas-56% | 60 |
| Cholelithiases | Duodenum/jejunum-44% | |||
| Diarrhea | ||||
| Steatorrhea | ||||
| GRFoma | Growth hormone releasing factor (GRF) | Acromegaly | Pancreas-30% | 30 |
| Lung-54% | ||||
| Jejunum-7% | ||||
| Other-13% (adrenal, foregut, retroperitoneum) | ||||
| ACTHoma [Cushing's syndrome] | Adrenocorticotropic hormone (ACTH) | Cushing's syndrome | Pancreas - 4-16% all ectopic Cushing's | >90 |
| PET causing the carcinoid syndrome [Carcinoid syndrome] | Serotonin s tachykinins prostaglandins | Diarrhea Flushing | Pancreas-100% | 68-88 |
| PET causing hypercalcemia | PTH-RP | Symptoms due to increased calcium | Pancreas-100% | 80-90 |
| Nonfunctioning [PPoma, Nonfunctional][PP,CgAAbdomi | None al] [PP, CgA NSE, etc *] | Weight loss, hepatomegaly Abdominal mass Occasionally asymptomatic | Pancreas-100% | 60-90 |
but no symptoms due to product hypersecretion; other peptides not causing symptoms include ghrelin, neurotensin, calcitonin, subunits of human chorionic gonadotropin, etc
PETs share pathological features with carcinoids; both are considered to arise from the diffuse neuroendocrine cell system, uncommonly demonstrate mitotic figures, commonly demonstrate electron-dense granules containing various peptides, chromogranins, NSE and synaptophysin, and they many similarities in biological behavior14, 18. The latter properties particularly the presence of chromogranin are widely used to identify GI NETs14, 18. Both functional and NF-PETs frequently (>50%) synthesize more than one peptide14, 18. However, in most cases, these multiple peptides are not associated with specific syndromes. For this reason the diagnosis of a functional syndrome (Table 1) depends not on immunocytochemistry, but is diagnosed clinically9, 14, 18.
A recent standard WHO classification has proposed GI NETs be assigned to one of three categories (well-differentiated tumor, well-differentiated carcinoma, and poorly differentiated carcinoma) based on histology, size and proliferative indices14. In general histological classifications of PETs have failed to predict growth patterns for a given tumor. However this classification will allow more standardized comparison of results of different studies. For the first time a TNM classification for PETs has also been proposed19 which is based on the WHO classification of GI NETs and which may provide a more standardized assessment of patients and have important prognostic clinical value.
Molecular pathogenesis
Little is known about the molecular pathogenesis of PETs1, 2, 8. This has occurred in part because alterations in common oncogenes (fos, jun, myc, k-ras, etc) or common tumor suppressor genes (p53, retinoblastoma, etc) are not generally implicated in their pathogenesis1, 2, 8. Some of the most important insights have come from studies of inherited PET syndromes1, 2, 8, 20. Altered genes causing these syndromes are important in some cases of sporadic PETs (i.e. nonfamilial cases)1, 2, 8, 20. MEN1 is caused by mutations in the MEN1 gene on chromosome 11q13 which encodes for a 610 amino acid protein, menin, a nuclear protein that bind to numerous transcription factors13, 21, 22. However, the exact mechanism leading to development of PETs still remains unclear. Sporadic PETs show an acquired loss of heterozygosity (LOH) at this locus in 20-90% and 27-39% have a mutation1, 2, 8, 23, 24. In addition, recent studies show alterations in the p16/MTS1 tumor suppressor gene, the DP64/SMAD4 gene, amplification of the HER-2/neu proto-oncogene, and loss of an unknown tumor suppressor gene on chromosome 1 or 3p could also be important in the molecular pathogenesis of PETs1, 2, 8, 20. Genome-wide allelotyping and comparative genomic hybridization demonstrate that chromosomal gains (especially 7q,17q,17p,20q) and losses (especially 1p,3p,3,6p,22q) frequently occur in PETs and carcinoids, however their frequency varies markedly in these two GI NETs, providing evidence that they have a different pathogenesis1, 2, 8, 20, 25. Gene expression-profiling using microarray analysis has recently identified in PETs numerous additional altered genes26-29. In comparison to normal islets in one study29, 66 genes were over-expressed [particularly genes for some growth factors (IFGFBP3), cell migration/adhesion molecules (fibronectin) and putative oncogenes (MLLT 10/AF10)] and 119 under-expressed [particularly genes involved in cell cycle regulation (p21cip1), transcription factors (JunD) and a putative metastasis suppressor gene (NME3)]. In a second study26, when gene expression patterns in NF-PETs were compared to normal islets and three neuroendocrine tumor cell lines, 667 genes were up-regulated (particularly SERPINA10, BIN1, LCK, BST2) and 323 down-regulated. At present a clear concordance amongst studies is still lacking, but this approach is leading to the identification of numerous new candidate genes that may prove important in the pathogenesis of PETs or in determining growth behavior, which may have prognostic implications.
Tumor biology, prognosis and tumor markers
PETs differ in their malignant potential and location (Table 1). Some PETs (insulinomas, glucagonomas,VIPomas in adults) are found almost entirely within the pancreas, whereas others, although still referred to as PETs, are actually extrapancreatic [duodenal gastrinomas (60-80%)30-32, small intestinal somatostatinomas (40-50%), GRFomas primarily in the lung (>70%)] (Table 1). Insulinomas are malignant in 5-15%, whereas the other PETs are malignant in 50-90%, with metastases usually developing initially in regional lymph nodes, later in the liver and subsequently in distant sites such as bone6, 8, 14, 30, 33, 34. PETs in different patients may show different growth patterns33, 35-38. For example, in patients with gastrinomas, 75% demonstrate no growth/indolent growth whereas 25% demonstrate aggressive growth35, 36. Furthermore, even in patients with liver metastases, aggressive growth occurred in less than one-half of patients37. Therefore, identification of prognostic factors is particularly important in patients with PETs33. In almost every study, the presence or development of liver metastases, but not lymph node metastases, is a very important prognostic factor11, 33, 35, 36, 38-41. In one study35 the 15-year survival in patients with liver metastases was 26%, whereas without liver metastases it was 96%. The extent or rate of growth of liver metastases, presence of bone metastases, primary tumor size or location (duodenal vs. pancreatic gastrinomas), development of ectopic Cushing's syndrome, various histological features, high tumor marker levels, various flow cytometric features and high proliferative indices (Ki67, mitotic index) are important prognostic factors11, 19, 33, 35-38, 42-44. Survival is related to PET extent such that patients with primary tumors so small they are not found at surgery or with complete resections have survivals of 90-100%, those with incomplete resections 15-75%, and those with diffuse unresectable liver metastases 25-50%33, 35, 45-47. In some studies48 but not others11, 43, 49 patients with functional PETs have better survivals than those with NF-PETs. Recently, two studies50, 51 demonstrated for the first time that complete resection of the primary PET decreases the rate of development of liver metastases and/or improves survival50.
In addition to the specific hormone released by a functional PET (Table 1), other putative tumor markers have been proposed which could be useful for diagnosis/prognosis. This is particularly the case for NF-PETs. The marker most widely used is plasma chromogranin A (CgA) (elevated in 88-100%), although also proposed is plasma NSE (elevated in 83-100%), PP, pancreastatin and α or β subunits of human chorionic gonadotropin (elevated in 25-40%)52-54. Chromogranins (A,B,C) are acidic soluble proteins (MW-49kDa) found in large secretory granules of neuroendocrine cells and assessment of CgA level is now being increasingly used to diagnosis and monitor changes in NF-PETs, carcinoids and other PETs52, 54-56. CgA has an overall diagnostic sensitivity of 60-100% in patients with metastatic disease, but <50% in patients with localized/early disease56-58. CgA levels reflect tumor burden and it has been used to assess recurrences, tumor growth and changes in tumor size52, 55, 58.
Clinical Features and Diagnosis of PETs
Gastrinoma-clinical features/diagnosis
Gastrinomas secrete gastrin which causes hyperchlorhydria, thereby producing the Zollinger-Ellison syndrome(ZES)31, 45, 59, 60. With a long mean delay (6.1 years) in presentation/diagnosis45, 61, 62, patients generally present with acid-peptic conditions including complicated and uncomplicated ulcers and/or GERD [Table 2 (Top)]. Occasionally other manifestations such as diarrhea, malabsorption or in MEN1/ZES patients, various other endocrine features predominate [Table 2(Top)]61, 63, 64.
Table 2.
Presenting Features of ZES (recent series) and Causes of hypergastrinemia
| Data are from60,61,74,177,297-299 |
| Presenting Features of ZES (recent series) |
| Abdominal pain (75-100%) |
| Diarrhea (35-73%) (isolated in up to 35%) |
| Pain and diarrhea (55-60%) |
| Heartburn (44-64% |
| Duodenal (and prepyloric) ulcers (71-91%) |
| Ulcer complications [bleeding (1-17%), perforation (0-5%) or obstruction (0-5%)] |
| With MEN1 (22-24%) |
|
Causes of hypergastrinemia |
| Appropriate |
| Antisecretory therapy (especially proton pump inhibitors) |
| Atrophic gastritis (autoimmune pernicious anemia) |
| H. pylori pangastritis with atrophy |
| Vagotomy |
| Fundectomy |
| Chronic renal failure |
| Inappropriate |
| Zollinger-Ellison syndrome |
| Retained antrum syndrome |
| Antral predominant H. pylori infection (antral G-cell hyperfunction) |
| Chronic renal failure |
| Gastric outlet obstruction |
| Massive intestinal resection |
In constrast to the normal circumstance65, with gastrinomas, the tumor secretion of gastrin is not physiologically regulated and sustained inappropriate hypergastrinemia occurs.
Basal acid hypersecretion (present in >90% of patients) or after stimulation59, is a consequence of the inappropriate hypergastrinemia. Because a fasting serum gastrin (FSG) level is often the initial determination done in the United States in patients suspected of having ZES, it is important to remember that elevated levels can also be due to an appropriate physiological response to hypo/achlorhydria or an inappropriate response in other disease states [Table 2(Bottom)]. With the dramatic increase in proton pump inhibitor (PPI) use in the population, a recent study raises concern66 about the impact this is having on the diagnosis/presentation of ZES (Fig. 1). This study66 reported a 49% decrease in referrals of patients with possible ZES to two centers in the US and Italy since the widespread use of PPIs, a 40% decrease in the number of patients with ZES diagnosed (Fig. 1) and a 3-fold increase in the number of false positive diagnosis of ZES. This occurred because PPIs can control the symptoms of acid hypersecretion in almost all ZES patients, in contrast to conventional doses of H2 blockers, and thus mask the diagnosis. The increased false positive rate occurred because treatment with PPIs in non-ZES patients can cause hypergastrinemia to a level seen in 60% of ZES patients31, 53, 67, 68. This delay in diagnosis may lead to more patients with ZES presenting with advanced disease66. Diagnosis of ZES requires a typical clinical syndrome together with the demonstration of inappropriate hypergastrinemia31, 45, 53, 59, 67, 69. Fasting hypergastrinemia occurs in 97-99% of patients so this is usually the initial study raising suspicion of the disease31, 67. No absolute level of elevation of FSG alone is diagnostic31, 53, 67, 68. In the 40% of ZES patients with a FSG level >10-fold elevated, the diagnosis can be made with certainty (after excluding retained gastric-antrum syndrome by history) if the gastric pH is <259, 67, 70. In the 60% of patients with a FSG <10-fold elevated and a gastric pH <2, assessment of BAO and a secretin test should be performed. A BAO>15 mEq/hr with an elevated FSG in the absence of antisecretory therapy and a positive secretin test firmly establishes ZES. A recent study shows that the best criterion for a positive secretin test for ZES is an increase in FSG after subcutaneous secretin injection (0.4ug/kg) of >120 pg/ml above baseline producing a sensitivity of 94% and specificity of 100% (a significantly improved accuracy over the older criterion of >200 pg/ml increase)71, 72. It is important to remember that hypo/achlorhydria can cause a false-positive secretin test. Because of this PPIs need to be stopped to adequately assess for the presence of ZES and because of their long duration of action they generally need to be stopped for at least one week. PPI withdrawal should be done with care by a group familiar with establishing the diagnosis of ZES because abrupt withdrawal in patients with ZES can potentially lead to serious consequences. The diagnosis of ZES in MEN1 can be complicated by the fact that successful treatment of the hyperparathyroidism, which is almost invariably present at the time of the presentation of ZES64, can decrease FSG, acid secretion and reverse a previously positive secretin test, thereby masking the disease73-75.
Figure 1.

Effect of widespread use of PPIs on diagnosis and referral of ZES patients in two centers (Italian-La Sapienza University [Rome, Italy] and NIH [Bethesda, Maryland]). The left panel shows the annual number of referrals of new cases before and after the widespread use of PPIs. The right panel shows the result for diagnosis of ZES at the NIH center. (Modified from66)
Insulinoma-clinical features/diagnosis
Insulinomas ectopically secrete insulin resulting in inappropriate hyperinsulinemia which causes hypoglycemic episodes characterized by neuroglycopenic symptoms and sympathetic overdrive [Table 3(Top)]. Symptoms classically develop during periods of relative substrate deficiency (fasting or exercise)76, 77.
Table 3.
Features of the insulinoma and glucagonoma syndromes
| Data are from9,47,80-82,76,300-303 |
| Features of the insulinoma syndrome |
| Neuroglycopenia (90%) |
| Amnesia or coma (47%) |
| Confusion (80%) |
| Visual changes (59%) |
| Convulsions (17%) |
| Altered consciousness (38%) |
| Sympathetic overdrive (60-70%) |
| Weakness (56%) |
| Sweating (69%) |
| Tremors (24%) |
| Palpitations (12%) |
| Hyperphagia (14%) |
| Obesity (<50%) |
| Features of the glucagonoma syndrome |
| Migratory necrolytic erythema (70- 90%) |
| Weight loss (80%) |
| Glucose intolerance (40-90%) |
| Normochromic, normocytic anemia (35-90%) |
| Hypoaminoacidemia (80%) |
| Diarrhea (25%) |
| Thromoboembolism (15-25%) |
| Glossitis, chelitis (15-40%) |
| Psychiatric symptoms (0-17%) |
Similar to ZES, there is a delay in diagnosis (mean 4 yrs)76. Elevated serum insulin levels may be appropriate (a consequence of elevated blood glucose levels such as in type 2 diabetes mellitus) or inappropriate (with insulinomas, nesidioblastosis [MEN1-associated or post-bariatric surgery] or exogenous insulin administration. A serum glucose level <2.5 mmol/l (45 mg/dL) with an insulin level >6 uU/ml (43pmol/L by radioimmunoassay [RIA], ≥3 uU/ml by immunochemoluminescent assay [ICMA]) combined with an elevated C-peptide level (≥200 pmol/L) and the absence of sulfonylurea in the plasma, establishes the diagnosis76. The gold standard for establishing the diagnosis of insulinoma remains the 72 hour fast76. One-third of patients will develop symptoms within 12 hrs, 80% at 24 hrs, 90% at 48 hrs and 100% at 72 hrs76. Insulin levels are being increasingly determined using ICMAs or insulin-specific IRMAs that have no cross-reactivity with proinsulin and give lower values, resulting in up to 60 % of patients with insulinomas having plasma insulin levels <6uU/mL78, 79. In one recent study using these specific assays the most sensitive criterion for diagnosing insulinoma was the combination of an elevated proinsulin level with a fasting glucose <45mg/dL79.
Glucagonoma-clinical features/diagnosis
Glucagonomas ectopically secrete glucagon resulting in hyperglucagonemia. Glucagonomas cause glucose intolerance, weight loss and a pathognomonic rash called migratory necrolytic erythema (MNE) characterized by erythematous macules that develop into papules, become necrotic and heal with pigmented scarring9, 47, 80-82 (Table 3,Bottom). As with gastrinomas and insulinomas, glucagonomas present with a long history of symptoms (mean delay in diagnosis of 7 yrs with reports of up to 18 years) and tumors are commonly large at presentation (mean 6 cm)9, 47, 80, 81.
Despite controversy in the past regarding the specific cause of MNE, recent studies show glucagon infusions can lead directly to MNE83-85. However, MNE is not specific for glucagonoma occurring also in celiac disease, cirrhosis or pancreatitis81, 85, 86.
Diagnosis of a glucagonoma requires demonstration of an inappropriately elevated serum glucagon level (diagnostic at levels above 500-1000 pg/ml). Lower elevations may be associated with glucagonomas, but can also be caused by cirrhosis, pancreatitis, diabetes mellitus, prolonged fasting, sepsis, burns, renal failure, familial hyperglucagonemia and acromegaly9, 47, 80, 81.
