

Abstract
A mechanism for proton pumping is described that is based on chemiosmotic principles and the detailed molecular structures now available for cytochrome oxidases. The importance of conserved water positions and a step-wise gated process of proton translocation is emphasized, where discrete electron transfer events are coupled to proton uptake and expulsion. The trajectory of each pumped proton is the same for all four substrate electrons. An essential role for the His-Tyr cross-linked species is discussed, in gating of the D-and K-channels and as an acceptor/donor of electrons and protons at the binuclear center.
Keywords: Proton pumping, Heme–copper oxidases, Chemiosmotic principles
Introduction
Some three decades ago it was demonstrated that cytochrome c oxidase was a proton pump (Wikstrom and Krab 1979; Wikstrom 1977) and this pumping activity was later shown to be a property of other members of the heme-copper oxidase superfamily (Wikstrom and Verkhovsky 2007; Wikstrom et al. 1994; Wikstrom 2004; Mills and Ferguson-Miller 2003; Gennis 2004; Branden et al. 2006). There have been many reaction schemes postulated for how these enzymes function; however, no model has been able to explain how all the members of this evolutionarily related, but diverse, family of oxidases are able to pump protons. An examination of recent high resolution crystal structures has led us to a focus on the role of water in the facilitation of proton movement (Sharpe et al. 2005), given the absence of many conserved amino acid residues. We have compared the structure and sequence data of a large number of heme-copper oxidases and have proposed a role for conserved water molecules in oxidase function, along with individual amino acid residues. Based on this comparative analysis, and on previous work (Sharpe et al. 2005), we present a model for a common mechanism of proton pumping in the family of heme-copper oxidases. The key features of our model include:
Water based
The protonation and deprotonation of “conserved” water molecules or clusters is a central component of the pumping mechanism. These waters are critical in the function of the recognized K-channel and D-channels (Konstantinov et al. 1997), as well as in additional hydrated cavities: the X-pathway (the eXit pathway), that facilitates the movement of H+(P) (pumped protons) out of the enzyme; and the vestibule, which is found above the heme propionates at the interface of subunits I and II and links the binuclear center (BNC) to the exit channel.
Electrostatically driven
The driving force for the uptake, internal movement and ejection of H+(P) consists of a series of discrete electrostatic potentials. As each of the four electrons and H+(S) (substrate protons) migrates through the enzyme they generate electrostatic potentials, in each case a movement of H+(P) collapses the potential generated. Ours could be described as a ‘Richelian’ mechanism (Rich 1995) initially proposed by Artzatbanov (Artzatbanov et al. 1978) in which entry of a substrate electron into the protein’s low dielectric causes the (electrophoretic) uptake of a H+(P). Subsequent transfer of the electron from heme a to the BNC forms a protonatable anion, inducing (electrogenic) uptake of a H+(S) into the BNC, that causes the expulsion of the pumped proton from the enzyme (Pisliakov et al. 2008; Fadda et al. 2008).
Multiple gated proton pathways
The movements of charges (H+(S) and H+(P)) in response to electron transfer, are strictly controlled by the use of gates that only open and close at the appropriate time. We describe three gating mechanisms, likely common to all the heme–Cu type oxidases, and an additional gate in CuA/Mg type oxidases. The necessity of proton gates has long been recognized in electrostatic pumping mechanisms, and our model is consistent with recently reported evidence of location (Pisliakov et al. 2008; Fadda et al. 2008; Busenlehner et al. 2008).
Use of the histidine-tyrosine crosslinked species
The presence of a covalent bond between the two aromatic rings of H284 and Y288 (His-Tyr) provides a CuB ligand with some unusual properties (Wikstrom et al. 1994; Bu and Cukier 2005; Buse et al. 1999; Cappuccio et al. 2002; Muramoto et al. 2007; Rauhamaki et al. 2006). These properties are proposed to allow: (1) changes in the His-Tyr geometry that can gate H+(S) movements into the BNC from the K and D-channels (Sharpe et al. 2005; Bu and Cukier 2005); and (2) the change from coordinate to ionic bonding of His-Tyr to CuB which facilitates that protonation and deprotonation of H334.
