

Abstract
Transfer-messenger RNA (tmRNA) and protein SmpB facilitate trans-translation, a quality-control process that tags truncated proteins with short peptides recognized by a number of proteases and recycles ribosomes stalled at the 3′ end of mRNA templates lacking stop codons. The tmRNA molecule is a hybrid of tRNA- and mRNA-like domains that are usually connected by four pseudoknots (pk1–pk4). Replacement of pk1 with a single-stranded RNA yields pk1L, a mutant tmRNA that tags truncated proteins very poorly in vitro but very efficiently in vivo. However, deletion of the whole pk1 is deleterious for protein tagging. In contrast, deletion of helix 4 yields Δh4, a fully functional tmRNA derivative containing a single hairpin instead of pk1. Further deletions in the pk1 segment yield two subclasses of mutant tmRNAs that are unable to tag truncated proteins, but some of them bind to stalled ribosomes. Our studies demonstrate that pk1 is not essential for tmRNA functions but contributes to the stability of the tmRNA structure. Our studies also indicate that the length of this RNA segment is critical for both tmRNA binding to the ribosome and resumption of translation.
Keywords: pseudoknot, SmpB, tmRNA, protein tagging, trans-translation
INTRODUCTION
Translation of mRNA templates that lack stop codons produces truncated proteins. In bacteria, the process of trans-translation evolved to remove these proteins and release stalled ribosomes from defective mRNA templates (Moore and Sauer 2007). Both reactions are facilitated by transfer-messenger RNA (tmRNA), a small stable RNA molecule encoded by the ssrA gene. To date, ssrA genes have been identified in bacterial, chloroplast, and mitochondrial genomes (Gueneau de Novoa and Williams 2004; Andersen et al. 2006). Some ssrA genes are circularly permuted and encode tmRNAs composed of two RNA molecules (Keiler et al. 2000; Williams 2002).
Structure probing of Escherichia coli tmRNA and comparative analyses of available tmRNA sequences revealed the presence of three distinct domains (Fig. 1; Williams and Bartel 1996; Felden et al. 1997; Zwieb et al. 1999). The 3′ and 5′ termini of tmRNA form the tRNA-like domain (TLD). The resume and stop codons demarcate an mRNA-like domain (MLD) that encodes tag peptides varying in length from 10 to 35 amino acid residues. A considerable portion of tmRNA folds into H-type pseudoknots. Most bacterial tmRNAs, such as the prototypical E. coli tmRNA, have four pseudoknots (pk1–pk4).
FIGURE 1.

Structural features of the E. coli tmRNA(H8hp). Schematic representation of the secondary structure of tmRNA(H8hp) composed of the tRNA-like domain (TLD), the mRNA-like domain (MLD) encoding the ANH8 peptide, and four pseudoknots (pk1–pk4). Helices are numbered 1–12. Open and black circles highlight the resume codon (GCA) and two stop codons (UAA–UAA).
The TLD, MLD, and pk1 contact the ribosome, whereas the segment consisting of pk2, pk3, and pk4 forms an arc that remains outside the ribosome during the initiation step of trans-translation (Valle et al. 2003). The ribosome readily translates the pseudoknot-rich segment consisting of pk2, pk3, and pk4 when the natural stop codons of tmRNA are replaced with sense codons (Wower et al. 2005). The functionalities of tmRNA on the ribosome are facilitated by three protein factors. Elongation factor Tu (EF-Tu) delivers tmRNA to the ribosome as an EF-Tu·GTP:aminoacyl-tmRNA ternary complex (Rudinger-Thirion et al. 1999; Barends et al. 2000). Protein SmpB binds to the TLD and stalled ribosomes (Karzai et al. 1999; Wower et al. 2002; Hallier et al. 2004; Konno et al. 2007). The crystal structure of SmpB in complex with the entire TLD domain of the tmRNA from Thermus thermophilus suggests that SmpB substitutes for the missing D and anticodon stems (Bessho et al. 2007). Ribosomal protein S1 binds to the MLD and the pk2–pk4 segment, but its functions in trans-translation are not well characterized (Wower et al. 2000; McGinness and Sauer 2004).
Many lines of experimental evidence indicate that the TLD and MLD are essential for tmRNA functions in trans-translation (Moore and Sauer 2007). While pk2, pk3, and pk4 are implicated in the proper overall folding of tmRNA, they are dispensable for trans-translation (Nameki et al. 2000; Wower et al. 2004). Individual disruptions of pk2, pk3, and pk4 in E. coli tmRNA decrease its tagging activity only moderately. Because replacement of pk1 with a single-stranded RNA renders tmRNA inactive in the tagging of truncated proteins both in vivo and in vitro, pk1 was suggested to be essential for tmRNA functions (Nameki et al. 1999b; Tanner et al. 2006). This suggestion is not supported by comparative analyses of tmRNA sequences, which indicate that pk1 is reduced to a single hairpin in some bacterial tmRNAs (Zwieb et al. 1999; Gaudin et al. 2002; Sharkady and Williams 2004). Moreover, the replacement of pk1 with a stable hairpin yields a mutant tmRNA with nearly wild-type tagging activity (Tanner et al. 2006).
We have developed very efficient and sensitive assays for testing functionality of the tmRNA domains both in vitro and in vivo (Wower et al. 2004). In these assays, we use tmRNA(H8hp), a fully functional derivative of E. coli tmRNA in which the naturally occurring tag coding region has been replaced by a series of histidine codons (Fig. 1). TmRNA(H8hp) tags truncated ribosomal protein L27 with a histidine-rich polypeptide (ANH8) to yield a fusion protein that is resistant to proteolysis. This fusion protein can be easily detected in fractionated E. coli lysates by staining with Coomassie Blue or Western blot analysis with anti-His-tag antibodies. Here, we have used these assays to investigate the contributions of pk1 to tmRNA structure and functions. Our studies demonstrated that pk1, like the other three pseudoknots, is dispensable in E. coli tmRNA. However, pk1 may contribute significantly to the structural stability of tmRNA by sequestering a significant portion of an RNA segment that connects the TLD and MLD and thus preventing formation of alternative structures. Moreover, our studies revealed that the length of this RNA segment plays a very important role in the binding of tmRNA to the ribosome and mediating its protein tagging activity.