VIPomas-clinical features/diagnosis
VIPomas ectopically secrete vasoactive intestinal polypeptide (VIP) leading to large volume diarrhea (90-100%) (100%>700 mL/day, 70-80%>3L/day), electrolyte disturbances [notably hypokalemia (70-100%)], dehydration (45-95%), hyperglycemia (20-50%), hypercalcemia (25-50%), hypochlorhydria (35-76%) and flushing (15-30%)9, 39, 87-89. The large volume diarrhea often results in dehydration without an osmolar gap because it is secretory in nature9, 39, 87-90. The diagnosis is confirmed by the presence of large volume secretory diarrhea with an elevated serum VIP level together with imaging evidence of a PET (in children the tumor commonly arises in extrapancreatic ganglioneuromas). However, even in the absence of imageable tumor, an elevated serum VIP level (>500pg/ml) in the presence of a documented secretory diarrhea is highly suggestive of VIPoma9, 39, 87-89.
Somatostatinoma-clinical features/diagnosis
Somatostatinomas are somatostatin (SS)-secreting tumors primarily occurring in the duodenum or pancreas which can produce the somatostatinoma syndrome, characterized by diabetes mellitus, gallbladder disease, weight loss, diarrhea, steatorrhea and anemia9, 40, 91-93. In the literature there is no general agreement on the definition of a somatostatinoma with most cases (55-89%) described as a PET with somatostatin present by immunohistochemistry, but with no associated clinical syndrome. It has been proposed that the term somatostatinoma syndrome should be reserved for cases with the specific clinical syndrome only. Duodenal somatostatinomas uncommonly produce the somatostatinoma syndrome (<20%) whereas pancreatic tumors often do (>90%)9, 40, 91-93. Because of the subtle nature of the syndrome, these tumors have an even later presentation than other PETs. They can occur in association with MEN1 (0-1% of all MEN1 patients) or in up to 10% of VRD patients 13. The diagnosis is best confirmed by the presence of a pancreatic or duodenal mass together with an elevated serum SS level in a patient with typical symptoms and a tumor staining for SS. However, serum levels should be interpreted with caution in individuals without concomitant masses. Unfortunately, there is no reliable provocative test to confirm the presence of a somatostatinoma in individuals with typical symptoms and no observable mass.
GRFoma-clinical features/diagnosis
GRFomas ectopically secrete growth hormone-releasing factor (GRF) leading to uncontrolled pituitary release of growth hormone resulting in acromegaly9, 94-96. Most cases of acromegaly are due to pituitary tumors and only a small fraction (<2%) to GRFomas. At least 50% of GRFomas arise in the lung (Table 1). Important clues to the presence of a GFRoma producing acromegaly are the absence of a pituitary tumor on imaging, the concomitant presence of MEN1 or the presence of an elevated prolactin level9, 94-96. GRFomas are diagnosed by the presence of an elevated GRF level (>300 pg/ml)9, 94-96. There are no reliable provocative tests for the GRFomas.
Nonfunctional PETs (NF-PET) -clinical features/diagnosis
NF-PETs are not associated with a hormonal syndrome (Table 1). Because of this, they are frequently found by chance and patients generally present late in the disease course with large primaries (70% >5 cm) and advanced disease (>60% have liver metastases)9, 97-101. NF-PETs produce symptoms due to tumor growth/spread [i.e., abdominal pain (40-60%), weight loss (25-50%), or jaundice (30-40%)]. In recent years, NF-PETs are increasingly being identified by chance [up to 35% of patients in one series99] as individuals undergo imaging studies for non-specific symptoms. Asymptomatic detection results in lower rates of metastases, increased resectability and improved survival102
A NF-PET is suggested by elevated levels of serum chromogranin A (69-100%) or PP (50-100%) or positive somatostatin-receptor scintigraphy (octreoscan) with a pancreatic mass. In the absence of a mass, other potential causes of elevated serum PP levels (e.g., old age, alcoholism, inflammatory conditions, renal failure and bowel resection) need to be considered. A confirmed diagnosis for NF-PET requires histological confirmation9, 97-101.
Tumor Localization/Staging
Imaging studies are essential for the management of patients with PETs. They are needed to localize the primary as well as for staging to guide management, including surgical plans (curative resection, debulking or medical management only), to monitor tumor growth, and for follow-up after therapy6, 9, 103-108.
Conventional cross-sectional imaging studies (MRI, CT, US)
Older studies evaluated various conventional imaging techniques [ultrasonography (US), computed tomographic (CT) scanning, or magnetic resonance imaging (MRI)] for localization/staging of PETs104, 105, 107, 109-111. PET detection with these techniques (which may be suggestive of a PET specifically) is size-dependent with <20% of PETs <1 cm identified, 30-40% 1-3 cm in diameter and >75% of PETs >3 cm45, 112. Most pancreatic VIPomas, glucagonomas and somatostatinomas are large and therefore detectable with conventional studies. However, many gastrinomas, insulinomas and duodenal somatostinomas are frequently <1 cm and will not be detected by these modalities104, 105, 109, 110. For identifying patients with liver metastases, US is the least sensitive (identifies 40% of patients with metastases), whereas CT and MRI are positive in 70-80%104, 105, 109, 110. Figure 2 (Top) shows liver metastases in a patient with gastrinoma by both CT and MRI scanning. As newer generations of scanners are being made available, these sensitivites may change105, 107. At present both high resolution spiral CT and modern MRI are highly effective at identifying liver metastases (sensitivity of up to 94%) but somewhat less effective in identifying primary tumors (sensitivity 55-78%), because the more common functional tumors (insulinomas or gastrinomas) are often small 113.
Figure 2.


CT, MRI, EUS in patients with PETs. Panel A illustrates CT (top) and MRI(bottom) images of the abdomen in a patient with a metastatic gastrinoma. Liver metastases are indicated by arrowheads. Panel B illustrates an endoscopic ultrasound image of a pancreatic body insulinoma confirmed at subsequent surgery. The tumor is indicated by the black arrowheads.
Endoscopic ultrasonography (EUS)
While standard upper endoscopy is occasionally of value in identifying PETs which arise within the luminal GI tract (gastrinomasm somatostatinomas), EUS with fine needle aspiration (FNA) has become part of the standard armamentarium for evaluating pancreatic masses114-118. EUS/FNA is useful to distinguish PETs (especially NF-PETs) from adenocarcinomas and also to localize tumors not imaged with conventional studies117-120. EUS/FNA is reported to have a diagnostic accuracy of 80% for pancreatic adenocarcinoma and 46% for PETs117. FNA is rarely needed with functional PETs (especially insulinomas/gastrinomas) because the diagnosis is made by biochemical/functional testing. EUS is more effective at localizing intrapancreatic PETs than extrapancreatic PETs such as duodenal gastrinomas117, 121. EUS plays an especially important role in localizing primary insulinomas because they are pancreatic, commonly small (<1 cm), frequently missed by conventional studies, and are frequently (>70%) negative on somatostatin receptor scanning (SRS, see below), because of low density or lack of somatostatin receptor subtypes that bind radiolabeled octreotide analogues with high affinity106, 122-124. EUS is able to identify intrapancreatic primary PETs in approximately 90% of cases. Figure 2 (Bottom) shows an EUS image of an insulinoma located in the body of the pancreas.
EUS is playing an increasingly important role in patients with MEN113, 125-128. MEN1 patients have NF-PETs in 80-100% of cases histologically, although often they are small (<0.5 cm)13, 125-128. EUS is able to detect PETS in MEN1 patients not seen on either SRS or conventional studies, especially in the size range from 0.4-1.1 cm, with the result that 55-100% of asymptomatic patients had NF-PETs identified126, 129. The management of these small asymptomatic NF-PETs is controversial because their natural history is largely unknown13. However, because EUS has been shown to have excellent specificity and reproducibility for small NF-PETs (<10 mm), it has been proposed that serial EUS studies could be used to monitor growth and determine when intervention should be considered125-127, 130.
Similarly in patients with VHL, PETs develop in 10-17% and they are almost invariably NF-PETs13, 131-135. Their management is also controversial because these patients are almost invariably asymptomatic, especially if the PET is small (<2 cm). In various studies because no patient with a NF-PET <3 cm had hepatic metastases, it has been recommended that PETs <3 cm not be routinely resected135-137. EUS is the most accurate method to assess PET size in these patients and could be used for serial studies similar to that proposed above in MEN1.
Angiography and selective hormone sampling
Prior to the development of functional imaging studies (see below), angiography and sampling for hormone gradients were widely used and extremely helpful in patients with PETs138-141. Originally, selective sampling for hormonal gradients was performed by portal-venous-sampling (PVS) 139, 142. This method was largely replaced by selective-arterial injection of secretin (gastrinomas) or calcium (other functional PETs) with assessment of hepatic venous hormone concentrations, because it can be performed at the time of angiography, has less complications and requires less expertise, but is similarly sensitive to PVS139, 140, 143, 144. This approach can also be utilized to identify liver metastases after selective hepatic artery cannulation141. In recent years, with advancement in other functional tumor localization methods, the utilization of these invasive localization techniques has declined. The three remaining areas in which these studies are still used are: 1) for localizing insulinomas following a negative octreoscan/EUS, 2) for preoperative evaluation of the liver prior to debulking surgery and 3) for localizing a functional PET in MEN1 patients with multiple lesions140, 145. Numerous studies have shown that intra-arterial injection of calcium with hepatic venous insulin sampling is a sensitive method of localizing insulinomas, even in imaging negative cases, being positive in 88-100 %139, 140, 145-149
Functional Imaging (SRS and positron-emission tomography)
Most PETs demonstrate high densities of sst2 or sst5 receptors, two of the 5 somatostatin receptor subtypes (designated sst1-sst5) which have high affinity for the SS analogues: octreotide and lanreotide150-153. Radiolabeled forms of these synthetic SS analogues with high affinity for sst2/sst5 receptors have proved sensitive and useful for localizing both the primary PET as well as metastases104, 151, 154, 155. [111In-DPTA-DPhe1]-octreotide is approved in the United States. Somatostatin receptor scanning (SRS or octreoscanning) identifies 50-70% of primary PETs but < 25% of insulinomas (which have absent or lower sst2/5 densities)104, 122-124, 151, 154, 155. In one prospective study, SRS was as sensitive as all conventional studies and angiography combined155. SRS is particularly useful for demonstrating liver metastases with the best sensitivity of any imaging modality (almost 90%)104, 155-157. The imaging results shown in Figure 3 in two patients with ZES demonstrate the greater sensitivity of SRS than conventional studies in localizing both the primary as well as metastatic disease to the liver/lymph nodes. SRS allows whole body scanning and it is therefore also useful to identify tumors beyond the liver (e.g., lungs/bone)34, 154, 158. To achieve high sensitivity it is essential that single photon emission tomography (SPECT imaging) be used to isolate possible lesions from the renal background106, 151, 159. Studies have shown that SRS changes the management in 24-47% of patients with PETs160-162. Although SRS has high specificity it is important to remember that a number of normal and abnormal tissues express increased densities of sst2/5 receptors that can result in false-positive scans. False positives can occur particularly with thyroid disease, breast disease, lymphoma, cholangiocarcinoma, hemangiomas, sites of inflammation and granulomatous disease151, 153, 161. In one prospective study 161, 12% of SRSs were false-positive for a PET, however when results were interpreted in the clinical context, the false positive rate was only 3%. Detection of PETs by SRS is also size-dependent with appropriately 50% of gastrinomas <1 cm in diameter not detected163. Therefore, there is a need for even more sensitive imaging methods154, 163.
Figure 3.

Comparison of conventional imaging (CT, MRI) and SRS to localize a primary gastrinoma (left) or metastatic disease (right) in two patients with ZES. In the left panel the patient had negative preoperative conventional imaging studies (CT, MRI) and angiography, but SRS showed a lesion in the pancreatic head area. At surgery a 2 cm tumor was resected and the patient has remained disease-free. In the right panel neither the MRI nor CT showed recurrent disease in this patient post resection of a gastrinoma, however the fasting gastrin was elevated and the SRS showed extensive metastases in lymph nodes and the liver. Both of these results show the greater sensitivity of SRS for localizing primary PETs as well as metastatic disease.
Positron-emission tomographic scanning is receiving increasing attention for PET localization106, 164. Standard substrates such as 18F-deoxyglucose (18FDG) are not useful for most PETs because of their slow glucose turnover and are only useful for the small subset with high proliferative rates and low differentiation106. 11C-5-hydroxytryptophan or 68Gallium-labeled SS analogues have greater sensitivity than SRS or conventional studies106, 164-166 and therefore may prove to be clinically useful in the future. Particularly important for the increased use of position-emission tomographic scanning in PET patients is the ability to make 68Gallium using a generator, similar to what is now used for 99mTC in most nuclear medicine departments, rather than requiring a cyclotron as is the case for these other isotopes106. In a recent study165 involving 84 patients with various GI NETs (carcinoids, 23 PETs), positron-emission tomographic scanning using 68Gallium-DOTA-Tyr3-octreotide had a sensitivity of 97% compared to 55% for SRS and a greater accuracy (96% vs. 58%, p<0.01) with equal specificity for the two techniques. One particular benefit of this scanning is the potential for image fusing (i.e., overlaying CT with PET images). It is likely that such scanning will play an increasing important role in the future for imaging PETs. Figure 4 demonstrates the increased sensitivity of positron-emission tomographic scanning with 11C-5-HTP for detecting liver metastases compared to CT scanning in a patient with a malignant PET.
Figure 4.
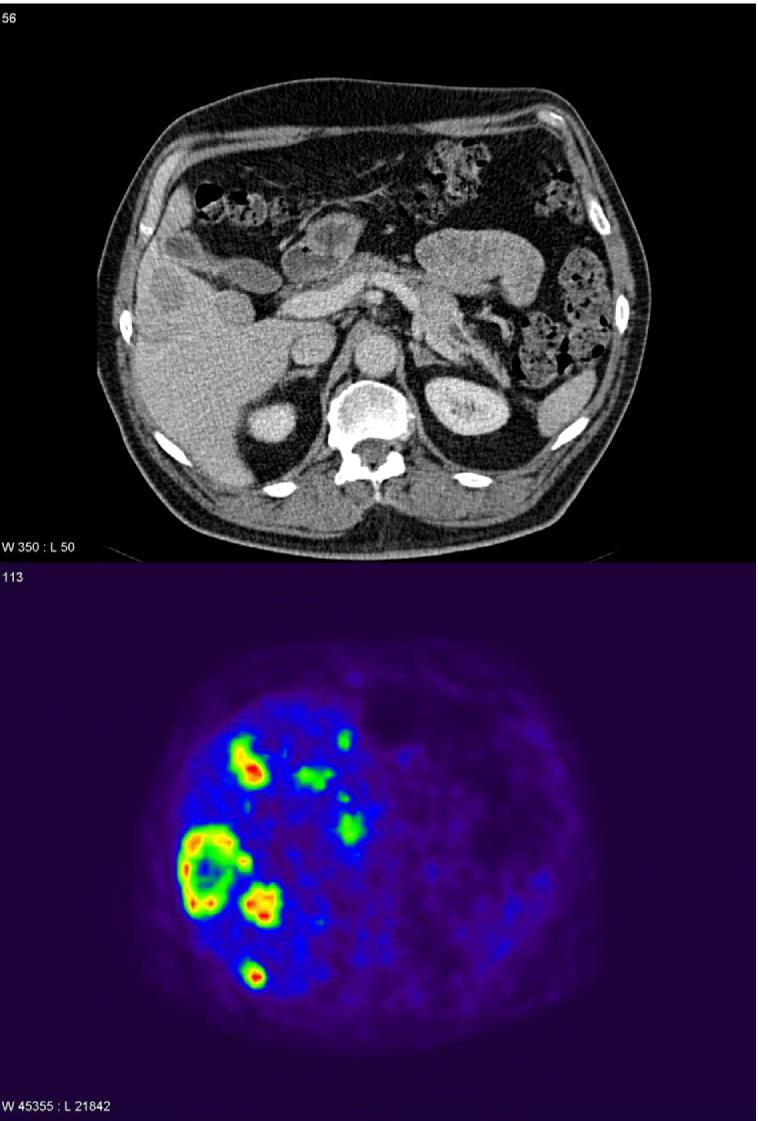
Comparison of the extent of liver metastases in a patient with a malignant PET on CT scanning (top panel) and positron emission tomographic scanning (bottom panel). This patient with a malignant PET had a few liver metastases seen on CT scanning (top) and SRS (not shown) but much more extensive disease on positron emission tomographic scanning with 11C-5-HTP demonstrating its greater sensitivity. (Images kindly provided by Prof. Anders Sundin, Department of Radiology, Uppsala University Hospital, Uppsala, Sweden).
Medical Management of the Hormonal Excess-State
Gastrinoma-medical management
In ZES acid hypersecretion is the most important clinical effect45, 62, 167, 168. Because of their potency and long-duration of action, proton pump inhibitors (PPIs) are the agents of choice for management31, 45, 53, 167, 169, 170. Histamine H2 receptor antagonists or SS analogs are effective, but the former drug class is limited by the need for frequent, high-dose administration167, 169, whereas the latter class is limited by the need for parenteral therapy.