Generality
The model, with minor changes as to the identity of some semi-conserved amino acids, can be used to describe the mechanism of proton pumping in members of the superfamily regardless of the heme in the BNC, the substrate, or amino acid sequence. The model also explains the phenotypes observed in the large number of site-directed mutants that have been generated in this enzyme.
Molecular consistency
All four H+(P) are taken up and ejected from oxidase in the same manner.
The model
1. Role of water
Table 1 shows the route taken by H+(P) from the inner to outer bulk phases, and the six storage sites at which the H+(P) rests during the course of the reaction cycle. The route taken by the H+(P) is independent of which particular one of the four substrate electrons is being consumed by the enzyme. There are six proposed storage sites, include two conserved residues and four water sites. They are, in order of protonation, (1−)D132 (Gennis 2004; Qin et al. 2007; Mills and Hosler 2005; Smirnova et al. 1999, Brzezinski and Adelroth 1998; Fetter et al. 1996), the Protonatable Water Cluster (PWC)(Xu et al. 2007; Xu and Voth 2006), (1−)H334 (Ji et al. 2008; Fadda et al. 2005; Popovic et al. 2005; Popovic and Stuchebrukhov 2004), W1a3(Sharpe et al. 2005), (1−)HO-W1Mg and W1E254II.
Table 1.
The route taken by H+ (P) in the catalytic cycle
| H+(P) Site | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Donor | HD132 | H+-PWC | HH334 | H+-W1a3 | W1Mg | H+-W1E254II |
| Acceptor | (1-)D132 | PWC | (1-)H334 | W1a3 | (1−)HO-W1Mg | W1E254II |
| Location | D-Channel | Binuclear Center(BNC) | Vestibule | X-Path | ||
There are also two proposed H+(S) binding sites, one located in the K-Channel, H+-K362, and one at the apex of the D-channel, HE286. The H+-K362 supplies protons (H+(SK)) to the binuclear center following the entry of the 1st and 2nd electrons into the BNC (Mills et al. 2000; Vygodina et al. 1998; Zaslavsky and Gennis 1998; Hosler et al. 1996); it is replenished from the inner bulk. Protons are supplied to the BNC on entry of the 3rd and 4th electrons (Busenlehner et al. 2008), (Vakkasoglu et al. 2006; Olsson and Warshel 2006; Seibold et al. 2005; Heberle et al. 2004; Nyquist et al. 2003; Bailey et al. 2002; Verkhovskaya et al.1997) by HE286 (H+(SD)) and the (1−)E286 that is generated is reprotonated by the inner bulk phase.
2. Operation of the CuA–Mg gate
The reduction of CuA by cytochrome c and its oxidation by heme a is proposed to be linked to the ejection of a H+(P) from the enzyme, providing an extra level of gating and thus efficiency to the oxidases that contain the Mg (Sharpe et al. 2008).
The key to the CuA/Mg gate is the common ligand, E254II, which is bonded to Mg2+ so that it is always in the carboxylate state, while the peptidyl carbonyl oxygen is bonded to one of the Cu atoms of the binuclear CuA (Fig. 1). Changes in the redox state of CuA alter in charge of the CuA binding site and affect the ligation of the Mg2+ binding site. We propose that upon reduction of CuA a pair of neutral waters undergo disproportionation and a H+(P) is transferred from a Mg2+ water ligand, leaving a (1−)OH–Mg2+, to form a hydronium ion that is ionically bonded to E254II. The reason for this disproportion is the change in the electron density on the carboxylic group of E254II upon CuA redox cycling. When CuA becomes reduced, the Cu atom bonded to the carbonyl oxygen of the peptidyl oxygen of E254II is no longer electron withdrawing and additional electron density is forced onto the carboxylate. Because of its bonding situation, direct protonation is not an option. Instead, this carboxylate forms a salt with a hydronium ion, created by the disproportion reaction to achieve electrostatic neutrality.
Fig. 1.