RESULTS
TmRNAs bearing mutations in pk1
Comparative sequence analyses of numerous tmRNAs and chemical probing of E. coli tmRNA revealed that the segment encompassing nucleotides 49–78 folds into an H-type pseudoknot denoted as pk1 in Figure 1 (Williams and Bartel 1996; Felden et al. 1997; Zwieb et al. 1999). The extent to which pk1 contributes to tmRNA structure and functions is not yet fully understood. A number of studies suggested that pk1 is essential for tmRNA folding and protein tagging because the replacement of pk1 with single-stranded RNAs yielded tmRNA derivatives that failed to tag both in vitro (Nameki et al. 1999a,b) and in vivo (Tanner et al. 2006). However, comparative sequence analyses identified tmRNAs that lack pk1, whereas genetic screens identified hairpin structures that functionally replace pk1 (Zwieb et al. 1999; Gueneau de Novoa and Williams 2004; Burks et al. 2005; Tanner et al. 2006). Whether the tmRNAs lacking pk1 are active in protein tagging in vivo and to what extent alterations of pk1 affect tmRNA processing, aminoacylation, and binding to proteins and ribosomes remain unknown.
To determine the roles of pk1 in these important functions, we introduced a number of mutations into tmRNA(H8hp) (see Fig. 1), an E. coli tmRNA derivative designed to direct the synthesis of ribosomal protein L27 with a protease-resistant peptide tag ANH8 (Wower et al. 2004). The secondary structures of the wild-type pk1 and its variants are illustrated in Figure 2. The most extensive alterations involved deletions of the entire pk1 (nucleotides 49–78), the central part of pk1 (nucleotides 54–72), the 5′ part of helix 4 (nucleotides 54–62), and the remaining 3′ part of helix 4 (nucleotides 69–78). These alterations produced tmRNA derivatives denoted henceforth as Δpk1, pk1L, Δh4, and Δh4/Δ69–78, respectively (Fig. 2). To promote formation of helix 3 (h3) in the Δh4 derivative, h3 was capped with a GAGA tetraloop. Primer extension of Δpk1, pk1L, and Δh4 probed with dimethyl sulfate (DMS), which modifies Watson–Crick pairing positions in adenine and cytosine, revealed that the RNA segments replacing pk1 remained single stranded as predicted. Probing data for the TLD domain in Δpk1, pk1L, Δh4 (Fig. 3), and in tmRNA(H8hp) (not shown) were identical and strongly suggested that, despite extensive alterations in the pk1 region, the 3′ and 5′ termini of mutant tmRNAs were properly folded.
FIGURE 2.

Pk1 of the E. coli tmRNA and its mutants. Pk1 is shown with abutting helix 2d. An open square and an open circle denote nucleotides C44 and C66, respectively. Open rectangles highlight mutations of G50C to C50G base pair. Open ovals mark the resume codon (GCA). Arrows indicate deletion sites of nucleotides 54–72 and 69–78 in the pk1L and Δh4/Δ69–78 mutants, respectively. Slanted lines mark deletions of a single base pair, 2 bp, and 3 bp in Δh4/Δ1p, Δh4/Δ2p, and Δh4/Δ3p, respectively. Numbering is according to E. coli tmRNA.
FIGURE 3.

Chemical probing of the E. coli tmRNA(H8hp) derivatives bearing mutations in pk1. Examples of sequencing gels displaying nucleotides modified by DMS in (A) Δpk1, (B) pk1L, and (C) Δh4 mutants. (Lanes C,U,A,G) Sequencing ladders generated by reverse transcription of tmRNA mutants in the presence of ddGTP, ddATP, ddTTP, and ddCTP, respectively. (Lane K) Incubation controls; (lane D) probing under semidenaturing conditions; (lane N) probing under native conditions. Nucleotides modified under native conditions are indicated by black bars both on the autoradiograms and in the secondary structures of the tmRNA mutants.
Recently, Tanner et al. (2006) identified 6–8 base-pair (bp) hairpin structures that could functionally replace pk1. The investigators suggested that the increased stability of the hairpin replacing pk1 plays a key role in the function of tmRNA mutants, whereas the specific nucleotide sequence of the hairpin is not important. However, hairpins that replace pk1 in the presumably functional tmRNAs from Mycoplasma pneumoniae, Prochlorococcus marinus, Gloeobacter violaceus, and Synechococcus WH8102 are predicted to contain only 5 bp (Andersen et al. 2006). To test whether a stable hairpin is indeed important for tmRNA functions, we sequentially deleted 1, 2, and 3 bp from helix 3 in mutant Δh4 to generate tmRNA derivatives denoted as Δh4/Δ1p, Δh4/Δ2p, and Δh4/Δ3p, respectively (Fig. 2). The deleted base pairs correspond to G50–C66, A51–U65, and G52–C64 in E. coli tmRNA.
A tertiary interaction between the C44 nucleotide in helix 2d and the C66 nucleotide in helix 3 of pk1 was proposed by covariation analysis (Kelley et al. 2001). C44 and C66 are also thought to form canonical base pairs with G307 and G50, respectively (Zwieb et al. 1999; Kelley et al. 2001). To investigate the importance of C44 and C66 residues in the tmRNA structure, we prepared a tmRNA mutant denoted as G50C–C66G, in which the base pair G50–C66 was replaced with C50–G66 (Fig. 2). Because the C66 residue is absent in pk1L and replaced by U in Δh4/Δ1p, functional analysis of these two mutant tmRNA was expected to yield additional information about the proposed C44:C66 tertiary interaction.
Monitoring tagging activity of mutant tmRNAs in vivo and in vitro
Tagging of truncated ribosomal protein L27 by tmRNA derivatives bearing mutations in pk1 was tested in IW363, an ssrA − derivative of E. coli strain BL21(DE3)pLysS (Wower et al. 2004). When expressed from plasmid pETrpmA-At-3, containing ssrA and smpB under the control of their natural promoters, all tmRNA mutants except Δpk1, Δh4/Δ69–78, and Δh4/Δ2p were able to tag the truncated ribosomal protein L27 in vivo as efficiently as tmRNA(H8hp) (Fig. 4A; see also Table 1).
FIGURE 4.