Once or twice daily oral PPIs (i.e., omeprazole (40 mg), lansoprazole (30 mg), rabeprazole (20 mg), pantoprazole (40 mg) or esomeprazolea (40 mg) are effective in virtually all ZES patients167, 169, 171-175. It is important to document control of acid output (i.e., <10mEq/hr in the last hour before the next dose of drug [intact stomachs] or < 5 mEq/hr [prior gastric resections] in patients with uncomplicated disease (i.e., no MEN1, mild GERD, and no prior Billroth 2 resection) rather than to titrate drug dosages to symptoms, since asymptomatic individuals may still have uncontrolled acid hypersecretion45, 167, 176. Patients with complicated disease (i.e., MEN1, moderate-severe GERD, Billroth 2 resection) often need higher doses and are usually best treated with at least BID dosing177-179. It is recommended that patients with uncomplicated disease be initially started on 40-60 mg of omeprazole (or equivalent) to adequately control acid output acutely180, however with time the dosage can be decreased in up to 60% of the patients179. Long-term follow-up of patients receiving PPI's demonstrates no tachyphylaxis and an excellent safety profile170-172, 181, although drug-induced achlorhydria may lead to substrate deficiencies (vitamin B12 is more of a concern than iron)181, 182. Even though in animal studies long-term high dose PPI treatment can lead to the development of gastric carcinoids, there is no evidence of an increased rate of their development with chronic PPI treatment in ZES patients167, 183-185. Almost every ZES patient demonstrates some degree of ECL hyperplasia183, 185-187 which is more severe in MEN1 patients7, 183, 186. Patients with MEN1/ZES develop gastric carcinoids in 23-33 % of cases183, 185, 186 however the rate in patients with sporadic ZES is <1% 183, 185-187 and there is no evidence that PPIs alter this rate in either group.
Intermittent intravenous PPI treatment (with pantoprazole (80 mg), lansoprazole (60 mg) or esomeprazole (80 mg) given two or three times daily effectively substitutes for oral therapy for brief periods in patients who cannot take oral drug188, 189. Three times daily therapy is generally recommended as this more frequent administration precludes the requirement to document effective control of acid in situations when the patients may be quite ill. There is no longer a role for gastric surgery to reduce acid output in ZES patients.
Insulinoma-medical management
Most patients (>85%) have a single small benign insulinoma76, 77, 190, except for those with MEN1 where multiple tumors frequently occur13, and therefore they are treated surgically soon after diagnosis with an excellent cure-rate76, 77, 190. However, prior to surgery and for the 5-15% (Table 1) with malignant disease, treatment for the hypoglycemia is needed. In addition to frequent small feedings the initial drug generally used is diazoxide (200-600 mg/day in divided doses), a benzothiadiazide, which directly inhibits insulin release and causes adrenergic stimulation promoting glycogenolysis9, 76. Diazoxide controls hypoglycemia in 50-60% of patients and has been used effectively for >20 years76, 77, 190, 191. Diazoxide frequently results in sodium/fluid retention requiring diuretics, as well as nausea and occasional hirsutism76, 77, 190, 191. Long-acting SS analogues (octreotide, lanreotide) are effective in 35-50% of patients with insulinomas, however they need to be used with care, because in some cases they worsen the hypoglycemia, presumably by inhibiting counter-regulatory mechanisms123, 153, 190. Therapy with other agents such as verapamil, propanolol or phenytoin has also been described though these agents are generally not first-line choices.
Other functional PET tumor syndromes-medical management
Until the availability of octreotide (see below), specific therapy for PETs included blood transfusions; insulin, zinc and amino acid transfusions for glucagonomas; replacement of volume losses and correction of acid-base disturbances for VIPomas; nutritional repletion and insulin administration for the somatostatinoma syndrome; and administration of adrenolytic agents (such as ketoconazole, aminoglutethimide, metyrapone or orthopara-DDD) or adrenalectomy for ectopic ACTH-producing tumors. However, octreotide availability has largely supplanted the need for many of these approaches.
Somatostatin (SS) is a widely distributed 14-amino acid cyclic paracrine peptide which exerts multiple inhibitory effects on secretory and motor functions150, 153. Its effects are mediated by binding to one of 5-receptor subtypes designated sst1-sst5, which are all G protein-coupled receptors150. SS has a short serum half life of about 2 minutes precluding its use clinically, but its synthetic analog, octreotide, with a serum half life of at least 1 hour has been used successfully to inhibit secretion from a variety of cell types including PETs, which usually exhibit high sst2 receptor densities153, 192, 193.
Octreotide is approved for use in patients with acromegaly, VIPomas and the carcinoid syndrome, but it is also useful off label to lower portal pressure in patients with bleeding from esophageal varices due to portal hypertension, to control diarrhea in patients with AIDS enteropathy and short bowel syndrome, and to control hormonal syndromes in patients with other NETs153. Octreotide is usually prescribed at doses ranging from 100-500 ug three times daily by subcutaneous injection initially but this form of administration can then be overlapped with once monthly depot injections of an even longer-acting formulation, octreotide LAR at doses of up to 30 mg/month7, 8, 194. Lanreotide sustained-release or autogel is another depot somatostatin analog available in Europe195.
In VIPomas, octreotide reduces serum VIP levels in >80% of patients and improves diarrhea in >75% but the response is often short-lived (<1 year) without dose increases. In glucagonomas, octreotide decreases plasma glucagon levels in >80% and improves MNE in 90% (with complete resolution in 30%). There are anecdotal reports of efficacy of octreotide in somatostatinoma syndrome as well as therapy for GRFomas7-9, 153. Octreotide therapy is not recommended for hormonal control of gastrinoma. Octreotide should be used with care in patients with insulinomas (as discussed above). The mean duration of octreotide treatment in studies is one year and frequently tachyphylaxis develops which may be overcome with higher doses8
Adverse effects of SS analogs are generally mild and include diarrhea/steatorrhea, flatulence, fluid retention, nausea, gallstones and glucose intolerance. Such side-effects are reported in 50% of patients treated with octreotide, but have rarely been serious enough to stop treatment153. In long-term treatment of patients with acromegaly only 5% developed side-effects severe enough to stop treatment194, 196. During long-term treatment concern has been raised about the possibility of an increased rate of gallstone evelopment. This has been particularly well-studied in patients with acromegaly with a mean incidence of 29%, however only 1% develop symptomatic gallbladder disease194.
Surgical therapy for cure
Surgery is the only treatment-modality with the potential to cure patients with PETs. However, surgery is only likely to be effective in patients without diffuse metastatic disease who are able to tolerate the intervention and, in the case of ZES specifically, only in those with sporadic disease13, 113, 197-199. Negative preoperative localization should not be considered a contraindication to surgery in patients with proven functional PETs as an experienced PET surgeon will very frequently localize the tumor (>95% of insulinomas or gastrinomas)76, 113, 198, 200. On the other hand, preoperative identification of diffuse disease beyond regional lymph nodes precludes attempts at curative surgery, though many authorities favor debulking surgery in cases where ≥90% of identifiable disease is thought resectable (see below). In the 5-15% of patients with limited hepatic metastases, many authorities attempt resection because this approach may result in extended disease free-survival in selected patients46, 201-204. Patients with MEN1 develop potentially curable PETs of various types (insulinomas, VIPomas, somatostatinomas, glucagonomas, GRFomas)13, 205-210, however both the NF-PETs and gastrinomas are invariably multiple arising throughout the pancreas or the proximal duodenum30, 127, 211, 212. At present, most authorities do not recommend subjecting patients with MEN1/ZES to a Whipple's resection or patients with multiple NF-PETs to total pancreatectomy, because these operations are extensive, the long-term consequences are unclear, post-operative morbidity can be significant and the long-term prognosis of these patients without such treatment remains excellent121, 127, 198, 206, 209, 213. In MEN1 patients the surgical treatment of NF-PETs (80-100% of patients) and gastrinomas (40-60% of patients), remains controversial because of multiplicity of primary tumors and failure of enucleation to result in cure121, 127, 198, 206, 209, 213. Potential approaches in these patients include not performing routine surgery, performing surgery with aggressive removal of all larger PETs or only operating in patients with imageable tumors >2cm121, 127, 198, 206, 209, 213, 214. This latter approach stems from a number of studies which demonstrated that patients with MEN1 and NF-PETs or gastrinomas <2cm in diameter have an excellent prognosis (survival equal to patients without PETs or 100% at 15 years) and they rarely develop advanced disease127, 197, 198, 206, 215.
In advance of surgery patients should be vaccinated against encapsulated microorganisms (pneumococcus, H. influenza, meningococcus) in anticipation of a possible splenectomy and they should receive a bowel preparation in anticipation of an expected enterotomy (mandatory in the case of gastrinomas and other hormonal syndromes with a predilection for duodenal primaries)198, 216-219. In general, all PETs (except imaged insulinomas) should be approached by laparotomy to permit an extensive exploration of the entire abdomen113, 203, 219-221. An exception to this rule is surgery for insulinoma in non-MEN1 individuals, because at least 85% of these tumors are benign, there usually is a single primary and if they can be localized preoperatively, laparoscopic resection is successful in 70-100% of cases and its use hastens postoperative recovery121, 222-224. It is also important to examine the entire pancreas which requires complete mobilization of the duodenum and exposure of the pancreatic tail32, 198, 216-219. Surgical exploration is assisted by intraoperative ultrasonography using appropriate transducers for evaluation of the liver (5 MHz) and pancreas (7.5-10MHz). Intraoperative endoscopic transillumination plus duodenotomy is required for tumors with a predilection for the duodenum (GRFomas, somatostatinomas, and especially gastrinomas), because they are frequently small (<0.5 cm), not detected by ultrasound or palapation and are primarily localized in the 1st and 2nd part of the duodenum113, 198, 216-220, 225-227. Some authorities favor intraoperative hormonal localization as well228.
The aims of surgical resection for cure are to remove the primary tumor and regional lymph nodes (if affected) with minimal disruption to the underlying anatomy. Enucleation is advised for insulinomas because they are generally benign as well as for localized tumors of the pancreatic head. Duodenal tumors are generally resected unless small and then may be removed endoscopically in some cases, conversely if they are large they may require a duodenectomy30, 229. Tumors in the pancreatic tail are generally resected (with splenic preservation if possible) as opposed to enucleated unless they are insulinomas113, 198, 216-220. MEN1 patients who come to surgery should have a careful exploration of the entire pancreas with enucleation or resection of all dominant masses, realizing that the largest lesion identified may not necessarily be the lesion causing the functional syndrome. In general, blind pancreatectomy in the rare case of no identifiable tumor after a careful exploration of the entire abdomen is not felt to be an acceptable approach.
In appropriate hands, cure rates for insulinomas approach 100%76, 230. For sporadic gastrinomas the figure is 60% immediately postoperatively and 30-40% at five years198, 216. In general, cure rates for other PETs are lower because they are generally larger at presentation, often with metastases. Surgical resection of the primary PET should be attempted whenever possible if the patient does not have another medical problem limiting life-expectancy, substantially increasing surgical risk or diffuse metastatic disease, because studies in patients with ZES show resection of the primary both decreases the rate of development of liver metastases and extends survival by preventing the development of progressive disease50, 51.
Treatment of metastatic disease
General Treatment of metastatic disease
In recent studies the long term outcome in patients with PETs is increasingly dependent on tumor growth. However, even with widespread liver metastases many patients remain relatively well with slow progression, especially early on in the disease process, such that many authorities advocate delaying the introduction of disease modifying agents until there is clear development of enlarging tumor burden or symptoms develop. Furthermore, standard antitumor therapies are not curative and frequently have limited efficacies.
Biotherapy
1. Octreotide/Interferon
Biotherapy with long-acting somatostatin (SS) analogs [octreotide LAR or lanreotide SR (autogel)] is frequently instituted first in patients with enlarging tumor burdens, especially patients with slow-growing tumors without extensive (<50%) liver involvement8, 231. This approach is commonly used even though the results are controversial and there are no studies that have clearly demonstrated it prolongs survival due to inhibition of tumor-related growth103, 232. SS analogues are frequently used first because these agents are well-tolerated and numerous studies suggest they have a tumoristatic effect, causing a decrease or cessation of growth in 30-80% of cases, without tumor regression in most cases (<15%) that demonstrated growth prior to treatment103, 206, 232-235. It is presumed this tumoristatic effect will result in improved survival, but at present this remains unproven. The tumoristatic effect can be prolonged (>2 years) and is more frequently seen in slow-growing tumors with a low proliferative index; therefore some recommend that rapidly growing tumors or tumors with high proliferative indices be treated with other modalities8, 206, 231, 233, 236. The exact mechanism of SS analogue action in PETs is not completely clear, however, they induce apoptosis and in various cells activate phosphatases, suppress release of growth factors, inhibit IGF-1 signaling, have immuno-modulatory effects and inhibit angiogenesis150
Interferon therapy (human leukocyte/alpha-interferon) is also frequently used for the treatment of metastatic disease but, as with octreotide, its major effect is tumor growth stablization rather than inducing regression (<20% of cases)8, 103, 232, 234. Similar to SS analogues it is hoped that this tumoristatic effect will result in improved survival, but at present this is also unproven232. The mechanism of interferon's anti-proliferative effect in PETs is not completely known, however it increases tumor expression of bcl-2 resulting in decreased cell proliferation and in other cells inhibits protein and hormone synthesis and angiogenesis and stimulates the immune system8. Unfortunately, interferon therapy causes frequent side-effects including flu-like symptoms (which may improve with prolonged therapy), fatigue, weight loss, lipid, thyroid and liver enzyme abnormalities and cytopenias including leucopenia which may persist and interfere with the acceptability of long-term treatment232, 233.
Since both interferon and octreotide therapy are tumoristatic by different mechanisms, combination therapy was felt to have promise. Non-randomized studies were suggestive of additive effects232, 237, but a recent prospective study238 showed no additivity, however a number of reservations have been raised about this study, primarily methodological issues239.
2. Peptide receptor radionuclide therapy(PRRT)
PRRT utilizes the fact that PETs almost uniformly overexpress SS receptors and internalize radiolabeled SS agonist analogues thereby facilitating the delivery of cytotoxic doses of localized radiation to the PET153, 233, 240-244. Three different radiolabeled SS analogues have been developed and investigated in patients with malignant NETs including analogues labeled with 111Indium (emits conversion and auger electrons, γ-rays), 90Yttrium (strongly emits β-particles) and 177Lutetium (emits β-particles and γ-rays)240-244. The effect of 111In-DPTA-octreotide was examined in two studies240, 245 including 52 patients with malignant progressive NETs and complete tumor regression were seen in 0%, partial regression in 0-8% and tumor stabilization in 42-81%. [90Y-DOTA,Tyr3]-octreotide, [90Y-DOTA]lanreotide or [90Y-DOTA-,Tyr3]octreotate were examined in 7 studies involving >280 patients with malignant NETs and complete tumor responses occurred in 0-3%, partial responses in 6-37% % and stabilization in 44-88%240, 245. In one study a longer survival was reported in patients treated with [90Y-DOTA-,Tyr3]octreotate than those previously treated with 111In-DPTA-octreotide (mean 37 mos vs. 12.5 mos)240, 246. One study reported results with 129 patients with malignant NETs treated with [177Lu-DOTA,Tyr3]octreotate and found a complete tumor response in 2%, a partial response in 32% and stabilization in 34% 240, 247. To date, no controlled studies have demonstrated that PRRT extends survival. In general PRRT with the different isotopes has been safe with severe side-effects uncommon240, 244-246. Approximately 30% of the patients develop acute side-effects (nausea, pain, vomiting) that are usually mild, can be controlled with symptomatic therapy and do not interfere with continued treatment240. More severe side-effects include hematological toxicity (15%-usually transient, 0.3% develop myelodysplastic syndrome) and renal toxicity (which occurs almost entirely in patients given 90Y-labeled SS analogues and can be limited by co-administration with amino acids)240, 244, 245. Although not yet approved for use in any country, the promising results described above have led to PRRT undergoing evaluation in a number of centers in the world to clearly establish its exact utility.
Liver-directed therapy (embolization, chemoembolization)
Most malignant PETs metastasize to the liver where they derive their blood supply from hepatic artery branches (75-80%), in contrast to native liver tissue, which derives the majority of its blood supply from the portal vein9, 248, 249. Recent studies demonstrate that liver metastases demonstrate rapid growth in <50% of patients and up to 30% demonstrate no growth on follow-up33, 37, 250. Consequently, the usual approach to palliative therapy for liver metastases is to delay therapy until symptoms supervene due to the metastases per se, the tumor shows rapid growth, or the patient develops refractory symptoms from a functional PET.
Selective deprivation of blood supply to metastases for the palliative management of metastatic disease can be achieved by surgical ligation, but interventional radiological approaches via intra-arterial catheterization of the iliac/brachial arteries without (hepatic artery embolization [HAE]) or with co-administration of chemotherapeutic agents (HACE) permits a similar result9, 248, 249, 251. Absolute contra-indications to HAE/HACE are portal venous thrombosis, liver failure and biliary reconstruction (Whipple resection), whereas relative contra-indications are hepatic tumor loads >50%, contrast allergy, extensive extrahepatic disease and poor performance status249, 252. There are no randomized studies comparing embolization alone (HAE) to those with embolization combined with chemotherapeutic agents (HACE) such as 5-fluorouracil, cisplatin, mitomycin C or streptozotocin.