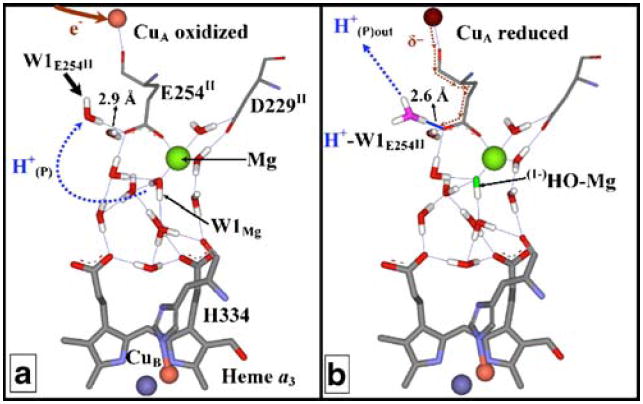
Shows two R.s. crystal structures a oxidized and b reduced, in which the protonation/redox states of the CuA, Mg and associated waters are depicted. Mg(2+) has three waters in its inner hydration sphere when CuA is oxidized; only one of these water molecules, W1Mg, is able to deprotonate and form a hydroxide, as shown in the reduced CuA state. The other two water molecules are hydrogen bonded to the carboxylate group of D229II and cannot deprotonate. The major difference between the two CuA redox states is shown as the formation of an ionic bond between W1E245II and E254II. This transition is can be deduced from the contraction of the hydrogen bond between them, from 2.9 Å in the oxidized structure to 2.6 Å in the reduced. The presence of an ionic bond is evidence for the formation of a hydronium ion, H+-W1E245II (magenta) from. Spectroscopic analysis of the Mg(Mn) site is consistent with the formation of H+-W1E245II upon CuA reduction, and concomitant deprotonation of W1Mg, with the formation of (1−)HO-W1Mg (green) and movement of H+(P) as indicated, first to W1E245II and then to the outside when CuA is reoxidized
The basis of this model was a detailed comparison of the high resolution crystal structures of both bovine and R.s. oxidases, to establish the protonation status of the water molecules in the vicinity of CuA and Mg2+. Figure 1 shows the arrangement and protonation status in the oxidized and reduced states, based on R.s. structures. In the previously published 2GSM structure, CuA is oxidized (Fig. 1A), whereas in a reduced structure (Fig. 1B) CuA is reduced. Since bond length is a function of bond strength, ionic bonds (<2.7 Å) are shorter than hydrogen bonds (> 2.8 Å). An examination of the distances between E254II and W1E254II, in the CuA oxidized state gives the distance as 2.9 Å, indicating the presence of a hydrogen bond. In the reduced structure the average distance between E254II and W1E254II is only 2.6 Å, indicative of ionic bonding. Similar measurements in the oxidized and reduced structures of bovine oxidase show an even more dramatic change in this bond, >3.7 Å (2DYR, oxidized) versus 2.6 Å (2EIJ, reduced) (Sharpe et al. 2008). The placement of hydrogen and ionic bonds in Fig. 1 is based on the standard geometry of water, hydronium and hydroxide ions, and is fully consistent with the formation of a hydronium/hydroxide pair.
Spectroscopic evidence also supports the existence of (1−)OH–Mg2+. Using the Mn substituted R.s. oxidase, the Mg/Mn site could be interrogated by a large number of electron spin resonance techniques. We found that the cyanide anion binds to the Mn ion only when CuA is reduced, consistent with the displacement of a hydroxide ion but not water. Also consistent with the model are D2O exchange experiments in combination with ENDOR and ESEEM, which show the reduced enzyme has one less exchangeable inner sphere proton at the Mn site in the reduced, cyanide-bound state (Sharpe et al. 2008).
After CuA reduction, the next step in the oxidase cycle is the oxidation of CuA by heme a. When CuA is oxidized, the E254II-hydronium salt form is no longer favored. The end result will be the movement of the H+(P) towards the cytochrome c binding site into the bulk aqueous phase (Fig. 1).
3. Operation of the D-gate: Reduction of heme a and H+ (P) uptake
The reduction of heme a by CuA is electrostatically linked to the uptake of a H+(P) from the inner bulk phase into the oxidase interior, the energetics of which was described by Rich and others. Simply, forcing an electron into a region with a low dielectric, is thermodynamically costly unless there is the movement of a counter ion. This counter ion flux can consist of a cation entry into the dielectric or the extrusion of an anion out of the dielectric (Rich 1995; Artzatbanov et al. 1978).