In vivo and in vitro tagging of truncated ribosomal protein L27 by tmRNA(H8hp) and its derivatives bearing mutations in the pk1. (A) In vivo tagging: Cell lysates from IPTG-induced E. coli IW363 cells coexpressing truncated protein L27, protein SmpB, and mutant tmRNAs were fractionated on a 12.5% SDS-polyacrylamide gel. Tagged proteins were detected by staining the gel with Coomassie Blue. (B) In vitro tagging: Reactions were assembled by addition of circular plasmid pETrpmA-At-1, tmRNA(H8hp) or its mutants, His-tagged E. coli protein SmpB, and E. coli alanyl-tRNA synthetase to the T7 transcription/translation mixture (Promega). After 60 min of incubation at 37°C, His-tagged proteins were captured on Ni-NTA-Sepharose and then fractionated on a 12.5% SDS-polyacrylamide gel. Tagging was visualized by Western blotting with anti-His-tag antibodies. SmpB-H6, ΔL27, and L27* denote His-tagged SmpB, truncated protein L27, and tagged protein L27, respectively.
TABLE 1.
Activities of mutant tmRNAs

Our earlier studies demonstrated that certain tmRNA mutations abolish in vivo tagging indirectly by inhibiting the processing of tmRNA precursors (Wower et al. 2004). To exclude this possibility, we monitored tagging activities of tmRNA(H8hp) and some of its mutants in vitro. Purified transcripts of tmRNA derivatives were added to a transcription–translation system that produced truncated ribosomal protein L27 by decoding an mRNA template lacking stop codons. Because in vitro tagging was less efficient than the in vivo reaction, formation of the tagged protein L27 was monitored by fractionation of the reaction products on SDS-polyacrylamide gels followed by Western blotting with anti-His-tag antibodies.
Preliminary experiments demonstrated that tmRNA(H8hp) and all its mutant derivatives, except pk1L, displayed the same tagging activities both in vitro and in vivo. Only pk1L, which tags in our assay efficiently in vivo (Fig. 4A), did not tag in vitro (not shown). The tagging inactivity that pk1L displayed in vitro was consistent with earlier studies of (Nameki et al. 1999b, 2000). In contrast, the strong tagging activity of pk1L in vivo was in conflict with studies that used the phage efficiency of plating assay to demonstrate that pk1L is completely inactive in vivo (Tanner et al. 2006).
In our previous studies we demonstrated that the addition of protein SmpB to the in vitro transcription/translation system increases the tagging activity of mutant tmRNAs pk2L, pk3L, and pk4L, in which pk2, pk3, and pk4 were replaced with single-stranded RNAs (Wower et al. 2004). In the same studies, we also demonstrated that coexpression of these mutant tmRNAs with SmpB encoded in plasmid pETrpmA-At-3 improves the tagging activity significantly. Therefore, we hypothesized that tagging inactivity of pk1L observed by Tanner and Nameki may be affected by the limited availability of SmpB in their experimental systems (Nameki et al. 1999b, 2000; Tanner et al. 2006). To test our hypothesis, we supplemented the in vitro trans-translational reaction mixture with a purified His-tagged SmpB protein. The addition of SmpB only partially ameliorated defects of pk1L (see Fig. 4B). As in our previous studies (Wower et al. 2004), the most efficient in vitro tagging was observed when pk1L and SmpB were provided in equimolar quantities. In contrast, the addition of SmpB had no effect on Δpk1, Δh4/Δ69–78, and Δh4/Δ2p, which remained inactive.
The observations described above are consistent with the essential role of SmpB in trans-translation (Karzai et al. 1999) and provide a plausible explanation for the inactivity of pk1L reported in earlier studies (Nameki et al. 1999b, 2000; Tanner et al. 2006). Our experiments also show that protein SmpB alone cannot compensate for deficiencies of this mutant.
Processing of tmRNA mutants
In E. coli, the ssrA gene encodes a 457-nucleotide (nt) precursor that is processed to the mature 363-nt tmRNA by RNases BN, D, E, III, P, PH, and T (Li et al. 1998; Nameki et al. 2000). To test whether modifications in the pk1 region affect tmRNA maturation, we isolated total RNA from IW363 cells before and after induction of the synthesis of truncated protein L27 by IPTG. The RNAs were examined by Northern blot analysis with a [32P]-labeled oligonucleotide probe complementary to nucleotides 125–139 in E. coli tmRNA. As shown in Figure 5, E. coli was able to process all tmRNA derivatives bearing mutations in the pk1 region, albeit with different efficiencies. Processing of precursor Δh4/Δ69–78 was particularly efficient (>95%). Because the levels of mature tmRNA(H8hp) and mutant tmRNAs were similar, it seems unlikely that the tagging inactivity displayed by the Δpk1, Δh4/Δ2p, and Δh4/Δ69–78 mutant tmRNAs in vivo was caused by defective processing.
FIGURE 5.

Processing of tmRNA(H8hp) and its derivatives bearing mutations in the pk1. Northern blot analysis of 1 μg of total RNA extracted from uninduced (−) and IPTG-induced (+) E. coli IW363 cells, fractionated on a 5% denaturing polyacrylamide gel and blotted to a Zeta-probe membrane. [32P]-Labeled oligonucleotide 5′-GGCATCATCATCGT-3′ complementary to E. coli tmRNA residues 125–139 was hybridized to both precursor tmRNAs (p) and mature tmRNAs (t).
Aminoacylation of mutant tmRNAs
To bind to stalled ribosomes, E. coli tmRNA has to be aminoacylated by alanyl-tRNA synthetase and form a ternary complex with EF-Tu and GTP. We tested whether modifications of pk1 inhibit aminoacylation of tmRNA mutants using an in vitro assay developed by Varshney et al. (1991). TmRNA transcripts were [32P]-labeled at their 3′ end with yeast ATP/CTP tRNA nucleotidyl transferase in the presence of [α-32P]ATP and fractionated on a nondenaturing polyacrylamide gel to remove unincorporated [α-32P]ATP. The vast majority of Δpk1 and Δh4 RNAs migrated like tmRNA(H8hp). In contrast, a significant fraction of pk1L migrated more slowly than tmRNA(H8hp), suggesting formation of an alternative stable conformer (Fig. 6A,B). This conformational heterogeneity might account for the poor tagging activity that pk1L displayed in vitro (Fig. 4B).
FIGURE 6.

Fractionation of tmRNA mutants. (A) TmRNAs were transcribed in vitro, radioactively labeled at the 3′ end by incubation with [α-32P]ATP and ATP/CTP tRNA nucleotidyl transferase, and fractionated by electrophoresis on a native 5% polyacrylamide gel. α and β denote tmRNA conformers. (B) Graphical representation of data derived from the polyacrylamide gel shown in A.