The usual approach to HAE/HACE is sequential catheterization of peripheral radicals of the hepatic artery in one liver lobe followed by repeated administration of therapy on the other side about 6-8 weeks later248, 249, 253. In various studies 55-100% of patients with malignant NETs treated by HAE/HACE have symptomatic improvement and 20-80% an objective response with tumor shrinkage9, 248, 249, 251, 253-256. The mean duration of response is 6-42 months248, 254-256. A lower response rate has been reported in patients with >75% of the liver involved and in patients with an intact primary tumor or extrahepatic metastases254.
HAE/HACE is not without side-effects with an overall mortality of <3%, but pain develops in 50-100%, nausea and vomiting in 50-90% and fever/leukocytosis in 30-60%. In 5-15% of patients serious side-effects can occur including hepatic failure, bleeding, gallbladder necrosis, hepatic abscess formation and renal failure9, 248, 253.
At present there is no uniform agreement on when HAE/HACE should be used in patients with malignant PETs. In patients with functional PETs not responding to other therapies or malignant PETs with diffuse hepatic metastases only which are increasing in size or causing local symptoms due to tumor bulk, this procedure may be considered and may be quite helpful in controlling symptoms248, 251, 254.
Surgical debulking (cytoreductive surgery)/radiofrequency ablation of hepatic metastases(RFA)
The role of cytoreductive surgery in patients with malignant PETs with incompletely resectable metastatic disease is controversial. Whereas numerous studies show surgery may help control symptoms in patients with advance metastatic functional PETs and likely prolong life expectancy in patients with malignant PETs, in most studies the patient groups are not strictly comparable and no randomized studies have examined this approach9, 46, 201, 257-261. In an analysis of 63 patients with malignant PETs from five different surgical series who underwent surgical resection, the operative mortality averaged 6%, symptom control was achieved in 85% and 5-year survival was 60-80% 257. The authors of this review, as well as those in a number of other surgical series, concluded that surgical resection should be attempted in patients with malignant PETs whenever it is determined that at least 90% of the visible tumor could likely be removed201, 202, 255, 257, 259-262. In one recent 255 retrospective comparison of results with cytoreduction or embolization in 120 patients with malignant NETs (33-PETs, 87-carcinoids), patients undergoing cytoreductive surgery had longer survival and greater reduction in symptoms.
RFA is being increasing used in patients with PETs with hepatic metastases either alone or in combination with other treatments248, 263-266. RFA can be performed at the time of surgery for isolated hepatic metastases or laparoscopically248, 264, 265. Factor limiting its application include tumor size (usually used in tumors <3.5 cm) and number (usually used in cases with<5 lesions)248, 264, 265. RFA morbidity is low (<15%), although occasional cases of hemorrhage or abscess formation occur. Response rates from 80-95% are reported and responses have lasted up to 3 years248, 264-266. Although RFA has not been shown to extend life, its ability to control local metastases with low morbidity has led to it being increasingly used for the treatment of limited small metastases and it may be particularly helpful for patients with limited metastases from a functional PET, especially at the time of surgery232, 263, 266.
Chemotherapy
Traditional chemotherapeutic approaches
If biotherapy fails or the PET is rapidly growing or poorly-differentiated, chemotherapy is frequently employed249, 267, 268. A large number of regimens have been utilized in patients with metastatic PETs with some success, in contrast to carcinoid tumors, where they have been generally unsuccessful249. Streptozotocin was the first agent shown to have significant benefit in a prospective study as monotherapy for malignant PETs269. However, this approach provided limited benefit with significant renal/hematological toxicity269. Combination therapy with streptozotocin and 5-fluorouracil or doxorubicin was subsequently employed to permit lower doses of streptozotocin to potentially limit side effects without sacrificing efficacy. In the 1992 Eastern Cooperative Oncology Group (ECOG) study of 105 patients who received one of three regimens (streptozotocin-doxorubicin - response rate [RR] 70%, streptozotocin-5-fluorouracil - RR 45% and chlorozotocin monotherapy - RR 30%), the streptozotocin-doxorubicin regimen was shown to improve overall survival with a mean duration of response of 18 months270. Later studies utilizing only imaging assessments and better imaging modalities, have not found this degree of success. In later studies utilizing streptozotocin in various combinations with 5-fluorouracil and doxorubicin, the overall survival was either not impacted at all or only minimally impacted, the response rate was 6-40% with no complete responses and the median response was short (9-18 mos)249, 268, 271, 272. Particularly poor RRs were seen in patients with replacement of >75% of the liver by tumor or in those who had previous received chemotherapy268. Streptozotocin is associated with significant side-effects with 74-100% of patients developing nausea/vomiting, and 20-40% with long-term treatment developing renal toxicity249, 268-270.Studies utilizing other chemotherapeutic agents including etoposide, DTIC (Dacarbazine) and cisplatin or carboplatin alone or in combination have in general also been rather disappointing9, 249, 267. In poorly-differentiated PETs chemotherapy with cisplatin, ectoposide or its derivatives is the recommended treatment with RRs of 40-70% reported, however the RRs are relatively short249, 273-275.
Angiogenesis inhibitors and other new, novel approaches
GI NETs frequently produce multiple growth factors including vascular endothelial-growth factor (VEGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF-1), basic fibroblast growth factor (bFGF), and transforming growth factor (TGF) as well as expressing receptors for these (VEGFR, PDGFR, IGF-1R) and other growth factors(epidermal growth factor receptor [EGFR])276-280. A number of new, novel therapies are now available that are directed at these growth factors or their receptors and are being investigated in GI NETs including a monoclonal antibody to VEGF (bevacizumab) as well as small-molecule inhibitors of the intracellular tyrosine kinase domain of VEGFR or other growth factor receptors (sunitinib [SU11248], sorafenib, vatalanib, imatinib (gleevac), gefitinib)280-284(Fig. 5). In one study reported in abstract form285 sunitinib, was evaluated in a phase II study of 61 patients with PETs. The treatment was well tolerated and a response occurred in 13%, tumor stabilization in 68% and the median time to tumor progression was 33 weeks. In another phase II trial286 of gefitinib, a tyrosine kinase inhibitor targeting EGFR, in 31 PET patients a tumor response of only 6% was noted. Other novel approaches to the management of metastatic PETs have focused on targeting downstream targets of tyrosine kinase receptor activation (Fig 5). For example, mammalian target of rapamycin (mTor) is a threonine kinase that is involved in the regulation of cell cycle progression and its inhibition has showed promising anti-tumor activity in a number of neoplasms280-282, 287. However, temsirolimus, an mTor inhibitor, when evaluated in a phase II trial of 15 patients with PETs showed a low response rate of 7% 287. Another mTor inhibitor, everolimus (RAD001) yielded a response rate of 15% when administered in combination with octreotide LAR in 13 patients with PETs280-282.
Figure 5.

Schematic diagram of a theoretical pancreatic endocrine tumor cell, smooth muscle cell (pericyte) or endothelial cell demonstrating the sites and mechanism of action of novel agents for the management of metastatic PETs. These cellular components of PETs all exhibit surface growth factor receptors (e.g., VEGFR, PDGFR, IGF-1R, c-KITR, etc) which when occupied by their respective growth factors (in an autocrine or paracrine manner) lead to autophosphorylation of the intracellular tyrosine kinase component of the receptor. Tyrosine kinase phosphorylation activates the PI3K-AKT-mTOR pathway (amongst others) ultimately promoting protein synthesis, cell cycle progression and cell survival which causes increased cellular proliferation, inhibition of apoptosis, cellular invasion, metastasis and tumor angiogenesis. This pathway can be inhibitied by monoclonal antibodies to growth factor receptors, tyrosine kinase inhibitors with specific activity against various growth factor receptors, or downstream mTOR inhibitors. Whilst mTOR inhibitors are active against both the tumor directly as well as its blood supply, tyrosine kinase inhibitors or antibodies directed against specific growth factors may predominantly effect the tumor itself or secondarily inhibit tumor cell growth by altering its blood supply304-308.
Although response rates in these initial studies are low, these agents represent new approaches to treatment. It is hoped that these novel antitumor agents may play a future role alone or in combination with other agents in the management of patients with metastatic PETs.
Palliative radiotherapy
NET cells are sensitive to standard external beam irradiation. Unfortunately, liver tissue has similar sensitivity such that the therapeutic index for radiation of liver metastases is prohibitive. On the other hand, palliative radiation to bone metastases in the spine and even brain metastases has been shown to be effective288, 289. Proton-beam radiation holds promise for effective palliation of many different types of cancers. To date, no information is available regarding the use of this potentially promising modality in NET patients.
Liver transplantation
In contrast to most other neoplasms, liver transplantation continues to be used for selected patients with metastatic PETs9, 290-293. Conclusions about its potential value or guidelines regarding which patients would most benefit are difficult because the available literature comprises <150 patients with malignant PETs treated with liver transplantation, the individual series are small (largest single center-19 cases) and long-term follow-up data are limited290. In a recent report involving 15 patients with malignant GI NETs (11-PETs) the 5-year disease-free survival was 20% and total survival 90%, which is in contrast, to the results of a review 293 of 103 patients from multiple small series with NETs (including 48 PETs), which demonstrated 2-and 5-year total survival rates of 60% and 47%. Younger patients (<50 years old), patients without extensive other surgical procedures (cluster operations) and with disease limited to the liver, appeared to fare best290, 293. Recent reviews suggest that liver transplantation should be considered in selected young patient with metastases limited to the liver and a previously resected primary PET who require relief from incapacitating hormonal or tumor symptoms290, 291, 293.
Future directions and unsettled problems
Even though there have been many advances in recent years in the diagnosis/management of PETs, it is not clear that survival in patients with advanced disease has improved. In fact in a recent review 294 of survival for all gastrointestinal NETs (both carcinoids and PETs), no change in survival was reported over a 30 year period. Numerous factors contribute to this including their continued delay in diagnosis (mean-4-6 years), the lack of general availability to most patients of the expertise and experience necessary to diagnose and manage them, the lack of good prognostic factors to stage disease extent and tailor treatment accordingly, and the lack of controlled trials, new treatments and a standardized approach to care so that approaches can be compared in different centers. These problems arise not only because PETs are uncommon, but also because large gaps in our knowledge remain regarding their molecular pathogenesis and there are no widely accepted animal models or PET cell lines that can be used to evaluate innovative treatments. Furthermore, it is difficult for young physicians who may want to acquire the necessary expertise to treat patients with PETs because of a paucity of well-rounded centers that have expertise in all facets of these tumors. Furthermore, comparison of results from study to study is difficult, because of a lack of uniformity in the United States in the pathological classification of these tumors or standardization of the minimum criteria for histological diagnosis. A number of recent consensus conferences statements have been published by the European Neuroendocrine-tumor Network Society (ENETS)6, 7, 295 which attempt to begin to standardize the approach to diagnosis/management including, for the first time, a proposed TNM classification19, 296. In addition, the National Cancer Institute recently mandated a summit conference on GI NETs and it has been proposed in another recent consensus conference that centers of excellence should be established dealing with all aspects of the diagnosis, management, and basic /clinical research needs related to PETs294.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Duerr EM, Chung DC. Molecular genetics of neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab. 2007;21:1–14. doi: 10.1016/j.beem.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Corleto VD, Delle Fave G, Jensen RT. Molecular insights into gastrointestinal neuroendocrine tumors: importance and recent advances. Dig Liver Dis. 2002;34:668–680. doi: 10.1016/s1590-8658(02)80212-2. [DOI] [PubMed] [Google Scholar]
- Arnold R, editor. Endocrine Tumors of the Gastrointestinal Tract: Part 11. 2005. [Google Scholar]
- Arnold R, editor. Endocrine tumors of the Gastrointestinal Tract: Part 1. 2005. [Google Scholar]
- Oberg K, Eriksson BE. Neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab. 2007;21:1–172. doi: 10.1016/j.beem.2006.12.001. [DOI] [PubMed] [Google Scholar]
- de Herder WW, O'Toole D, Rindi G, et al. ENETS consensus guidelines for the management of patients with Digestive Neuroendocrine tumors Part 1-Stomach, Duodeneum and Pancreas. (84 ed.) 2006:151–216. doi: 10.1159/000098006. [DOI] [PubMed] [Google Scholar]
- Plockinger U, Rindi G, Arnold R, et al. Guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumours. A consensus statement on behalf of the European Neuroendocrine Tumour Society (ENETS) Neuroendocrinology. 2004;80:394–424. doi: 10.1159/000085237. [DOI] [PubMed] [Google Scholar]
- Oberg K, Eriksson B. Endocrine tumours of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753–781. doi: 10.1016/j.bpg.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Jensen RT. Endocrine Neoplasms of the Pancreas. In: Yamada T, Alpers DH, Kaplowitz N, Owyang C, Powell DW, Kalloo AN, editors. Textbook of Gastroenterology. Fifth ed. Blackwell; Oxford, England: 2008. in press. [Google Scholar]
- Ito T, Tanaka M, Sasano H, et al. Preliminary results of a Japanese nationwide survey of neuroendocrine gastrointestinal tumors. J Gastroenterol. 2007;42:497–500. doi: 10.1007/s00535-007-2056-6. [DOI] [PubMed] [Google Scholar]
- Panzuto F, Nasoni S, Falconi M, et al. Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer. 2005;12:1083–1092. doi: 10.1677/erc.1.01017. [DOI] [PubMed] [Google Scholar]
- Alexakis N, Connor S, Ghaneh P, et al. Hereditary pancreatic endocrine tumours. Pancreatology. 2004;4:417–435. doi: 10.1159/000079616. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Berna MJ, Bingham MD, et al. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management and controversies. Cancer. 2008 doi: 10.1002/cncr.23648. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppel G. Tumour biology and histopathology of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:15–31. doi: 10.1016/j.beem.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Brignardello E, Manti R, Papotti M, et al. Ectopic secretion of LH by an endocrine pancreatic tumor. J Endocrinol Invest. 2004;27:361–365. doi: 10.1007/BF03351063. [DOI] [PubMed] [Google Scholar]