The arrangement of amino acids, including alcohols and the carbonyl oxygen atoms of peptide bonds, at the top of the D-channel allows water molecules to form a protonated water cluster (H+-PWC). Modeling studies suggest that, in vivo, the proton in the H+-PWC is more delocalized and more Eigen-like rather then Zundel-like. In a previous paper we and colleagues conclude from computational analysis that oxidase is capable of using water molecules to stabilize a proton at the top of the D-channel, but did not explicitly state how this capacity might be linked to oxidase turnover (Xu et al. 2007; Xu and Voth 2006). The most obvious linkage is to the reduction of heme a, where electron entry into the low dielectric interior can be electostatically compensated by the movement of a H+(P) into the D-channel from HD132, with the formation of the H+-PWC, while (1−)D132 is rapidly reprotonated.
4. Reactions at the binuclear center
A key difference of our model, in comparison with previous models, concerns the eventual fate of each of the four substrate electrons of the oxygen reduction reaction. We postulate that the 1st/2nd and the 3rd/4th electron pairs take part in different chemistry; this allows the overall catalytic cycle to be split into two distinct subcycles, the metal reduction phase and the oxygen reduction phase. In the metal reduction phase, the 1st/2nd electrons sequentially enter the BNC, via CuA and heme a, reducing CuB and heme a3. In the oxygen reduction phase, the reduced BNC reacts with molecular oxygen forming a ferryl heme a3 and a protein radical. Reduction of the ferryl and the protein radical by the 3rd/ 4th electrons, completes the cycle.
i) Initial State Before we can describe the movements of protons and electrons within oxidase we must describe the starting state of the oxidase, including the occupancy of both H+(P)/H+(S) donor and acceptor sites within the enzyme. Our starting point is the fully oxidized R.s. oxidase that has just completed a reaction cycle and is in the state we define as OH; pre-primed to pump protons. There are likely a large number of oxidized oxidase states, given the number of protons sites that may or may not be occupied, but only one is fully catalytically competent (Brand et al. 2007; Belevich and Verkhovsky 2008; Jancura et al. 2006; Wrigglesworth et al. 1988; Brunori et al. 1981, 1979; Antonini et al. 1977). This state is defined by Wikstrom as O~ or OH (Wikstrom 2004; Verkhovsky et al. 1999; Bloch et al. 2004).
The two initial H+(S) acceptors of OH are two hydroxides in the BNC. The two oxygen atoms in the BNC of “resting” oxidase (the “O” or “resting” state) were initially identified as a peroxy-bridge (Mochizuki et al. 1999; Yoshikawa et al. 1998), but that remains controversial (Aoyama et al. 2008). OH has one less proton in its BNC than does “O”, so the BNC of OH contains a pair of hydroxides bonded to each other. OH has a short half-life because of the ease at which this dihydroxide can protonate and form a hydroxide hydrate. This protonation status, where the BNC in the OH state contains a dihydroxide, has been recently proposed independent of ourselves (Fadda et al. 2008).
The internal H+(S) donors are H+-K362, HE286, both protonated in OH. In OH, an H+(P) is also on the water molecule coordinated to the Mg2+ ion, deposited there during the last catalytic cycle and on HD132.
Below is an outline of the redox reactions that occur in the BNC involving the 1st and 2nd electrons of the metal reduction phase (Fig. 2A) and the 3rd and 4th electrons of the oxygen reduction phase (Fig. 2B).
Fig. 2.