Each 3′-[32P]-labeled tmRNA conformer was extracted from polyacrylamide gel (Fig. 6), charged with alanine in the presence of E. coli alanyl-tRNA synthetase, and then digested with RNase T1. This treatment released radioactively labeled CUCCACCA-Ala and CUCCACCA oligonucleotides, which were separated by electrophoresis on a denaturing 15% polyacrylamide gel at acidic pH to inhibit the deaminoacylation. Figure 7 shows examples of aminoacylation patterns for tmRNA(H8hp) and Δpk1. Typically, 60%–70% of the [32P]-labeled tmRNA(H8hp) could be charged in vitro. Δpk1α and Δh4α always aminoacylated more efficiently than tmRNA(H8hp) (Fig. 7B, ∼80%–95%). In contrast, both conformers of the pk1L mutant aminoacylated poorly. Only up to 20% of pk1L could be charged under standard conditions (Fig. 7B). However, the charging of both pk1L conformers increased up to 65%, when the aminoacylation reaction was carried out in the presence of SmpB (Fig. 7C). This observation was consistent with our earlier studies, which demonstrated that SmpB stimulates tmRNA aminoacylation when present at a 1:1 ratio with tmRNA (Barends et al. 2000, 2001).
FIGURE 7.
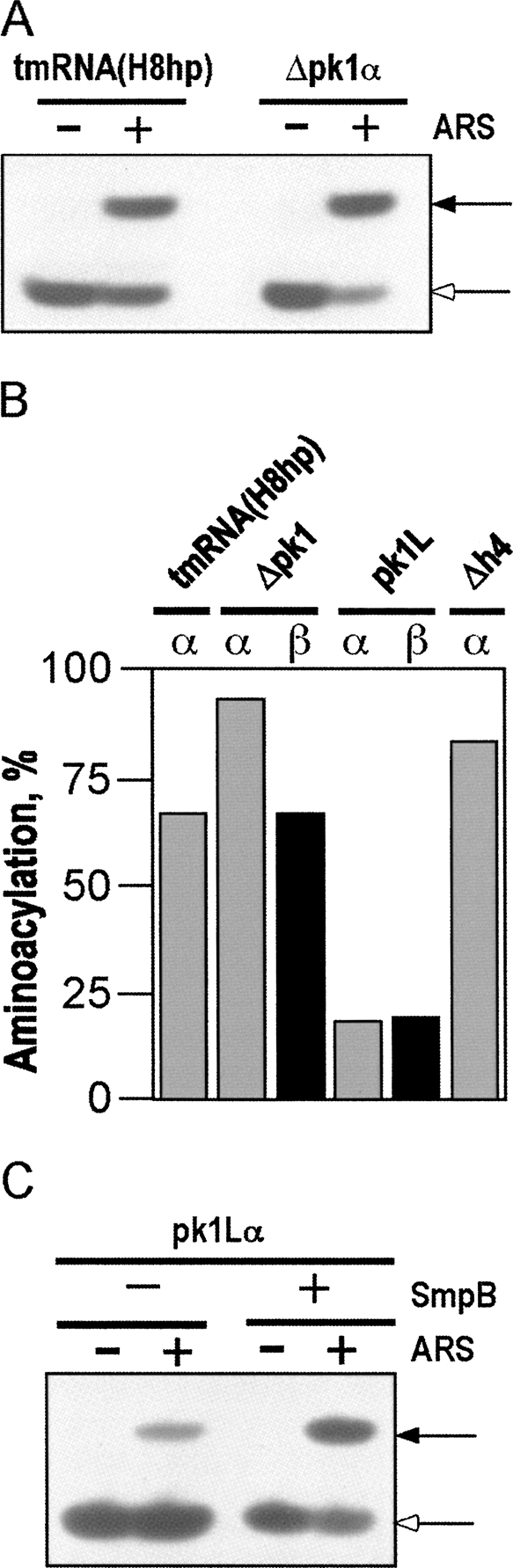
Aminoacylation of tmRNA(H8hp) and its derivatives bearing mutations in the pk1. The aminoacylated and nonaminoacylated [32P]-labeled tmRNAs were digested with RNase T1 to yield CUCCACCA-Ala and CUCCACCA, respectively, and were separated by PAGE according to Varshney et al. (1991). (A) Aminoacylation of tmRNA(H8hp) and Δpk1α (representative examples). (B) Graphical representation of PhosphorImager-derived data from the analysis of aminoacylation of selected tmRNA mutants. (C) Aminoacylation of pk1Lα in the presence and in the absence of His-tagged protein SmpB. Aminoacylation was quantified using a Typhoon PhosphorImager. (+) and (−) indicate the presence and the absence of the alanyl-tRNA synthetase (ARS) and protein SmpB. Solid and open arrows show the positions of [32P]-labeled CUCCACCA-Ala and CUCCACCA, respectively.
Binding of protein SmpB to tmRNAs
Protein SmpB is an indispensable component of the trans-translational apparatus (Karzai et al. 1999). SmpB binds to TLD to compensate for the missing D and anticodon arms, enhances tmRNA processing and aminoacylation, and facilitates tmRNA binding to ribosomes (Barends et al. 2001; Wower et al. 2004; Bessho et al. 2007; Sundermeier and Karzai 2007). We investigated the ability of mutant tmRNAs to bind to the purified E. coli protein SmpB using gel mobility shift assays. Increasing amounts of SmpB protein were added to [32P]-labeled tmRNA and held at a constant concentration of 0.5 × 10−9 M until binding was saturated. Figure 8A shows, as an example, the binding of protein SmpB to pk1Lß.
FIGURE 8.

Gel-shift analysis of binding the SmpB protein to tmRNAs. The 3′-[32P]-labeled tmRNAs (10−9 M) were titrated with the E. coli SmpB protein. Aliquots of binding mixture were analyzed by electrophoresis on a 5% polyacrylamide gel in TGE buffer. Binding of SmpB to tmRNA derivatives was quantified using a Typhoon PhosphorImager. (A) Gel-shift analysis of pk1Lβ binding to SmpB. (B) Graphical representation of PhosphorImager-derived data illustrating the binding of pkL1β (open circles) and tmRNA(H8hp) (solid circles) to SmpB. Binding curves for other mutant tmRNAs were very similar to the tmRNA(H8hp) curve and therefore are not shown.