- Samyn I, Fontaine C, Van Tussenbroek F, et al. Paraneoplastic syndromes in cancer: Case 1. Polycythemia as a result of ectopic erythropoietin production in metastatic pancreatic carcinoid tumor. J Clin Oncol. 2004;22:2240–2242. doi: 10.1200/JCO.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Kawano K, Ushijima K, Fujimoto T, et al. Peptide YY producing strumal carcinoid of the ovary as the cause of severe constipation with contralateral epithelial ovarian cancer. J Obstet Gynaecol Res. 2007;33:392–396. doi: 10.1111/j.1447-0756.2007.00544.x. [DOI] [PubMed] [Google Scholar]
- Kloppel G, Anlauf M. Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2005;19:507–517. doi: 10.1016/j.bpg.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395–401. doi: 10.1007/s00428-006-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindi G, Bordi C. Aetiology, molecular pathogenesis and genetics. Best Pract Res Clin Gastroenterol. 2005;19:519–534. doi: 10.1016/j.bpg.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-Type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Busygina V, Bale AE. Multiple Endocrine Neoplasia Type 1 (MEN1) as a Cancer Predisposition Syndrome: Clues into the Mechanisms of MEN1-related Carcinogenesis. Yale J Biol Med. 2006;79:105–114. [PMC free article] [PubMed] [Google Scholar]
- Goebel SU, Heppner C, Burns AD, et al. Geneotype/phenotype correlations of MEN1 gene mutations in sporadic gastrinoma. J Clin Endocrinol Metab. 2000;85:116–123. doi: 10.1210/jcem.85.1.6260. [DOI] [PubMed] [Google Scholar]
- Debelenko LV, Zhuang ZP, Emmert-Buck MR, et al. Allelic deletions on chromosome 11q13 in Multiple Endocrine Neoplasia Type-I-associated sporadic gastrinomas and pancreatic endocrine tumors. Cancer Res. 1997;57:2238–2243. [PubMed] [Google Scholar]
- Perren A, Komminoth P, Heitz PU. Molecular genetics of gastroenteropancreatic endocrine tumors. Ann N Y Acad Sci. 2004;1014:199–208. doi: 10.1196/annals.1294.021. [DOI] [PubMed] [Google Scholar]
- Capurso G, Lattimore S, Crnogorac-Jurcevic T, et al. Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr Relat Cancer. 2006;13:541–558. doi: 10.1677/erc.1.01153. [DOI] [PubMed] [Google Scholar]
- Dilley WG, Kalyanaraman S, Verma S, et al. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol Cancer. 2005;4:9. doi: 10.1186/1476-4598-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel DE, Rahman A, House M, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10:6152–6158. doi: 10.1158/1078-0432.CCR-04-0285. [DOI] [PubMed] [Google Scholar]
- Maitra A, Hansel DE, Argani P, et al. Global expression analysis of well-differentiated pancreatic endocrine neoplasms using oligonucleotide microarrays. Clin Cancer Res. 2003;9:5988–5995. [PubMed] [Google Scholar]
- Hoffmann KM, Furukawa M, Jensen RT. Duodenal neuroendocrine tumors: Classification, functional syndromes, diagnosis and medical treatment. Best Pract Res Clin Gastroenterol. 2005;19:675–697. doi: 10.1016/j.bpg.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Niederle B, Mitry E, et al. Gastrinoma (duodenal and pancreatic) Neuroendocrinology. 2006;84:173–182. doi: 10.1159/000098009. [DOI] [PubMed] [Google Scholar]
- Thom AK, Norton JA, Axiotis CA, et al. Location, incidence and malignant potential of duodenal gastrinomas. Surgery. 1991;110:1086–1093. [PubMed] [Google Scholar]
- Jensen RT. Natural history of digestive endocrine tumors. In: Mignon M, Colombel JF, editors. Recent advances in pathophysiology and management of inflammatory bowel diseases and digestive endocrine tumors. John Libbey Eurotext Publishing Co.; Paris, France: 1999. pp. 192–219. [Google Scholar]
- Gibril F, Doppman JL, Reynolds JC, et al. Bone metastases in patients with gastrinomas: a prospective study of bone scanning, somatostatin receptor scanning, and MRI in their detection, their frequency, location and effect of their detection on management. J Clin Oncol. 1998;16:1040–1053. doi: 10.1200/JCO.1998.16.3.1040. [DOI] [PubMed] [Google Scholar]
- Yu F, Venzon DJ, Serrano J, et al. Prospective study of the clinical course, prognostic factors and survival in patients with longstanding Zollinger-Ellison syndrome. J Clin Oncol. 1999;17:615–630. doi: 10.1200/JCO.1999.17.2.615. [DOI] [PubMed] [Google Scholar]
- Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637–1649. doi: 10.1016/0016-5085(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Sutliff VE, Doppman JL, Gibril F, et al. Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J Clin Oncol. 1997;15:2420–2431. doi: 10.1200/JCO.1997.15.6.2420. [DOI] [PubMed] [Google Scholar]
- Madeira I, Terris B, Voss M, et al. Prognostic factors in patients with endocrine tumours of the duodenopancreatic area. Gut. 1998;43:422–427. doi: 10.1136/gut.43.3.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga J, Yakuwa Y. Vipoma/diarrheogenic syndrome: a statistical evaluation of 241 reported cases. J Exp Clin Cancer Res. 1998;17:389–400. [PubMed] [Google Scholar]
- Nesi G, Marcucci T, Rubio CA, et al. Somatostatinoma: Clinico-pathological features of three cases and literature reviewed. J Gastroenterol Hepatol. 2007 doi: 10.1111/j.1440-1746.2007.05053.x. [DOI] [PubMed] [Google Scholar]
- Chu QD, Hill HC, Douglass HO, Jr., et al. Predictive factors associated with long-term survival in patients with neuroendocrine tumors of the pancreas. Ann Surg Oncol. 2002;9:855–862. doi: 10.1007/BF02557521. [DOI] [PubMed] [Google Scholar]
- La Rosa S, Sessa F, Capella C, et al. Prognostic criteria in nonfunctioning pancreatic endocrine tumours. Virchows Arch. 1996;429:323–333. doi: 10.1007/BF00198436. [DOI] [PubMed] [Google Scholar]
- Hochwald SN, Zee S, Conlon KC, et al. Prognostic factors in pancreatic endocrine neoplasms: an analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol. 2002;20:2633–2642. doi: 10.1200/JCO.2002.10.030. [DOI] [PubMed] [Google Scholar]
- Maton PN, Gardner JD, Jensen RT. Cushing's syndrome in patients with Zollinger-Ellison syndrome. N Engl J Med. 1986;315:1–5. doi: 10.1056/NEJM198607033150101. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Gardner JD. Gastrinoma. In: Go VLW, DiMagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA, editors. The Pancreas: Biology, Pathobiology and Disease. 2 ed. Raven Press Publishing Co.; New York: 1993. pp. 931–978. [Google Scholar]
- Norton JA, Sugarbaker PH, Doppman JL, et al. Aggressive resection of metastatic disease in selected patients with malignant gastrinoma. Ann Surg. 1986;203:352–359. doi: 10.1097/00000658-198604000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillausseau PJ, Guillausseau-Scholer C. Glucagonomas: clinical presentation, diagnosis, and advances in management. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Volume Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 183–193. (Series: Frontiers in Gastrointestinal Research). [Google Scholar]
- Thompson GB, Van Heerden JA, Grant CS, et al. Islet cell carcinomas of the pancreas: a twenty-year experience. Surgery. 1988;104:1011–1017. [PubMed] [Google Scholar]
- Lo CY, Van Heerden JA, Thompson GB, et al. Islet cell carcinoma of the pancreas. World J Surg. 1996;20:878–884. doi: 10.1007/s002689900134. [DOI] [PubMed] [Google Scholar]
- Norton JA, Fraker DL, Alexander HR, et al. Surgery increases survival in patients with gastrinoma. Ann Surg. 2006;244:410–419. doi: 10.1097/01.sla.0000234802.44320.a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraker DL, Norton JA, Alexander HR, et al. Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann Surg. 1994;220:320–330. doi: 10.1097/00000658-199409000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Herder WW. Biochemistry of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:33–41. doi: 10.1016/j.beem.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Gibril F, Jensen RT. Zollinger-Ellison syndrome revisited: diagnosis, biologic markers, associated inherited disorders, and acid hypersecretion. Curr Gastroenterol Rep. 2004;6:454–463. doi: 10.1007/s11894-004-0067-5. [DOI] [PubMed] [Google Scholar]
- Nobels FR, Kwekkeboom DJ, Coopmans W, et al. Chromogranin A as serum marker for neuroendocrine neoplasia: comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J Clin Endocrinol Metab. 1997;82:2622–2628. doi: 10.1210/jcem.82.8.4145. [DOI] [PubMed] [Google Scholar]
- Campana D, Nori F, Piscitelli L, et al. Chromogranin A: is it a useful marker of neuroendocrine tumors? J Clin Oncol. 2007;25:1967–1973. doi: 10.1200/JCO.2006.10.1535. [DOI] [PubMed] [Google Scholar]
- Zatelli MC, Torta M, Leon A, et al. Chromogranin A as a marker of neuroendocrine neoplasia: an Italian Multicenter Study. Endocr Relat Cancer. 2007;14:473–482. doi: 10.1677/ERC-07-0001. [DOI] [PubMed] [Google Scholar]
- Perren A, Anlauf M, Henopp T, et al. Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab. 2007;92:1118–1128. doi: 10.1210/jc.2006-1944. [DOI] [PubMed] [Google Scholar]
- Baudin E. Gastroenteropancreatic endocrine tumors: clinical characterization before therapy. Nat Clin Pract Endocrinol Metab. 2007;3:228–239. doi: 10.1038/ncpendmet0425. [DOI] [PubMed] [Google Scholar]
- Roy P, Venzon DJ, Feigenbaum KM, et al. Gastric secretion in Zollinger-Ellison syndrome: correlation with clinical expression, tumor extent and role in diagnosis - A prospective NIH study of 235 patients and review of the literature in 984 cases. Medicine(Baltimore) 2001;80:189–222. doi: 10.1097/00005792-200105000-00005. [DOI] [PubMed] [Google Scholar]
- Soga J, Yakuwa Y. The gastrinoma/Zollinger-Ellison syndrome: statistical evaluation of a Japanese series of 359 cases. J Hep Bil Pancr Surg. 1998;5:77–85. doi: 10.1007/pl00009955. [DOI] [PubMed] [Google Scholar]
- Roy P, Venzon DJ, Shojamanesh H, et al. Zollinger-Ellison syndrome: clinical presentation in 261 patients. Medicine (Baltimore) 2000;79:379–411. doi: 10.1097/00005792-200011000-00004. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Doppman JL, Gardner JD. Gastrinoma. In: Go VLW, Brooks FA, DiMagno EP, Gardner JD, Lebenthal E, Scheele GA, editors. The Exocrine Pancreas: Biology, Pathobiology and Disease. 1 ed. Raven Press; New York: 1986. pp. 727–744. [Google Scholar]
- Benya RV, Metz DC, Venzon DJ, et al. Zollinger-Ellison syndrome can be the initial endocrine manifestation in patients with multiple endocrine neoplasia-type 1. Am J Med. 1994;97:436–444. doi: 10.1016/0002-9343(94)90323-9. [DOI] [PubMed] [Google Scholar]
- Gibril F, Schumann M, Pace A, et al. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. A prospective study of 107 cases and comparison with 1009 patients from the literature. Medicine (Baltimore) 2004;83:43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- Hou W, Schubert ML. Gastric secretion. Curr Opin Gastroenterol. 2006;22:593–598. doi: 10.1097/01.mog.0000245538.43142.87. [DOI] [PubMed] [Google Scholar]
- Corleto VD, Annibale B, Gibril F, et al. Does the widespread use of proton pump inhibitors mask, complicate and/or delay the diagnosis of Zollinger-Ellison syndrome? Aliment Pharmacol Ther. 2001;15:1555–1561. doi: 10.1046/j.1365-2036.2001.01085.x. [DOI] [PubMed] [Google Scholar]
- Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore) 2006;85:295–330. doi: 10.1097/01.md.0000236956.74128.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszniewski P, Podevin P, Cadiot G, et al. Clinical, anatomical, and evolutive features of patients with the Zollinger-Ellison syndrome combined with type I multiple endocrine neoplasia. Pancreas. 1993;8:295–304. doi: 10.1097/00006676-199305000-00003. [DOI] [PubMed] [Google Scholar]
- Berna MJ, Hoffmann KM, Long SH, et al. Serum gastrin in Zollinger-Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore) 2006;85:331–364. doi: 10.1097/MD.0b013e31802b518c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibril F, Lindeman RJ, Abou-Saif A, et al. Retained gastric antrum syndrome. A forgotten, treatable cause of refractory peptic ulcer disease. Dig Dis Sci. 2001;46:610–617. doi: 10.1023/a:1005667719847. [DOI] [PubMed] [Google Scholar]
- McGuigan JE, Wolfe MM. Secretin injection test in the diagnosis of gastrinoma. Gastroenterology. 1980;79:1324–1331. [PubMed] [Google Scholar]
- Frucht H, Howard JM, Slaff JI, et al. Secretin and calcium provocative tests in the Zollinger-Ellison syndrome: A prospective study. Ann Intern Med. 1989;111:713–722. doi: 10.7326/0003-4819-111-9-713. [DOI] [PubMed] [Google Scholar]
- Norton JA, Cornelius MJ, Doppman JL, et al. Effect of parathyroidectomy in patients with hyperparathyroidism, Zollinger-Ellison syndrome and multiple endocrine neoplasia Type I: A prospective study. Surgery. 1987;102:958–966. [PubMed] [Google Scholar]
- Jensen RT. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. J Intern Med. 1998;243:477–488. doi: 10.1046/j.1365-2796.1998.00281.x. [DOI] [PubMed] [Google Scholar]
- Norton JA, Venzon DJ, Berna MJ, et al. Prospective study of surgery for primary hyperaparathyroidism (HPT) in Multiple Endocrine Neoplasia type 1 (MEN1), and Zollinger-Ellison syndrome (ZES): longterm outcome of a more virulent form of HPT. 247 ed. 2008. pp. 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CS. Insulinoma. Best Pract Res Clin Gastroenterol. 2005;19:783–798. doi: 10.1016/j.bpg.2005.05.008. [DOI] [PubMed] [Google Scholar]
- de Herder WW, Niederle B, Scoazec JY, et al. Well-differentiated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology. 2006;84:183–188. doi: 10.1159/000098010. [DOI] [PubMed] [Google Scholar]
- Vezzosi D, Bennett A, Fauvel J, et al. Insulin levels measured with an insulin-specific assay in patients with fasting hypoglycaemia related to endogenous hyperinsulinism. Eur J Endocrinol. 2003;149:413–419. doi: 10.1530/eje.0.1490413. [DOI] [PubMed] [Google Scholar]
- Vezzosi D, Bennet A, Fauvel J, et al. Insulin, C-peptide and proinsulin for the biochemical diagnosis of hypoglycaemia related to endogenous hyperinsulinism. Eur J Endocrinol. 2007;157:75–83. doi: 10.1530/EJE-07-0109. [DOI] [PubMed] [Google Scholar]
- Soga J, Yakuwa Y. Glucagonomas/diabetico-dermatogenic syndrome (DDS): a statistical evaluation of 407 reported cases. J Hep Bil Pancr Surg. 1998;5:312–319. doi: 10.1007/s005340050052. [DOI] [PubMed] [Google Scholar]
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531–537. doi: 10.1530/eje.0.1510531. [DOI] [PubMed] [Google Scholar]
- Kindmark H, Sundin A, Granberg D, et al. Endocrine pancreatic tumors with glucagon hypersecretion: a retrospective study of 23 cases during 20 years. Med Oncol. 2007;24:330–337. doi: 10.1007/s12032-007-0011-2. [DOI] [PubMed] [Google Scholar]
- Wald M, Lawrenz K, Luckner D, et al. Glucagon therapy as a possible cause of erythema necrolyticum migrans in two neonates with persistent hyperinsulinaemic hypoglycaemia. Eur J Pediatr. 2002;161:600–603. doi: 10.1007/s00431-002-1022-9. [DOI] [PubMed] [Google Scholar]
- Case CC, Vassilopoulou-Sellin R. Reproduction of features of the glucagonoma syndrome with continuous intravenous glucagon infusion as therapy for tumor-induced hypoglycemia. Endocr Pract. 2003;9:22–25. doi: 10.4158/EP.9.1.22. [DOI] [PubMed] [Google Scholar]
- Mullans EA, Cohen PR. Iatrogenic necrolytic migratory erythema: a case report and review of nonglucagonoma-associated necrolytic migratory erythema. J Am Acad Dermatol. 1998;38:866–873. doi: 10.1016/s0190-9622(98)70478-5. [DOI] [PubMed] [Google Scholar]
- Appetecchia M, Ferretti E, Carducci M, et al. Malignant glucagonoma. New options of treatment. J Exp Clin Cancer Res. 2006;25:135–139. [PubMed] [Google Scholar]
- Matuchansky C, Rambaud JC. VIPomas and endocrine cholera: clinical presentation, diagnosis, and advances in management. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 166–182. (Series: Frontiers in Gastrointestinal Research). [Google Scholar]
- Nikou GC, Toubanakis C, Nikolaou P, et al. VIPomas: an update in diagnosis and management in a series of 11 patients. Hepatogastroenterology. 2005;52:1259–1265. [PubMed] [Google Scholar]
- Ghaferi AA, Chojnacki KA, Long WD, et al. Pancreatic VIPomas: Subject Review and One Institutional Experience. J Gastrointest Surg. 2007 doi: 10.1007/s11605-007-0177-0. [DOI] [PubMed] [Google Scholar]
- Jensen RT. Diarrhea in neuroendocrine disorders. In: Domschke W, Stoll R, Brasitus TA, Kagnoff MF, editors. Intestinal Mucosa and Its Diseases: Pathophysiology and Clinics. 110 ed. Kluwer Academic Publishers BV; Dordrecht, The Netherlands: 1999. pp. 61–76. [Google Scholar]
- Krejs GJ, Orci L, Conlon JM, et al. Somatostatinoma syndrome. Biochemical, morphologic and clinical features. N Engl J Med. 1979;301:285–292. doi: 10.1056/NEJM197908093010601. [DOI] [PubMed] [Google Scholar]
- Moayedoddin B, Booya F, Wermers RA, et al. Spectrum of malignant somatostatin-producing neuroendocrine tumors. Endocr Pract. 2006;12:394–400. doi: 10.4158/EP.12.4.394. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Yamasaki S, Matsushita S, et al. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol Int. 2000;50:146–152. doi: 10.1046/j.1440-1827.2000.01016.x. [DOI] [PubMed] [Google Scholar]
- Losa M, von Werder K. Pathophysiology and clinical aspects of the ectopic GH-releasing hormone syndrome. Clin Endocrinol. 1997;47:123–135. doi: 10.1046/j.1365-2265.1997.2311048.x. [DOI] [PubMed] [Google Scholar]
- Sano T, Asa SL, Kovacs K. Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev. 1988;9:357–373. doi: 10.1210/edrv-9-3-357. [DOI] [PubMed] [Google Scholar]
- Gola M, Doga M, Bonadonna S, et al. Neuroendocrine tumors secreting growth hormone-releasing hormone: Pathophysiological and clinical aspects. Pituitary. 2006 doi: 10.1007/s11102-006-0267-0. [DOI] [PubMed] [Google Scholar]
- Falconi M, Plockinger U, Kwekkeboom DJ, et al. Well-differentiated pancreatic nonfunctioning tumors/carcinoma. Neuroendocrinology. 2006;84:196–211. doi: 10.1159/000098012. [DOI] [PubMed] [Google Scholar]
- Plockinger U, Wiedenmann B. Diagnosis of non-functioning neuro-endocrine gastro-enteropancreatic tumours. Neuroendocrinology. 2004;80(Suppl 1):35–38. doi: 10.1159/000080739. [DOI] [PubMed] [Google Scholar]
- Gullo L, Migliori M, Falconi M, et al. Nonfunctioning pancreatic endocrine tumors: a multicenter clinical study. Am J Gastroenterol. 2003;98:2435–2439. doi: 10.1111/j.1572-0241.2003.07704.x. [DOI] [PubMed] [Google Scholar]
- Eriksson B, Oberg K. PPomas and nonfunctioning endocrine pancreatic tumors: clinical presentation, diagnosis, and advances in management. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 208–222. (Series: Frontiers in Gastrointestinal Research). [Google Scholar]
- Hochwald SN, Conlon KC, Brennan MF. Nonfunctional pancreatic islet cell tumors. In: Doherty GM, Skogseid B, editors. Surgical Endocrinology. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 361–373. [Google Scholar]
- Brennan MF, Marx SJ, Doppman J, et al. Results of reoperation for persistent and recurrent hyperparathyroidism. Ann Surg. 1981;194:671–676. doi: 10.1097/00000658-198112000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plockinger U, Wiedenmann B. Management of metastatic endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:553–576. doi: 10.1016/j.bpg.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Gibril F, Jensen RT. Diagnostic uses of radiolabelled somatostatin-receptor analogues in gastroenteropancreatic endocrine tumors. Dig Liver Dis. 2004;36:S106–S120. doi: 10.1016/j.dld.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Rockall AG, Reznek RH. Imaging of neuroendocrine tumours (CT/MR/US) Best Pract Res Clin Endocrinol Metab. 2007;21:43–68. doi: 10.1016/j.beem.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Sundin A, Garske U, Orlefors H. Nuclear imaging of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:69–85. doi: 10.1016/j.beem.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Noone TC, Hosey J, Firat Z, et al. Imaging and localization of islet-cell tumours of the pancreas on CT and MRI. Best Pract Res Clin Endocrinol Metab. 2005;19:195–211. doi: 10.1016/j.beem.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Wiedenmann B, Jensen RT, Mignon M, et al. Preoperative diagnosis and surgical manangement of neuroendocrine gastroenteropancreatic tumors: general recommendations by a consensus workshop. World J Surg. 1998;22:309–318. doi: 10.1007/s002689900387. [DOI] [PubMed] [Google Scholar]
- Orbuch M, Doppman JL, Strader DB, et al. Imaging for pancreatic endocrine tumor localization: recent advances. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Frontiers of Gastrointestinal Research. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 268–281. [Google Scholar]
- Krudy AG, Doppman JL, Jensen RT, et al. Localization of islet cell tumors by dynamic CT: Comparison with plain CT, arteriography, sonography and venous sampling. Am J Roentgenol. 1984;143:585–589. doi: 10.2214/ajr.143.3.585. [DOI] [PubMed] [Google Scholar]
- Frucht H, Doppman JL, Norton JA, et al. Gastrinomas: Comparison of MR Imaging with CT, angiography and US. Radiology. 1989;171:713–717. doi: 10.1148/radiology.171.3.2655004. [DOI] [PubMed] [Google Scholar]
- Orbuch M, Doppman JL, Jensen RT, Jensen Robert T. Localization of pancreatic endocrine tumors. In: Sleisenger MH, Fordtran JS, editors. Pancreatic endocrine tumors. Seminars in Gastrointestinal Disease. Volume 6. W.B. Saunders Publishing Co.; Duluth, MN: 1995. pp. 90–101. [PubMed] [Google Scholar]
- Akerstrom G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:87–109. doi: 10.1016/j.beem.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Ho JM, Darcy SJ, Eysselein VE, et al. Evolution of fine needle aspiration cytology in the accurate diagnosis of pancreatic neoplasms. Am Surg. 2007;73:941–944. [PubMed] [Google Scholar]
- Nichols MT, Russ PD, Chen YK. Pancreatic imaging: current and emerging technologies. Pancreas. 2006;33:211–220. doi: 10.1097/01.mpa.0000227912.71202.2c. [DOI] [PubMed] [Google Scholar]
- Pausawasdi N, Scheiman J. Endoscopic evaluation and palliation of pancreatic adenocarcinoma: current and future options. Curr Opin Gastroenterol. 2007;23:515–521. doi: 10.1097/MOG.0b013e3282ba5713. [DOI] [PubMed] [Google Scholar]
- McLean AM, Fairclough PD. Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:177–193. doi: 10.1016/j.beem.2004.11.012. [DOI] [PubMed] [Google Scholar]
- DeAngelis C, Carucci P, Repici A, et al. Endosonography in decision making and management of gastrointestinal endocrine tumors. Eur J Ultrasound. 1999;10:139–150. doi: 10.1016/s0929-8266(99)00054-3. [DOI] [PubMed] [Google Scholar]
- Chang F, Chandra A, Culora G, et al. Cytologic diagnosis of pancreatic endocrine tumors by endoscopic ultrasound-guided fine-needle aspiration: a review. Diagn Cytopathol. 2006;34:649–658. doi: 10.1002/dc.20503. [DOI] [PubMed] [Google Scholar]
- Stelow EB, Woon C, Pambuccian SE, et al. Fine-needle aspiration cytology of pancreatic somatostatinoma: the importance of immunohistochemistry for the cytologic diagnosis of pancreatic endocrine neoplasms. Diagn Cytopathol. 2005;33:100–105. doi: 10.1002/dc.20305. [DOI] [PubMed] [Google Scholar]
- Norton JA, Jensen RT. Resolved and unresolved controversies in the surgical management of patients with Zollinger-Ellison syndrome. Ann Surg. 2004;240:757–773. doi: 10.1097/01.sla.0000143252.02142.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer T, Stolzel U, Bader M, et al. Endoscopic ultrasonography and somatostatin receptor scintigraphy in the preoperative localisation of insulinomas and gastrinomas. Gut. 1996;39:562–568. doi: 10.1136/gut.39.4.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzosi D, Bennet A, Rochaix P, et al. Octreotide in insulinoma patients: efficacy on hypoglycemia, relationships with Octreoscan scintigraphy and immunostaining with anti-sst2A and anti-sst5 antibodies. Eur J Endocrinol. 2005;152:757–767. doi: 10.1530/eje.1.01901. [DOI] [PubMed] [Google Scholar]
- Bertherat J, Tenenbaum F, Perlemoine K, et al. Somatostatin receptors 2 and 5 are the major somatostatin receptors in insulinomas: an in vivo and in vitro study. J Clin Endocrinol Metab. 2003;88:5353–5360. doi: 10.1210/jc.2002-021895. [DOI] [PubMed] [Google Scholar]
- Langer P, Kann PH, Fendrich V, et al. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. 2004;28:1317–1322. doi: 10.1007/s00268-004-7642-7. [DOI] [PubMed] [Google Scholar]
- Wamsteker EJ, Gauger PG, Thompson NW, et al. EUS detection of pancreatic endocrine tumors in asymptomatic patients with type 1 multiple endocrine neoplasia. Gastrointest Endosc. 2003;58:531–535. doi: 10.1067/s0016-5107(03)01965-5. [DOI] [PubMed] [Google Scholar]
- Kann PH, Balakina E, Ivan D, et al. Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Endocr Relat Cancer. 2006;13:1195–1202. doi: 10.1677/erc.1.01220. [DOI] [PubMed] [Google Scholar]
- Hellman P, Hennings J, Akerstrom G, et al. Endoscopic ultrasonography for evaluation of pancreatic tumours in multiple endocrine neoplasia type 1. Br J Surg. 2005;92:1508–1512. doi: 10.1002/bjs.5149. [DOI] [PubMed] [Google Scholar]
- Langer P, Wild A, Celik I, et al. Prospective controlled trial of a standardized meal stimulation test in the detection of pancreaticoduodenal endocrine tumours in patients with multiple endocrine neoplasia type 1. Br J Surg. 2001;88:1403–1407. doi: 10.1046/j.0007-1323.2001.01874.x. [DOI] [PubMed] [Google Scholar]
- Kann PH, Kann B, Fassbender WJ, et al. Small neuroendocrine pancreatic tumors in multiple endocrine neoplasia type 1 (MEN1): least significant change of tumor diameter as determined by endoscopic ultrasound (EUS) imaging. Exp Clin Endocrinol Diabetes. 2006;114:361–365. doi: 10.1055/s-2006-924322. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. Gastroenterology. 2000;119:1087–1095. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- Libutti SK, Choyke PL, Alexander HR, et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery. 2000;128:1022–1027. doi: 10.1067/msy.2000.110239. [DOI] [PubMed] [Google Scholar]
- Binkovitz LA, Johnson CD, Stephens DH. Islet cell tumors in von Hippel-Lindau disease: increased prevalence and relationship to the multiple endocrine neoplasias. AJR Am J Roentgenol. 1990;155:501–505. doi: 10.2214/ajr.155.3.1974734. [DOI] [PubMed] [Google Scholar]
- Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recomendations. Surgery. 1998;124:1153–1159. doi: 10.1067/msy.1998.91823. [DOI] [PubMed] [Google Scholar]
- Marcos HB, Libutti SK, Alexander HR, et al. Neuroendocrine tumors of the pancreas in von Hippel-Lindau disease: spectrum of appearances at CT and MR imaging with histopathologic comparison. Radiology. 2002;225:751–758. doi: 10.1148/radiol.2253011297. [DOI] [PubMed] [Google Scholar]
- Blansfield JA, Choyke L, Morita SY, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs) Surgery. 2007;142:814–818. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. 1954;16:363–371. doi: 10.1016/0002-9343(54)90353-8. [DOI] [PubMed] [Google Scholar]
- Strader DB, Doppman JL, Orbuch M, et al. Functional localization of pancreatic endocrine tumors. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent Advances in Research and Management. Volume 23. Karger Publishing Co.; Basel, Switzerland: 1995. pp. 282–297. (Series: Frontiers of Gastrointestinal Research). [Google Scholar]
- Jackson JE. Angiography and arterial stimulation venous sampling in the localization of pancreatic neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:229–239. doi: 10.1016/j.beem.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Gibril F, Doppman JL, Chang R, et al. Metastatic gastrinomas: localization with selective arterial injection of secretin. Radiology. 1996;198:77–84. doi: 10.1148/radiology.198.1.8539410. [DOI] [PubMed] [Google Scholar]
- Miller DL, Doppman JL, Metz DC, et al. Zollinger-Ellison syndrome: technique, results and complications of portal venous sampling. Radiology. 1992;182:235–241. doi: 10.1148/radiology.182.1.1727289. [DOI] [PubMed] [Google Scholar]
- Thom AK, Norton JA, Doppman JL, et al. Prospective study of the use of intraarterial secretin injection and portal venous sampling to localize duodenal gastrinomas. Surgery. 1992;112(6):1002–1008. [PubMed] [Google Scholar]
- Doppman JL, Miller DL, Chang R, et al. Gastrinomas: localization by means of selective intraarterial injection of secretin. Radiology. 1990;174:25–29. doi: 10.1148/radiology.174.1.2294556. [DOI] [PubMed] [Google Scholar]
- Lo CY, Chan FL, Tam SC, et al. Value of intra-arterial calcium stimulated venous sampling for regionalization of pancreatic insulinomas. Surgery. 2000;128:903–909. doi: 10.1067/msy.2000.109729. [DOI] [PubMed] [Google Scholar]
- Piovesan A, Pia A, Visconti G, et al. Proinsulin-secreting neuroendocrine tumor of the pancreas. J Endocrinol Invest. 2003;26:758–761. doi: 10.1007/BF03347360. [DOI] [PubMed] [Google Scholar]
- Doppman JL, Chang R, Fraker DL, et al. Localization of insulinomas to regions of the pancreas by intra-arterial stimulation with calcium. Ann Intern Med. 1995;123:269–273. doi: 10.7326/0003-4819-123-4-199508150-00004. [DOI] [PubMed] [Google Scholar]
- Defreyne L, Konig K, Lerch MM, et al. Modified intra-arterial calcium stimulation with venous sampling test for preoperative localization of insulinomas. Abdom Imaging. 1998;23:322–331. doi: 10.1007/s002619900350. [DOI] [PubMed] [Google Scholar]
- Baba Y, Miyazono N, Nakajo M, et al. Localization of insulinomas. Comparison of conventional arterial stimulation with venous sampling (ASVS) and superselective ASVS. Acta Radiol. 2000;41:172–177. doi: 10.1080/028418500127345000. [DOI] [PubMed] [Google Scholar]
- Guillermet-Guibert J, Lahlou H, Pyronnet S, et al. Somatostatin receptors as tools for diagnosis and therapy: Molecular aspects. Best Pract Res Clin Gastroenterol. 2005;19:535–551. doi: 10.1016/j.bpg.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Krenning EP, Kwekkeboom DJ, Bakker WH, et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123ITyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716–731. doi: 10.1007/BF00181765. [DOI] [PubMed] [Google Scholar]
- Papotti M, Bonjiovanni M, Volante M, et al. Expression of somatostatin receptor types 1-5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. Virchows Arch. 2002;440:461–475. doi: 10.1007/s00428-002-0609-x. [DOI] [PubMed] [Google Scholar]
- Jensen RT. Peptide therapy. Recent advances in the use of somatostatin and other peptide receptor agonists and antagonists. In: Lewis JH, Dubois A, editors. Current Clinical Topics in Gastrointestinal Pharmacology. Blackwell Science, Inc.; Malden, MA: 1997. pp. 144–223. [Google Scholar]
- Virgolini I, Traub-Weidinger T, Decristoforo C. Nuclear medicine in the detection and management of pancreatic islet-cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:213–227. doi: 10.1016/j.beem.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Gibril F, Reynolds JC, Doppman JL, et al. Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas: a prospective study. Ann Intern Med. 1996;125:26–34. doi: 10.7326/0003-4819-125-1-199607010-00005. [DOI] [PubMed] [Google Scholar]
- Termanini B, Gibril F, Doppman JL, et al. Distinguishing small hepatic hemangiomas from vascular liver metastases in patients with gastrinoma: Use of a somatostatin-receptor scintigraphic agent. Radiology. 1997;202:151–158. doi: 10.1148/radiology.202.1.8988205. [DOI] [PubMed] [Google Scholar]
- Schillaci O, Spanu A, Scopinaro F, et al. Somatostatin receptor scintigraphy in liver metastasis detection from gastroenteropancreatic neuroendocrine tumors. J Nucl Med. 2003;44:359–368. [PubMed] [Google Scholar]
- Lebtahi R, Cadiot G, Delahaye N, et al. Detection of bone metastases in patients with endocrine gastroenteropancreatic tumors: bone scintigraphy compared with somatostatin receptor scintigraphy. J Nucl Med. 1999;40:1602–1608. [PubMed] [Google Scholar]
- Schillaci O, Corleto VD, Annibale B, et al. Single photon emission computed tomography procedure improves accuracy of somatostatin receptor scintigraphy in gastro-entero pancreatic tumours. Ital J Gastroenterol Hepatol. 1999;31:S186–S189. [PubMed] [Google Scholar]
- Lebtahi R, Cadiot G, Sarda L, et al. Clinical impact of somatostatin receptor scintigraphy in the management of patients with neuroendocrine gastroenteropancreatic tumors. J Nucl Med. 1997;38:853–858. [PubMed] [Google Scholar]