Our experiments demonstrated that all tmRNA mutants readily bound to protein SmpB. The binding of tmRNAs bearing mutations in pk1 to SmpB was saturable with half-maximal binding at free protein concentrations ranging approximately from 10 nM to 100 nM (Fig. 8). Similar K d was reported earlier for the binding of wild-type SmpB protein from E. coli to unmodified E. coli tmRNA (Karzai et al. 1999; Barends et al. 2001). These data provided strong evidence that alterations of pk1 do not disturb interactions between tmRNA and SmpB protein. Moreover, consistent with earlier studies that demonstrated that the TLD represents the primary SmpB binding site (Karzai et al. 1999; Barends et al. 2001; Wower et al. 2002), our studies suggested that the TLD is properly folded in all mutant tmRNAs.
TmRNA binding to ribosomes
TmRNA binds to E. coli ribosomes to form stable complexes. This process is carefully choreographed by the EF-Tu•GTP complex, which exclusively binds aminoacylated tmRNAs (Barends et al. 2001). SmpB also facilitates tmRNA binding to ribosomes, but this function of SmpB is less well understood (Karzai et al. 1999; Kaur et al. 2006). Because all tmRNAs bearing mutations in pk1 were aminoacylated and able to form complexes with SmpB protein, they were expected to bind to ribosomes.
To test whether alterations in pk1 affect tmRNA binding to ribosomes, we isolated ribosomes from IPTG-induced IW363 cells expressing either tmRNA(H8hp) or mutant tmRNAs by ultracentrifugation through a sucrose cushion as described in Materials and Methods. The ribosomes were extracted with phenol, and the RNAs were examined by Northern blot analysis with a [32P]-labeled oligonucleotide probe complementary to nucleotides 125–139 in E. coli tmRNA. As expected, tmRNA(H8hp) was able to bind to E. coli ribosomes (Fig. 9). Consistent with their tagging inactivity in vivo, the Δpk1 and Δh4/Δ2p mutants could not bind to ribosomes, suggesting that an unknown structural deficiency prevented these mutants tmRNAs from entering the A site of the ribosome. In contrast, Δh4/Δ69–78 bound to ribosomes as well as tmRNA(H8hp). However, because Δh4/Δ69–78 was unable to tag truncated proteins in vivo, this interaction was not functional.
FIGURE 9.

Binding of tmRNA(H8hp) and its derivatives bearing mutations in the pk1 to E. coli ribosomes in vivo. Northern blot analysis of 1 μg of RNA extracted from ribosomes isolated from IPTG-induced E. coli IW363 cells expressing either tmRNA(H8hp) or its mutant.
DISCUSSION
E. coli tmRNA does not require pk1 for its functions
Our assays revealed that the Δh4 mutant tags truncated protein both in vivo and in vitro as efficiently as wild-type tmRNA (Fig. 4, cf. A and B). Consistent with this observation, processing of precursor Δh4 was efficient (Fig. 5). Moreover, the mature Δh4 mutant aminoacylated better than tmRNA(H8hp) (Fig. 7B). These observations provided unambiguous proof that pk1 was not essential for trans-translation. Our finding is consistent with comparative analyses of tmRNA sequences that identified a number of bacterial tmRNAs, in which pk1 was replaced by a single hairpin (Zwieb and Wower 2000; Gaudin et al. 2002; Sharkady and Williams 2004; Andersen et al. 2006). In M. pneumoniae, P. marinus, and G. violaceus, the hairpin contained 5 bp. Our finding is also consistent with Tanner et al. (2006), who identified functional E. coli tmRNA mutants in which pk1 was replaced by a single hairpin containing either 6 or 8 bp. However, in the present study, we demonstrated that, contrary to earlier reports (Nameki et al. 1999b; Tanner et al. 2006), pk1 could be replaced either by a short hairpin (e.g., Δh4/Δ3p) or a single-stranded RNA (e.g., pk1L) without affecting the tagging activities of mutant tmRNAs.
Pk1 stabilizes tmRNA structure
Earlier studies suggested that pk1 is essential for tmRNA folding and protein tagging, because disruption of pk1 induced large structural rearrangements in tmRNA and inactivated protein tagging in vitro (Nameki et al. 1999a, b). In our studies, we were able to separate structural and functional effects caused by either the disruption or deletion of pk1. As shown in Figure 6A, the vast majority of Δh4 and Δpk1 migrated in a native polyacrylamide gel like tmRNA(H8hp), suggesting that the Δh4 and Δpk1 RNAs are as stable as wild-type tmRNA. In contrast, the pk1L mutant RNA displayed a propensity for misfolding. When fractionated on a native gel, one third of pk1L migrated more slowly than tmRNA(H8hp) (Fig. 6A). These observations indicated that, when the RNA segment connecting TLD and MLD was shortened as in Δpk1, the pseudoknot was dispensable for proper folding of the tmRNA molecule. In contrast, when the connecting RNA segment was as long as in tmRNA(H8hp) and Δh4, either a pseudoknot or a hairpin was required. Together, these observations suggested that the main role of pk1 is to sequester a part of the connecting RNA segment and prevent formation of alternative structures.
Tertiary interaction between C44 and C66 residues is not essential for E. coli tmRNA structure and functions
Covariation analysis of 131 tmRNA sequences provided evidence for a tertiary interaction involving the C44 residue in helix 2d and the C66 residue in pk1 (Kelley et al. 2001). The replacement of the 50G–C66 base pair with either C-G or A-U base pairs produced the G50C–C66G and Δh4/Δ1p mutants, respectively, that tagged both in vitro and in vivo as efficiently as wild-type tmRNA (Fig. 4A,B). Because the pk1L mutant, which lacks residue C66, was able to tag both in vivo and in vitro (Fig. 4, cf. A and B), we suggest that a proposed tertiary interaction between C44 and C66 is not essential for the function of E. coli tmRNA. However, more detailed investigations might explain the observed covariation in the future.
Resume codon-positioning segment is essential for tmRNA functions
The deletion of either pk1 (Δpk1) or the decanucleotide encompassing residues 69–78 (Δh4/Δ69–78) in E. coli tmRNA abolished protein tagging (see Fig. 4A) even though the resulting mutant tmRNAs were correctly processed (Fig. 5), efficiently aminoacylated (Fig. 6A,B) and readily associated with SmpB protein (Fig. 7). Further analysis revealed that Δpk1 could not bind to ribosomes in vivo (Fig. 9). This defect accounted for the tagging inactivity of Δpk1. In contrast, Δh4/69–78 bound readily to ribosomes. Because Δh4/69–78, despite binding to ribosomes, was unable to tag truncated proteins, this finding suggested that the interactions between Δh4/69–78 and the ribosome is nonfunctional.