- Termanini B, Gibril F, Reynolds JC, et al. Value of somatostatin receptor scintigraphy: A prospective study in gastrinoma of its effect on clinical management. Gastroenterology. 1997;112:335–347. doi: 10.1053/gast.1997.v112.pm9024287. [DOI] [PubMed] [Google Scholar]
- Cadiot G, Bonnaud G, Lebtahi R, et al. Usefulness of somatostatin receptor scintigraphy in the management of patients with Zollinger-Ellison syndrome. Gut. 1997;41:107–114. doi: 10.1136/gut.41.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander HR, Fraker DL, Norton JA, et al. Prospective study of somatostatin receptor scintigraphy and its effect on operative outcome in patients with Zollinger-Ellison syndrome. Ann Surg. 1998;228:228–238. doi: 10.1097/00000658-199808000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson B, Orlefors H, Oberg K, et al. Developments in PET for the detection of endocrine tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:311–324. doi: 10.1016/j.beem.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-Octreotide PET in Neuroendocrine Tumors: Comparison with Somatostatin Receptor Scintigraphy and CT. J Nucl Med. 2007;48:508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392–3400. doi: 10.1210/jc.2004-1938. [DOI] [PubMed] [Google Scholar]
- Metz DC, Jensen RT. Advances in gastric antisecretory therapy in Zollinger-Ellison syndrome. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 240–257. (Series: Frontiers of Gastrointestinal Research). [Google Scholar]
- Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem. 2007;7:1211–1231. doi: 10.2174/156802607780960519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RT. Use of omeprazole and other proton pump inhibitors in the Zollinger-Ellison syndrome. In: Olbe L, editor. Milestones in Drug Therapy. Birkhauser Verlag AG Publish. Co.; Basel, Switzerland: 1999. pp. 205–221. [Google Scholar]
- Metz DC, Strader DB, Orbuch M, et al. Use of omeprazole in Zollinger-Ellison: A prospective nine-year study of efficacy and safety. Aliment Pharmacol Ther. 1993;7:597–610. doi: 10.1111/j.1365-2036.1993.tb00140.x. [DOI] [PubMed] [Google Scholar]
- Metz DC, Pisegna JR, Ringham GL, et al. Prospective study of efficacy and safety of lansoprazole in Zollinger-Ellison syndrome. Dig Dis Sci. 1993;38:245–256. doi: 10.1007/BF01307541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschowitz BI, Simmons J, Mohnen J. Clinical outcome using lansoprazole in acid hypersecretors with and without Zollinger-Ellison syndrome: a 13-year prospective study. Clin Gastroenterol Hepatol. 2005;3:39–48. doi: 10.1016/s1542-3565(04)00606-8. [DOI] [PubMed] [Google Scholar]
- Metz DC, Comer GM, Soffer E, et al. Three-year oral pantoprazole administration is effective for patients with Zollinger-Ellison syndrome and other hypersecretory conditions. Aliment Pharmacol Ther. 2006;23:437–444. doi: 10.1111/j.1365-2036.2006.02762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morocutti A, Merrouche M, Bjaaland T, et al. An open-label study of rabeprazole in patients with Zollinger-Ellison syndrome or idiopathic gastric acid hypersecretion. Aliment Pharmacol Ther. 2006;24:1439–1444. doi: 10.1111/j.1365-2036.2006.03137.x. [DOI] [PubMed] [Google Scholar]
- Metz DC, Sostek MB, Ruszniewski P, et al. Effects of esomeprazole on Acid output in patients with zollinger-ellison syndrome or idiopathic gastric Acid hypersecretion. Am J Gastroenterol. 2007;102:2648–2654. doi: 10.1111/j.1572-0241.2007.01509.x. [DOI] [PubMed] [Google Scholar]
- Raufman JP, Collins SM, Pandol SJ, et al. Reliability of symptoms in assessing control of gastric acid secretion in patients with Zollinger-Ellison syndrome. Gastroenterology. 1983;84:108–113. [PubMed] [Google Scholar]
- Miller LS, Vinayek R, Frucht H, et al. Reflux esophagitis in patients with Zollinger-Ellison syndrome. Gastroenterology. 1990;98:341–346. doi: 10.1016/0016-5085(90)90823-j. [DOI] [PubMed] [Google Scholar]
- Maton PN, Frucht H, Vinayek R, et al. Medical management of patients with Zollinger-Ellison syndrome who have had previous gastric surgery: A prospective study. Gastroenterology. 1988;94:294–299. doi: 10.1016/0016-5085(88)90415-5. [DOI] [PubMed] [Google Scholar]
- Metz DC, Pisegna JR, Fishbeyn VA, et al. Currently used doses of omeprazole in Zollinger-Ellison syndrome are too high. Gastroenterology. 1992;103:1498–1508. doi: 10.1016/0016-5085(92)91170-9. [DOI] [PubMed] [Google Scholar]
- Termanini B, Gibril F, Stewart CA, et al. A prospective study of the effectiveness of low dose omeprazole as initial therapy in Zollinger-Ellison syndrome. Aliment Pharmacol Ther. 1996;10:61–71. doi: 10.1111/j.1365-2036.1996.tb00178.x. [DOI] [PubMed] [Google Scholar]
- Termanini B, Gibril F, Sutliff VE, et al. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am J Med. 1998;104:422–430. doi: 10.1016/s0002-9343(98)00087-4. [DOI] [PubMed] [Google Scholar]
- Stewart CA, Termanini B, Sutliff VE, et al. Assessment of the risk of iron malabsorption in patients with Zollinger-Ellison syndrome treated with long-term gastric acid antisecretory therapy. Aliment Pharmacol Ther. 1998;12:83–98. doi: 10.1046/j.1365-2036.1998.00274.x. [DOI] [PubMed] [Google Scholar]
- Peghini PL, Annibale B, Azzoni C, et al. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: insights from patients with sporadic gastrinomas. Gastroenterology. 2002;123:68–85. doi: 10.1053/gast.2002.34231. [DOI] [PubMed] [Google Scholar]
- Maton PN, Lack EE, Collen MJ, et al. The effect of Zollinger-Ellison syndrome and omeprazole therapy on gastric oxyntic endocrine cells. Gastroenterology. 1990;99:943–950. doi: 10.1016/0016-5085(90)90611-4. [DOI] [PubMed] [Google Scholar]
- Berna MJ, Annibale B, Marignani M, et al. A prospective study of gastric carcinoids and enterochromaffin-like cells changes in Multple Endocrine Neoplaisa Type 1 and Zollinger-Ellison syndrome: Identification of risk factors. J Clin Endocrinol Metab. doi: 10.1210/jc.2007-2279. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehy T, Cadiot G, Mignon M, et al. Influence of multiple endocrine neoplasia type 1 on gastric endocrine cells in patients with the Zollinger-Ellison syndrome. Gut. 1992;33:1275–1279. doi: 10.1136/gut.33.9.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi C, Azzoni C, Ferraro G, et al. Sampling strategies for analysis of enterochromaffin-like cell changes in Zollinger-Ellison syndrome. Am J Clin Pathol. 2000;114:419–425. doi: 10.1093/ajcp/114.3.419. [DOI] [PubMed] [Google Scholar]
- Lew EA, Pisegna JR, Starr JA, et al. Intravenous pantoprazole rapidly controls gastric acid hypersecretion in patients with Zollinger-Ellison syndrome. Gastroenterology. 2000;118:696–704. doi: 10.1016/s0016-5085(00)70139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Lubke HJ, Frieling T, et al. Prospective study on the detection of insulinomas by endoscopic ultrasonography. Endoscopy. 1996;28:273–276. doi: 10.1055/s-2007-1005452. [DOI] [PubMed] [Google Scholar]
- Creutzfeldt W. Insulinomas: Clinical presentation, diagnosis, and advances in management. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent Advances in Research and Management. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 148–165. (Series: Frontiers of Gastrointestinal Research). [Google Scholar]
- Gill GV, Rauf O, MacFarlane IA. Diazoxide treatment for insulinoma: a national UK survey. Postgrad Med J. 1997;73:640–641. doi: 10.1136/pgmj.73.864.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless J, Bauer W, Briner U, et al. Chemistry and pharmacology of SMS 201-995, a long-acting octapeptide analogue of somatostatin. Scand J Gastroenterol Suppl. 1986;119:54–64. doi: 10.3109/00365528609087432. [DOI] [PubMed] [Google Scholar]
- Maton PN, Gardner JD, Jensen RT. Use of the long-acting somatostatin analog, SMS 201-995 in patients with pancreatic islet cell tumors. Dig Dis Sci. 1989;34:28S–39S. doi: 10.1007/BF01536043. [DOI] [PubMed] [Google Scholar]
- McKeage K, Cheer S, Wagstaff AJ. Octreotide long-acting release (LAR): a review of its use in the management of acromegaly. Drugs. 2003;63:2473–2499. doi: 10.2165/00003495-200363220-00014. [DOI] [PubMed] [Google Scholar]
- Bajetta E, Procopio G, Catena L, et al. Lanreotide autogel every 6 weeks compared with Lanreotide microparticles every 3 weeks in patients with well differentiated neuroendocrine tumors: a Phase III Study. Cancer. 2006;107:2474–2481. doi: 10.1002/cncr.22272. [DOI] [PubMed] [Google Scholar]
- Newman CB, Melmed S, Snyder PJ, et al. Safety and efficacy of long-term octreotide therapy of acromegaly: results of a multicenter trial in 103 patients--a clinical research center study. J Clin Endocrinol Metab. 1995;80:2768–2775. doi: 10.1210/jcem.80.9.7673422. [DOI] [PubMed] [Google Scholar]
- Norton JA, Alexander HR, Fraker DL, et al. Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. Ann Surg. 2001;234:495–506. doi: 10.1097/00000658-200110000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton JA, Fraker DL, Alexander HR, et al. Surgery to cure the Zollinger-Ellison syndrome. N Engl J Med. 1999;341:635–644. doi: 10.1056/NEJM199908263410902. [DOI] [PubMed] [Google Scholar]
- Fishbeyn VA, Norton JA, Benya RV, et al. Assessment and prediction of long-term cure in patients with Zollinger-Ellison syndrome: the best approach. Ann Intern Med. 1993;119:199–206. doi: 10.7326/0003-4819-119-3-199308010-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramoto JS, Feldstein VA, LaBerge JM, et al. Intraoperative ultrasound and preoperative localization detects all occult insulinomas. Arch Surg. 2001;136:1020–1026. doi: 10.1001/archsurg.136.9.1020. [DOI] [PubMed] [Google Scholar]
- Norton JA. Surgical treatment of neuroendocrine metastases. Best Pract Res Clin Gastroenterol. 2005;19:577–583. doi: 10.1016/j.bpg.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Que FG, Sarmiento JM, Nagorney DM. Hepatic surgery for metastatic gastrointestinal neuroendocrine tumors. Adv Exp Med Biol. 2006;574:43–56. doi: 10.1007/0-387-29512-7_7. [DOI] [PubMed] [Google Scholar]
- Norton JA, Doherty GD, Fraker DL, et al. Surgical treatment of localized gastrinoma within the liver: A prospective study. Surgery. 1998;124:1145–1152. doi: 10.1067/msy.1998.93110. [DOI] [PubMed] [Google Scholar]
- Norton JA, Warren RS, Kelly MG, et al. Aggressive surgery for metastatic liver neuroendocrine tumors. Surgery. 2003;134:1057–1065. doi: 10.1016/j.surg.2003.07.025. [DOI] [PubMed] [Google Scholar]
- Levy-Bohbot N, Merle C, Goudet P, et al. Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol. 2004;28:1075–1081. doi: 10.1016/s0399-8320(04)95184-6. [DOI] [PubMed] [Google Scholar]
- Norton JA, Fang TD, Jensen RT. Surgery for gastrinoma and insulinoma in multiple endocrine neoplasia type 1. J Natl Compr Canc Netw. 2006;4:148–153. doi: 10.6004/jnccn.2006.0015. [DOI] [PubMed] [Google Scholar]
- Cougard P, Goudet P, Peix JL, et al. [Insulinomas in multiple endocrine neoplasia type 1. Report of a series of 44 cases by the multiple endocrine neoplasia study group]. Ann Chir. 2000;125:118–123. doi: 10.1016/s0001-4001(00)00112-4. [DOI] [PubMed] [Google Scholar]
- MacFarlane MP, Fraker DL, Alexander HR, et al. A prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia-Type 1. Surgery. 1995;118:973–980. doi: 10.1016/s0039-6060(05)80102-3. [DOI] [PubMed] [Google Scholar]
- Akerstrom G, Hessman O, Hellman P, et al. Pancreatic tumours as part of the MEN-1 syndrome. Best Pract Res Clin Gastroenterol. 2005;19:819–830. doi: 10.1016/j.bpg.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Sheppard BC, Norton JA, Doppman JL, et al. Management of islet cell tumors in patients with Multiple Endocrine Neoplasia: A prospective study. Surgery. 1989;106:1108–1117. [PubMed] [Google Scholar]
- Anlauf M, Garbrecht N, Henopp T, et al. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: distinct clinico-pathological and epidemiological features. World J Gastroenterol. 2006;12:5440–5446. doi: 10.3748/wjg.v12.i34.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triponez F, Cadiot G. Non-functioning tumours of the pancreas in MEN1 patients. J Gastrointestin Liver Dis. 2007;16:295–296. [PubMed] [Google Scholar]
- Fendrich V, Langer P, Waldmann J, et al. Management of sporadic and multiple endocrine neoplasia type 1 gastrinomas. Br J Surg. 2007;94:1331–1341. doi: 10.1002/bjs.5987. [DOI] [PubMed] [Google Scholar]
- Mignon M, Cadiot G, Rigaud D, et al. Management of islet cell tumors in patients with multiple endocrine neoplasia type 1. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Vol. 23. S. Karger; Basel, Switzerland: 1995. pp. 342–359. (Series: Frontiers in Gastrointestinal Research). [Google Scholar]
- Triponez F, Goudet P, Dosseh D, et al. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg. 2006;30:654–662. doi: 10.1007/s00268-005-0354-9. [DOI] [PubMed] [Google Scholar]
- Norton JA, Alexander HR, Fraker DL, et al. Does the use of routine duodenotomy (DUODX) affect rate of cure, development of liver metastases or survival in patients with Zollinger-Ellison syndrome (ZES)? Ann Surg. 2004;239:617–626. doi: 10.1097/01.sla.0000124290.05524.5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugg SL, Norton JA, Fraker DL, et al. A prospective study of intraoperative methods to diagnose and resect duodenal gastrinomas. Ann Surg. 1993;218:138–144. doi: 10.1097/00000658-199308000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton JA. Surgery and prognosis of duodenal gastrinoma as a duodenal neuroendocrine tumor. Best Pract Res Clin Gastroenterol. 2005;19:699–704. doi: 10.1016/j.bpg.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Norton JA, Jensen RT. Current surgical management of Zollinger-Ellison syndrome (ZES) in patients without multiple endocrine neoplasia-type 1 (MEN1) Surg Oncol. 2003;12:145–151. doi: 10.1016/s0960-7404(03)00035-5. [DOI] [PubMed] [Google Scholar]
- Norton JA. Intraoperative methods to stage and localize pancreatic and duodenal tumors. Ann Oncol. 1999;10:182–184. [PubMed] [Google Scholar]
- Maton PN, Mackem SM, Norton JA, et al. Ovarian carcinoma as a cause of Zollinger-Ellison syndrome. Natural history, secretory products and response to provocative tests. Gastroenterology. 1989;97:468–471. doi: 10.1016/0016-5085(89)90085-1. [DOI] [PubMed] [Google Scholar]
- Furihata M, Tagaya N, Kubota K. Laparoscopic enucleation of insulinoma in the pancreas: case report and review of the literature. Surg Laparosc Endosc Percutan Tech. 2001;11:279–283. doi: 10.1097/00129689-200108000-00011. [DOI] [PubMed] [Google Scholar]
- Ayav A, Bresler L, Brunaud L, et al. Laparoscopic approach for solitary insulinoma: a multicentre study. Langenbecks Arch Surg. 2005;390:134–140. doi: 10.1007/s00423-004-0526-3. [DOI] [PubMed] [Google Scholar]
- Toniato A, Meduri F, Foletto M, et al. Laparoscopic treatment of benign insulinomas localized in the body and tail of the pancreas: a single-center experience. World J Surg. 2006;30:1916–1919. doi: 10.1007/s00268-005-0645-1. [DOI] [PubMed] [Google Scholar]
- Frucht H, Norton JA, London JF, et al. Detection of duodenal gastrinomas by operative endoscopic transillumination: a prospective study. Gastroenterology. 1990;99:1622–1627. doi: 10.1016/0016-5085(90)90466-e. [DOI] [PubMed] [Google Scholar]
- Norton JA, Doppman JL, Jensen RT. Curative resection in Zollinger-Ellison syndrome: Results of a 10-year prospective study. Ann Surg. 1992;215:8–18. doi: 10.1097/00000658-199201000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton JA, Doppman JL, Collen MJ, et al. Prospective study of gastrinoma localization and resection in patients with Zollinger-Ellison syndrome. Ann Surg. 1986;204:468–479. doi: 10.1097/00000658-198610000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Imamura M, Hosotani R, et al. Curative resection of microgastrinomas based on the intraoperative secretin test. World J Surg. 2000;24:1425–1430. doi: 10.1007/s002680010235. [DOI] [PubMed] [Google Scholar]