Only two domains in tmRNA are known to interact with the ribosome (Valle et al. 2003; Ivanova et al. 2005; Kaur et al. 2006). The TLD-SmpB complex binds to the A and P sites as tmRNA transits the ribosome. The resume codon enters the A site part of the decoding site on the 30S ribosomal subunit at the beginning of trans-translation. A number of studies demonstrated that the resume codon and 5 nt upstream of the resume codon play a critical role in determining a reading frame in tmRNA and resuming trans-translation (Williams et al. 1999; Lee et al. 2001).
Because our experiments demonstrated that modification of pk1 did not affect the structure and functions of TLD, we speculated that deletions of pk1 and residues 69–78 affected the placement of the resume codon on the ribosome. This suggestion is consistent with earlier studies that revealed that mutations in the region upstream of the resume codon lead to the resumption of translation in the incorrect frame (Lee et al. 2001; Trimble et al. 2004; Miller et al. 2008). As highlighted in Figure 10, the RNA segment connecting the TLD to MLD in the tagging Δh4 and pk1L derivatives could span a longer distance than the analogous segment in wild-type E. coli tmRNA. In contrast, this RNA segment was considerably shorter in the nontagging Δpk1 and Δh4/Δ69–78 mutants. Together, these observations suggest that the length of the connecting RNA segment might be important for proper accommodation of the resume codon in the decoding site of the ribosome.
FIGURE 10.

Schematic representation of an RNA segment connecting the TLD and MLD in tmRNAs containing large deletions in the pk1 domain. The length of the connecting RNA segment is expressed as a number of nucleotides. Open ovals and a letter R mark the resume codon (GCA).
Comprehensive analysis is important for studying tmRNA mutants
Some tmRNA mutants (e.g., pk1L) were fully functional in vivo, but poorly mediated the tagging of truncated L27 in vitro (Fig. 4, cf. A and B). Our data suggest that SmpB was responsible for this effect. This suggestion is consistent with our earlier studies, which demonstrated that SmpB protein could stimulate the aminoacylation of tmRNA (Barends et al. 2001). However, when provided in excess, SmpB forms multimeric complexes with tmRNA and inhibits the aminoacylation of tmRNA in vitro (Barends et al. 2001; Wower et al. 2002). On the other hand, a number of studies indicated that two molecules of SmpB are required to facilitate tmRNA functions on the ribosomes (Hallier et al. 2004, 2006; Metzinger et al. 2005; Kaur et al. 2006; Sundermeier and Karzai 2007). To optimize the in vitro tagging assays, a better understanding of the way in which SmpB facilitates tmRNA functions will be required.
In this work, the contributions of pk1 for tmRNA structure were analyzed both in vitro and in vivo for the first time. We demonstrated unambiguously that pk1 is not an essential component of tmRNA but plays an important role in stabilizing the structure of tmRNAs with long RNA segments connecting the TLD and MLD. Our findings are consistent with comparative analyses of tmRNA sequences, which revealed that in some tmRNAs pk1 could be replaced with either a single-stranded RNA or a single hairpin (Zwieb et al. 1999; Gaudin et al. 2002; Gueneau de Novoa and Williams 2004; Sharkady and Williams 2004; Andersen et al. 2006).
MATERIALS AND METHODS
Chemicals and enzymes
Sources of enzymes, [32P]-labeled nucleotides, and antibodies were as in Wower et al. (2000). E. coli strain XL1-B was the host for purification and maintenance of plasmids. Expression strain IW363 was constructed by P1 transduction of gene ssrA deletion from W3110ΔssrA∷kan r into BL21(DE3) and BL21(DE3)/pLysS, respectively (Komine et al. 1994; Wower et al. 2004). TmRNA derivatives bearing mutations in pk1 were obtained by PCR-directed mutagenesis as described earlier (Wower et al. 2004).
In vitro tagging assay
Tagging in vitro was performed with the E. coli T7 transcription/translation reaction system for circular DNA (Promega) as described earlier (Wower et al. 2004). This assay used plasmid pETrpmA-At-1, which carried a copy of a truncated rpmA gene encoding a truncated ribosomal protein L27. Since mRNA for truncated protein L27 lacked a stop codon, truncated protein L27 was a substrate for tagging to be initiated by addition of the purified tmRNA to the in vitro translation reaction. To avoid degradation of the tagged L27, a tmRNA(H8hp) derivative encoding the peptide ANHHHHHHHH instead of the proteolysis signal ANDENYALAA was used. Formation of the tagged protein was monitored by fractionation of the tagging reaction products on SDS-polyacrylamide gels followed by Western blotting with anti-His-tag antibodies from Qiagen.
In vivo tagging assay
Plasmid pETrpmA-At-3 was used for the coexpression of tmRNA(H8hp), protein SmpB, and truncated protein L27 in E. coli strain IW363, which lacks the ssrA gene (Wower et al. 2004). SsrA and SmpB genes were under control of their natural promoters. The gene encoding truncated ribosomal protein L27 was under control of the T7 promoter. Cell cultures were incubated with 1 mM IPTG to induce the synthesis of truncated ribosomal protein L27. One-milliliter aliquots of cell cultures were collected for protein and RNA analyses. Cells were lysed and fractionated on 12.5% SDS-polyacrylamide gels. Tagged proteins were detected by staining gels with Coomassie Brilliant Blue R-250 (Sigma).
TmRNA processing and binding to ribosomes in vivo
Processing of tmRNA precursors and binding of mature tmRNAs to ribosomes was analyzed by Northern blotting as described earlier (Wower et al. 2004). Blots were probed with [32P]-labeled oligonucleotide 5′-GGCATCATCATCGT-3′ complementary to E. coli tmRNA residues 125–139. Hybridization signals were quantified using a Typhoon PhosphorImager and ImageQuant version 5.2 software (Amersham).
TmRNA synthesis and aminoacylation
TmRNAs were transcribed in vitro, [32P]-labeled at their 3′ ends by incubation with [α-32P]ATP and yeast ATP/CTP tRNA nucleotidyl transferase, and fractionated by electrophoresis on a native 5% polyacrylamide gel (Wower et al. 2000). Aminoacylation of 3′-[32P]-labeled tmRNA was carried out as described by Sampson and Uhlenbeck (1988) using partially purified E. coli alanyl-tRNA synthetase. The extent of aminoacylation was analyzed by electrophoresis on a 15% acid/urea polyacrylamide gel according to Varshney et al. (1991). Aminoacylation of tmRNA mutants was quantified using a Typhoon PhosphorImager and ImageQuant version 5.2 software (Amersham).
Isolation and analysis of tmRNA:70S ribosome complexes
IW363 cells transformed with pETrpmA-At-3 encoding either tmRNA(H8hp) or its derivatives bearing mutations in pk1 were grown at 37°C in 1 L of 2× YT broth supplemented with appropriate antibiotics (kanamycin, 50 μg/mL; chloramphenicol, 30 μg/mL; ampicillin, 200 μg/mL). At 0.3 A600, the tagging of truncated L27 was induced with 1 mM IPTG and the culture was incubated for an additional 2 h. Cells were harvested by centrifugation in a Sorvall GSA rotor (10 min, 16,000g). Pellets were resuspended in 100 mL of buffer A (20 mM Tris-HCl at pH 7.5, 70 mM NH4Cl, 10 mM magnesium acetate, 0.5 mM EDTA, 6 mM 2-mercaptoethanol) containing DNase I (RNase free; Worthington Biochemical Corporation) at 5 μg/mL and opened by passage through a chilled French press at 10,000 psi. Cell debris was removed by centrifugation in a Beckman 50.2 Ti rotor (45 min, 30,000g). The resulting S-30 fraction was layered in portions of 15 mL on 10 mL of 1.1 M sucrose in buffer A. After centrifugation for 12 h at 145,000g in a Beckman 50.2 Ti rotor, the ribosomes were resuspended in the same buffer, frozen in small aliquots in liquid nitrogen, and stored at −80°C.
Chemical probing of tmRNA
TmRNA derivatives were modified with dimethyl sulfate (DMS) under both native (supplemented with 10 mM magnesium acetate) and semidenaturing conditions (supplemented with 1 mM EDTA and without magnesium) (Felden et al. 1997). Sites of chemical modification were identified by primer extension and gel electrophoresis as described by Stern et al. (1988). Oligonucleotide 5′-GGCATCATCATCGT-3′ complementary to residues 125–139 in E. coli tmRNA was used as a primer.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health Grant GM58267. I.K.W. and J.W. were supported by an Auburn University Biogrant. Publication costs were supported in part by the Upchurch Fund for Excellence.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1192409.
REFERENCES
- Andersen E.S., Rosenblad M.A., Larsen N., Westergaard J.C., Burks J., Wower I.K., Wower J., Gorodkin J., Samuelsson T., Zwieb C. The tmRDB and SRPDB resources. Nucleic Acids Res. 2006;34:D163–D168. doi: 10.1093/nar/gkj142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barends S., Wower J., Kraal B. Kinetic parameters for tmRNA binding to alanyl-tRNA synthetase and elongation factor Tu from Escherichia coli . Biochemistry. 2000;39:2652–2658. doi: 10.1021/bi992439d. [DOI] [PubMed] [Google Scholar]
- Barends S., Karzai A.W., Sauer R.T., Wower J., Kraal B. Simultaneous and functional binding of SmpB and EF-Tu-TP to the alanyl acceptor arm of tmRNA. J. Mol. Biol. 2001;314:9–21. doi: 10.1006/jmbi.2001.5114. [DOI] [PubMed] [Google Scholar]
- Bessho Y., Shibata R., Sekine S., Murayama K., Higashijima K., Hori-Takemoto C., Shirouzu M., Kuramitsu S., Yokoyama S. Structural basis for functional mimicry of long-variable-arm tRNA by transfer-messenger RNA. Proc. Natl. Acad. Sci. 2007;104:8293–8298. doi: 10.1073/pnas.0700402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks J., Zwieb C., Muller F., Wower I., Wower J. Comparative 3D modeling of tmRNA. BMC Mol. Biol. 2005;6:14–29. doi: 10.1186/1471-2199-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B., Himeno H., Muto A., McCutcheon J.P., Atkins J.F., Gesteland R.F. Probing the structure of the Escherichia coli 10Sa RNA (tmRNA) RNA. 1997;3:89–103. [PMC free article] [PubMed] [Google Scholar]
- Gaudin C., Zhou X., Williams K.P., Felden B. Two-piece tmRNA in cyanobacteria and its structural analysis. Nucleic Acids Res. 2002;30:2018–2024. doi: 10.1093/nar/30.9.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueneau de Novoa P., Williams K.P. The tmRNA website: Reductive evolution of tmRNA in plastids and other endosymbionts. Nucleic Acids Res. 2004;32:D104–D108. doi: 10.1093/nar/gkh102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallier M., Ivanova N., Rametti A., Pavlov M., Ehrenberg M., Felden B. Pre-binding of small protein B to a stalled ribosome triggers trans-translation. J. Biol. Chem. 2004;279:25978–25985. doi: 10.1074/jbc.M314086200. [DOI] [PubMed] [Google Scholar]
- Hallier M., Desreac J., Felden B. Small protein B interacts with the large and the small subunits of a stalled ribosome during trans-translation. Nucleic Acids Res. 2006;34:1935–1943. doi: 10.1093/nar/gkl097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N., Pavlov M.Y., Bouakaz E., Ehrenberg M., Schiavone L.H. Mapping the interaction of SmpB with ribosomes by footprinting of ribosomal RNA. Nucleic Acids Res. 2005;33:3529–3539. doi: 10.1093/nar/gki666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai A.W., Susskind M.M., Sauer R.T. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA) EMBO J. 1999;18:3793–3799. doi: 10.1093/emboj/18.13.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S., Gillet R., Li W., Gursky R., Frank J. Cryo-EM visualization of transfer-messenger RNA with two SmpBs in a stalled ribosome. Proc. Natl. Acad. Sci. 2006;103:16484–16489. doi: 10.1073/pnas.0607438103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler K.C., Shapiro L., Williams K.P. tmRNAs that encode proteolysis-inducing tags are found in all known bacterial genomes: A two-piece tmRNA functions in Caulobacter . Proc. Natl. Acad. Sci. 2000;97:7778–7783. doi: 10.1073/pnas.97.14.