- Norton JA. Surgical treatment and prognosis of gastrinoma. Best Pract Res Clin Gastroenterol. 2005;19:799–805. doi: 10.1016/j.bpg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Nikfarjam M, Warshaw AL, Axelrod L, et al. Improved Contemporary Surgical Management of Insulinomas: A 25-year Experience at the Massachusetts General Hospital. Ann Surg. 2008;247:165–172. doi: 10.1097/SLA.0b013e31815792ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butturini G, Bettini R, Missiaglia E, et al. Predictive factors of efficacy of the somatostatin analogue octreotide as first line therapy for advanced pancreatic endocrine carcinoma. Endocr Relat Cancer. 2006;13:1213–1221. doi: 10.1677/erc.1.01200. [DOI] [PubMed] [Google Scholar]
- Shah T, Caplin M. Biotherapy for metastatic endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:617–636. doi: 10.1016/j.bpg.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Plockinger U, Wiedenmann B. Biotherapy. Best Pract Res Clin Endocrinol Metab. 2007;21:145–162. doi: 10.1016/j.beem.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25:458–511. doi: 10.1210/er.2003-0014. [DOI] [PubMed] [Google Scholar]
- Delaunoit T, Rubin J, Neczyporenko F, et al. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumors. Mayo Clin Proc. 2005;80:502–506. doi: 10.4065/80.4.502. [DOI] [PubMed] [Google Scholar]
- Panzuto F, Di Francesco V, Iannicelli E, et al. Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Ann Oncol. 2006;17:461–466. doi: 10.1093/annonc/mdj113. [DOI] [PubMed] [Google Scholar]
- Fallucca P, Delle Fave G, Giangrande L, et al. Effect of somatostatin on gastrin, insulin and glucagon secretion in two patients with Zollinger-Ellison syndrome. J Endocrinol Invest. 1981;4:451–453. doi: 10.1007/BF03348310. [DOI] [PubMed] [Google Scholar]
- Faiss S, Pape UF, Bohmig M, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors--the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. 2003;21:2689–2696. doi: 10.1200/JCO.2003.12.142. [DOI] [PubMed] [Google Scholar]
- Fazio N, de Braud F, Delle Fave G, et al. Interferon-alpha and somatostatin analog in patients with gastroenteropancreatic neuroendocrine carcinoma: single agent or combination? Ann Oncol. 2007;18:13–19. doi: 10.1093/annonc/mdl144. [DOI] [PubMed] [Google Scholar]
- Forrer F, Valkema R, Kwekkeboom DJ, et al. Neuroendocrine tumors. eptide receptor radionuclide therapy. Best Pract Res Clin Endocrinol Metab. 2007;21:111–129. doi: 10.1016/j.beem.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Van Essen M, Krenning EP, de Jong M, et al. Peptide Receptor Radionuclide Therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007;46:723–734. doi: 10.1080/02841860701441848. [DOI] [PubMed] [Google Scholar]
- Kwekkeboom DJ, Teunissen JJ, Kam BL, et al. Treatment of patients who have endocrine gastroenteropancreatic tumors with radiolabeled somatostatin analogues. Hematol Oncol Clin North Am. 2007;21:561–573. doi: 10.1016/j.hoc.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Virgolini I, Traub T, Novotny C, et al. Experience with indium-111 and yttrium-90-labeled somatostatin analogs. Curr Pharm Des. 2002;8:1781–1807. doi: 10.2174/1381612023393756. [DOI] [PubMed] [Google Scholar]
- Kaltsas GA, Papadogias D, Makras P, et al. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr Relat Cancer. 2005;12:683–699. doi: 10.1677/erc.1.01116. [DOI] [PubMed] [Google Scholar]
- Kwekkeboom DJ, Mueller-Brand J, Paganelli G, et al. Overview of results of peptide receptor radionuclide therapy with 3 radiolabeled somatostatin analogs. J Nucl Med. 2005;46(Suppl 1):62S–66S. [PubMed] [Google Scholar]
- Valkema R, Pauwels S, Kvols LK, et al. Survival and Response After Peptide Receptor Radionuclide Therapy With [(90)Y-DOTA(0),Tyr(3)]Octreotide in Patients With Advanced Gastroenteropancreatic Neuroendocrine Tumors. Semin Nucl Med. 2006;36:147–156. doi: 10.1053/j.semnuclmed.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754–2762. doi: 10.1200/JCO.2005.08.066. [DOI] [PubMed] [Google Scholar]
- O'Toole D, Ruszniewski P. Chemoembolization and other ablative therapies for liver metastases of gastrointestinal endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:585–594. doi: 10.1016/j.bpg.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Toumpanakis C, Meyer T, Caplin ME. Cytotoxic treatment including embolization/chemoembolization for neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:131–144. doi: 10.1016/j.beem.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Arnold R, Trautmann ME, Creutzfeldt W, et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430–438. doi: 10.1136/gut.38.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszniewski P, O'Toole D. Ablative therapies for liver metastases of gastroenteropancreatic endocrine tumors. Neuroendocrinology. 2004;80 Suppl 1:74–78. doi: 10.1159/000080746. [DOI] [PubMed] [Google Scholar]
- Ramage JK, Davies AH, Ardill J, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2005;54 Suppl 4:iv1–16. doi: 10.1136/gut.2004.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venook AP. Embolization and chemoembolization therapy for neuroendocrine tumors. Curr Opin Oncol. 1999;11:38–41. doi: 10.1097/00001622-199901000-00009. [DOI] [PubMed] [Google Scholar]
- Gupta S, Johnson MM, Murthy R, et al. Hepatic arterial embolization and chemoembolization for the treatment of patients with metastatic neuroendocrine tumors: variables affecting response rates and survival. Cancer. 2005;104:1590–1602. doi: 10.1002/cncr.21389. [DOI] [PubMed] [Google Scholar]
- Osborne DA, Zervos EE, Strosberg J, et al. Improved outcome with cytoreduction versus embolization for symptomatic hepatic metastases of carcinoid and neuroendocrine tumors. Ann Surg Oncol. 2006;13:572–581. doi: 10.1245/ASO.2006.03.071. [DOI] [PubMed] [Google Scholar]
- Ho AS, Picus J, Darcy MD, et al. Long-term outcome after chemoembolization and embolization of hepatic metastatic lesions from neuroendocrine tumors. AJR Am J Roentgenol. 2007;188:1201–1207. doi: 10.2214/AJR.06.0933. [DOI] [PubMed] [Google Scholar]
- Sarmiento JM, Que FG. Hepatic surgery for metastases from neuroendocrine tumors. Surg Oncol Clin North Am. 2003;12:231–242. doi: 10.1016/s1055-3207(02)00076-5. [DOI] [PubMed] [Google Scholar]
- Falconi M, Bettini R, Boninsegna L, et al. Surgical strategy in the treatment of pancreatic neuroendocrine tumors. JOP. 2006;7:150–156. [PubMed] [Google Scholar]
- Carty SE, Jensen RT, Norton JA. Prospective study of aggressive resection of metastatic pancreatic endocrine tumors. Surgery. 1992;112:1024–1031. [PubMed] [Google Scholar]
- Sarmiento JM, Heywood G, Rubin J, et al. Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival. J Am Coll Surg. 2003;197:29–37. doi: 10.1016/S1072-7515(03)00230-8. [DOI] [PubMed] [Google Scholar]
- House MG, Cameron JL, Lillemoe KD, et al. Differences in survival for patients with resectable versus unresectable metastases from pancreatic islet cell cancer. J Gastrointest Surg. 2006;10:138–145. doi: 10.1016/j.gassur.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Musunuru S, Chen H, Rajpal S, et al. Metastatic neuroendocrine hepatic tumors: resection improves survival. Arch Surg. 2006;141:1000–1004. doi: 10.1001/archsurg.141.10.1000. [DOI] [PubMed] [Google Scholar]
- Moug SJ, Leen E, Horgan PG, et al. Radiofrequency ablation has a valuable therapeutic role in metastatic VIPoma. Pancreatology. 2006;6:155–159. doi: 10.1159/000090257. [DOI] [PubMed] [Google Scholar]
- Mazzaglia PJ, Berber E, Milas M, et al. Laparoscopic radiofrequency ablation of neuroendocrine liver metastases: a 10-year experience evaluating predictors of survival. Surgery. 2007;142:10–19. doi: 10.1016/j.surg.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Gillams A, Cassoni A, Conway G, et al. Radiofrequency ablation of neuroendocrine liver metastases: the Middlesex experience. Abdom Imaging. 2005;30:435–441. doi: 10.1007/s00261-004-0258-4. [DOI] [PubMed] [Google Scholar]
- Elvin A, Skogseid B, Hellman P. Radiofrequency ablation of neuroendocrine liver metastases. Abdom Imaging. 2005;30:427–434. doi: 10.1007/s00261-004-0257-5. [DOI] [PubMed] [Google Scholar]
- Arnold R, Rinke A, Schmidt C, et al. Chemotherapy. Best Pract Res Clin Gastroenterol. 2005;19:649–656. doi: 10.1016/j.bpg.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Moertel CG, Hanley JA, Johnson LA. Streptozotocin alone compared with streptozotocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189–1194. doi: 10.1056/NEJM198011203032101. [DOI] [PubMed] [Google Scholar]
- Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozotocin-doxorubicin, streptozotocin-flourouracil or chlorozotocin in the treatment of advanced islet cell carcinoma. N Engl J Med. 1992;326:519–523. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- von Schrenck T, Howard JM, Doppman JL, et al. Prospective study of chemotherapy in patients with metastatic gastrinoma. Gastroenterology. 1988;94:1326–1334. doi: 10.1016/0016-5085(88)90670-1. [DOI] [PubMed] [Google Scholar]
- Cheng PN, Saltz LB. Failure to confirm major objective antitumor activity for streptozocin and doxorubicin in the treatment of patients with advanced islet cell carcinoma. Cancer. 1999;86:944–948. [PubMed] [Google Scholar]
- Ron D, Kazanietz MG. New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999;13:1658–1676. [PubMed] [Google Scholar]
- Moertel CG, Kvols LK, O'Connell MJ, et al. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68:227–232. doi: 10.1002/1097-0142(19910715)68:2<227::aid-cncr2820680202>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Hainsworth JD, Spigel DR, Litchy S, et al. Phase II trial of paclitaxel, carboplatin, and etoposide in advanced poorly differentiated neuroendocrine carcinoma: a Minnie Pearl Cancer Research Network Study. J Clin Oncol. 2006;24:3548–3554. doi: 10.1200/JCO.2005.05.0575. [DOI] [PubMed] [Google Scholar]
- Fjallskog ML, Lejonklou MH, Oberg KE, et al. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin Cancer Res. 2003;9:1469–1473. [PubMed] [Google Scholar]
- Wulbrand U, Wied M, Zofel P, et al. Growth factor receptor expression in human gastroenteropancreatic neuroendocrine tumours. Eur J Clin Invest. 1998;28:1038–1049. doi: 10.1046/j.1365-2362.1998.00397.x. [DOI] [PubMed] [Google Scholar]
- Peghini PL, Iwamoto M, Raffeld M, et al. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin Cancer Res. 2002;8:2273–2285. [PubMed] [Google Scholar]
- Furukawa M, Raffeld M, Mateo C, et al. Increased expression of insulin-like growth factor I (IGF-1) and/or its receptor (IGF-1R) in gastrinomas is associated with low curability, increased growth and development of metastases. 2004 doi: 10.1158/1078-0432.CCR-04-1915. [DOI] [PubMed] [Google Scholar]
- Yao JC. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab. 2007;21:163–172. doi: 10.1016/j.beem.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Strosberg JR, Kvols LK. A review of the current clinical trials for gastroenteropancreatic neuroendocrine tumours. Expert Opin Investig Drugs. 2007;16:219–224. doi: 10.1517/13543784.16.2.219. [DOI] [PubMed] [Google Scholar]
- Chan JA, Kulke MH. Emerging therapies for the treatment of patients with advanced neuroendocrine tumors. Expert Opin Emerg Drugs. 2007;12:253–270. doi: 10.1517/14728214.12.2.253. [DOI] [PubMed] [Google Scholar]
- Yao JC, Zhang JX, Rashid A, et al. Clinical and in vitro studies of imatinib in advanced carcinoid tumors. Clin Cancer Res. 2007;13:234–240. doi: 10.1158/1078-0432.CCR-06-1618. [DOI] [PubMed] [Google Scholar]
- Hopfner M, Sutter AP, Gerst B, et al. A novel approach in the treatment of neuroendocrine gastrointestinal tumours. Targeting the epidermal growth factor receptor by gefitinib (ZD1839). Br J Cancer. 2003;89:1766–1775. doi: 10.1038/sj.bjc.6601346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulke M, Lenz HJ, Meropol NJ, et al. A phase 2 study to evaluate the efficacy and safety of SU11248 in patients with unresectable neuroendocrine tumors (NET): a Phase 11 consortium (P2C) study. 23 ed. 2005. p. 310s. [Google Scholar]
- Hobday TJ, Holen K, Donehower R, et al. A phase II trial of gefitinib in patients with progressive metastatic neuroendocrine tumors: a Phase II consortium study. (24 ed.) 2006:A4043. [Google Scholar]
- Duran I, Kortmansky J, Singh D, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–1154. doi: 10.1038/sj.bjc.6603419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmig BN. Radiotherapy for gastroenteropancreatic neuroendocrine tumors. Ann N Y Acad Sci. 1994;733:488–495. doi: 10.1111/j.1749-6632.1994.tb17299.x. [DOI] [PubMed] [Google Scholar]
- Strosberg J, Hoffe S, Gardner N, et al. Effective treatment of locally advanced endocrine tumors of the pancreas with chemoradiotherapy. Neuroendocrinology. 2007;85:216–220. doi: 10.1159/000102969. [DOI] [PubMed] [Google Scholar]
- Pascher A, Klupp J, Neuhaus P. Transplantation in the management of metastatic endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:637–648. doi: 10.1016/j.bpg.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Olausson M, Friman S, Herlenius G, et al. Orthotopic liver or multivisceral transplantation as treatment of metastatic neuroendocrine tumors. Liver Transpl. 2007;13:327–333. doi: 10.1002/lt.21056. [DOI] [PubMed] [Google Scholar]
- Le Treut YP, Delpero JR, Dousset B, et al. Results of liver transplantation in the treatment of metastatic neuroendocrine tumors. A 31-case French multicentric report. Ann Surg. 1997;225:355–364. doi: 10.1097/00000658-199704000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert T. Liver transplantation for metastatic neuroendocrine carcinoma. Transplantation. 1998;66:1307–1312. doi: 10.1097/00007890-199811270-00007. [DOI] [PubMed] [Google Scholar]
- Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- de Herder WW, O'Toole D, Rindi G, et al. ENETS Consensus Guidelines for the Diagnosis and Treatment of Neuroendocrine Gastrointestinal Tumors: Part 2-Midgut and Hindgut tumors. Neuroendocrinology. 2008;87:1–63. [Google Scholar]
- Rindi G, Kloppel G, Couvelard A, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2007 doi: 10.1007/s00428-007-0452-1. [DOI] [PubMed] [Google Scholar]
- Kaplan EL, Horvath K, Udekwu A, et al. Gastrinomas: a 42-year experience. World J Surg. 1990;14:365–375. doi: 10.1007/BF01658530. [DOI] [PubMed] [Google Scholar]
- Farley DR, Van Heerden JA, Grant CS, et al. The Zollinger-Ellison syndrome. A collective surgical experience. Ann Surg. 1992;215:561–569. doi: 10.1097/00000658-199206000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignon M, Cadiot G. Diagnostic and therapeutic criteria in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. J Intern Med. 1998;243:489–494. doi: 10.1046/j.1365-2796.1998.00287.x. [DOI] [PubMed] [Google Scholar]
- Galbut DL, Markowitz AM. Insulinoma: diagnosis, surgical management and long-term follow- up. Review of 41 cases. Am J Surg. 1980;139:682–690. doi: 10.1016/0002-9610(80)90363-3. [DOI] [PubMed] [Google Scholar]
- Dizon AM, Kowalyk S, Hoogwerf BJ. Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med. 1999;106:307–310. doi: 10.1016/s0002-9343(99)00021-2. [DOI] [PubMed] [Google Scholar]
- Soga J, Yakuwa Y, Osaka M. Insulinomas/hypoglycemic syndrome: a statistical evaluation of 1085 reported cases of a Japanese series. J Exp Clin Cancer Res. 1998;17:379–388. [PubMed] [Google Scholar]
- Fajans SS, Vinik AI. Insulin-producing islet cell tumors. Endocrinol Metab Clin North Am. 1989;18:45–74. [PubMed] [Google Scholar]
- Wiedmann MW, Caca K. Molecularly targeted therapy for gastrointestinal cancer. Curr Cancer Drug Targets. 2005;5:171–193. doi: 10.2174/1568009053765771. [DOI] [PubMed] [Google Scholar]
- Smolewski P. Recent developments in targeting the mammalian target of rapamycin (mTOR) kinase pathway. Anticancer Drugs. 2006;17:487–494. doi: 10.1097/00001813-200606000-00001. [DOI] [PubMed] [Google Scholar]
- Dancey JE. Therapeutic targets: MTOR and related pathways. Cancer Biol Ther. 2006;5:1065–1073. doi: 10.4161/cbt.5.9.3175. [DOI] [PubMed] [Google Scholar]
- Rocha-Lima CM, Soares HP, Raez LE, et al. EGFR targeting of solid tumors. Cancer Control. 2007;14:295–304. doi: 10.1177/107327480701400313. [DOI] [PubMed] [Google Scholar]
- Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer. 2004;11:689–708. doi: 10.1677/erc.1.00600. [DOI] [PubMed] [Google Scholar]