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley S.T., Harris J.K., Pace N.R. Evaluation and refinement of tmRNA structure using gene sequences from natural microbial communities. RNA. 2001;7:1310–1316. doi: 10.1017/s1355838201010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine Y., Kitabatake M., Yokogawa T., Nishikawa K., Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli . Proc. Natl. Acad. Sci. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T., Kurita D., Takada K., Muto A., Himeno H. A functional interaction of SmpB with tmRNA for determination of the resuming point of trans-translation. RNA. 2007;13:1723–1731. doi: 10.1261/rna.604907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Ishii M., Tadaki T., Muto A., Himeno H. Determinants on tmRNA for initiating efficient and precise trans-translation: Some mutations upstream of the tag-encoding sequence of Escherichia coli tmRNA shift the initiation point of trans-translation in vitro. RNA. 2001;7:999–1012. doi: 10.1017/s1355838201010342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Pandit S., Deutscher M.P. 3′ Exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli . Proc. Natl. Acad. Sci. 1998;95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness K.E., Sauer R.T. Ribosomal protein S1 binds mRNA and tmRNA similarly but plays distinct roles in translation of these molecules. Proc. Natl. Acad. Sci. 2004;101:13454–13459. doi: 10.1073/pnas.0405521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzinger L., Hallier M., Felden B. Independent binding sites of small protein B onto transfer-messenger RNA during trans-translation. Nucleic Acids Res. 2005;33:2384–2394. doi: 10.1093/nar/gki534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.R., Heally D.W., Robison S.G., Dewey J.D., Buskirk A.R. The role of upstream sequences in selecting the reading frame on tmRNA. BMC Biol. 2008;6:29–38. doi: 10.1186/1741-7007-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S.D., Sauer R.T. The tmRNA system for translational surveillance and ribosome rescue. Annu. Rev. Biochem. 2007;76:101–124. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- Nameki N., Chattopadhyay P., Himeno H., Muto A., Kawai G. An NMR and mutational analysis of an RNA pseudoknot of Escherichia coli tmRNA involved in trans-translation. Nucleic Acids Res. 1999a;27:3667–3675. doi: 10.1093/nar/27.18.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nameki N., Felden B., Atkins J.F., Gesteland R.F., Himeno H., Muto A. Functional and structural analysis of a pseudoknot upstream of the tag-encoded sequence in E. coli tmRNA. J. Mol. Biol. 1999b;286:733–744. doi: 10.1006/jmbi.1998.2487. [DOI] [PubMed] [Google Scholar]
- Nameki N., Tadaki T., Himeno H., Muto A. Three of four pseudoknots in tmRNA are interchangeable and are substitutable with single-stranded RNAs. FEBS Lett. 2000;470:345–349. doi: 10.1016/s0014-5793(00)01349-1. [DOI] [PubMed] [Google Scholar]
- Rudinger-Thirion J., Giege R., Felden B. Aminoacylated tmRNA from Escherichia coli interacts with prokaryotic elongation factor Tu. RNA. 1999;5:989–992. doi: 10.1017/s135583829999101x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson J.R., Uhlenbeck O.C. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl. Acad. Sci. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkady S.M., Williams K.P. A third lineage with two-piece tmRNA. Nucleic Acids Res. 2004;32:4531–4538. doi: 10.1093/nar/gkh795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S., Moazed D., Noller H.F. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 1988;164:481–489. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- Sundermeier T.R., Karzai A.W. Functional SmpB-ribosome interactions require tmRNA. J. Biol. Chem. 2007;282:34779–34786. doi: 10.1074/jbc.M707256200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner D.R., Dewey J.D., Miller M.R., Buskirk A.R. Genetic analysis of the structure and function of transfer-messenger RNA pseudoknot 1. J. Biol. Chem. 2006;281:10561–10566. doi: 10.1074/jbc.M600167200. [DOI] [PubMed] [Google Scholar]
- Trimble M.J., Minnicus A., Williams K.P. tRNA slippage at the tmRNA resume codon. RNA. 2004;10:805–812. doi: 10.1261/rna.7010904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M., Gillet R., Kaur S., Henne A., Ramakrishnan V., Frank J. Visualizing tmRNA entry into a stalled ribosome. Science. 2003;300:127–130. doi: 10.1126/science.1081798. [DOI] [PubMed] [Google Scholar]
- Varshney U., Lee C.P., RajBhandary U.L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- Williams K.P. Descent of a split RNA. Nucleic Acids Res. 2002;30:2025–2030. doi: 10.1093/nar/30.9.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K.P., Bartel D.P. Phylogenetic analysis of tmRNA secondary structure. RNA. 1996;2:1306–1310. [PMC free article] [PubMed] [Google Scholar]
- Williams K.P., Martindale K.A., Bartel D.P. Resuming translation on tmRNA: A unique mode of determining a reading frame. EMBO J. 1999;18:5423–5433. doi: 10.1093/emboj/18.19.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wower I.K., Zwieb C.W., Guven S.A., Wower J. Binding and cross-linking of tmRNA to ribosomal protein S1, on and off the Escherichia coli ribosome. EMBO J. 2000;19:6612–6621. doi: 10.1093/emboj/19.23.6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wower J., Zwieb C.W., Hoffman D.W., Wower I.K. SmpB: A protein that binds to double-stranded segments in tmRNA and tRNA. Biochemistry. 2002;41:8826–8836. doi: 10.1021/bi0201365. [DOI] [PubMed] [Google Scholar]
- Wower I.K., Zwieb C., Wower J. Contributions of pseudoknots and protein SmpB to the structure and function of tmRNA in trans-translation. J. Biol. Chem. 2004;279:54202–54209. doi: 10.1074/jbc.M410488200. [DOI] [PubMed] [Google Scholar]
- Wower I.K., Zwieb C., Wower J. Transfer-messenger RNA unfolds as it transits the ribosome. RNA. 2005;11:668–673. doi: 10.1261/rna.7269305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C., Wower J. tmRDB (tmRNA database) Nucleic Acids Res. 2000;28:169–170. doi: 10.1093/nar/28.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C., Wower I., Wower J. Comparative sequence analysis of tmRNA. Nucleic Acids Res. 1999;27:2063–2071. doi: 10.1093/nar/27.10.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]