a The mechanism of H+(P) uptake and ejection from the binuclear center (BNC), upon entry of the 1st electron in the metal reduction phase. In the oxidized enzyme, the charge of CuB (2+) is neutralized by two counter ions, the anionic ligands, (1−)OH and (1−)H334 (1). Reduction of CuB (2+) to CuB(1+) by the 1st electron causes a localized charge imbalance on CuB so that one of its ionically bonded ligands must protonate (2). Proton gating of the D- and K-channels regulates proton movements into the BNC. The K-Gate is closed, so that electron entry into the BNC must be accompanied by (1-)H334 protonation, by H+(P) from the H+-PWC. Formation of HH334 triggers a change in the configuration of His-Tyr and opens the K-gate (3). This opening of the K-gate allows a H+(SK) to travel from H+-K362 into the BNC, protonating CuB (1+)-OH(1−) and forming CuB(1+)… OH2, and also closing the K-gate. The formation of CuB(1+)…OH2 has deprived CuB(1+) of an anionic ligand. The charge balance of CuB(1+) is restored by HH334 deprotonation, restoring the ionic ligand (1-)H334. The H+(P) is ejected from the BNC and is transferred to W1a3 (4). The subsequent reprotonation of K362 from the inner bulk generates an electrostatic potential that forces the H+(P) from H+-W1a3 into the ‘proton hole’ previously generated in the vestibule, (1-)HO-W1Mg. Before entry of the 2nd electron into the BNC there is a reorganization reaction, whereby an electron and proton are transferred from CuB to heme a3: (1-)HO-a3(3+) + (1−)H334-CuB(1+)…OH2 → H2O…a3 (2+) + (1-)H334-CuB (2+)-OH(1-), the pumping form of CuB in the metal reduction phase (1). The input of the 2nd electron then proceeds as the 1st. Fig. 2 b shows the mechanism of H+(P) uptake and ejection from the BNC upon entry of the 3rd electron in the oxygen reduction phase, initiated by the reaction of the two-electron reduced BNC with molecular oxygen. This generates a ferryl (a3 (4+)=O(2−)), a radical (His-•Tyr) and CuB in the form (1-)H334–CuB (2+)-OH(1−) (1). Reduction of His-•Tyr to (1−)His-Tyr by the 3rd electron causes a localized charge imbalance on CuB so that one of its ionically bonded ligands must protonate (2). Gating of the D- and K-channels ensures that electron entry is accompanied by the protonation of (1−)H334 with a H+(P) from the H+-PWC of the D-channel. Formation of HH334 triggers a change in the configuration of (1−)His-Tyr and opens up a pathway connecting HE286 to (1−)His-Tyr, allowing a H+(SD) to protonate (1−)His-Tyr (3). The protonation of (1−)His-Tyr deprives CuB(2+) of an anionic ligand, and so HH334 undergoes deprotonation so as to restore the charge balance of CuB (2+). The deprotonation of HH334 results in H+(P) ejection from the BNC, transferring it to W1a3 (4). The subsequent reprotonation of (1−)E286 by a H+(SD) from the inner bulk generates an electrostatic potential that forces the H+(P) from H+-W1a3 into (1-)HO-W1Mg. Before entry of the 4th electron into the BNC there is a reorganization reaction, whereby an electron and proton are transferred from His-Tyr to heme a3: a3(4+)=O(2−) +(His-Tyr)CuB(2+)→ (1−)HO-a3(3+) + (His-•Tyr)CuB(2+), the pumping form of CuB in the oxygen reduction phase (1). The input of the 4th electron then proceeds as the 3rd.
ii) The 1st and 2nd electrons In OH, CuB 2+ is charge balanced by two ionically bonded ligands, (1−)OH and (1−)H334, both of which are capable of protonation. The ability of H334 (H291 in bovine) to flip between the unprotonated imidazolate ((1−)H334) and neutral, protonated, imidazole (HH334) states in response to changes in CuB redox/ligation state was proposed by ourselves and others. CuB 2+ reduction is accompanied by the protonation of one of its ligands, keeping this metal electroneutral. However, gating (K-gate closed) ensures that (1−)H334 is the CuB 1+ ligand that undergoes protonation, ultilizing a H+(P) from the D-channel; the H+(P) donor is the protonated water cluster, H+–PWC.
Following the rapid uptake of H+(P) by (1−)H334, (1−)OH–CuB 1+ is protonated by H+(SK) entering the BNC from the K-channel, from H+–K362. As a result of the entry of H(SK), H+(P) is pushed from HH334 and deposited into the interface between the BNC and the vestibule H+–W1a3, Fig. 2A.
Preceding the entry of the second electron into the BNC, a rearrangement occurs involving the coordinated transfer of both a proton and electron from CuB to heme a3, generating ferrous aqua heme a3 and regenerating CuB as (1−)HO–Cu2+ to act again as the electron acceptor for the 2nd electron. The events which follow the entry of the second electron into the BNC are essentially the same as occur following the 1st, so that proton pumping occurs in an identical manner. The only difference in the chemistry of the BNC for uptake of the first and second electrons is that in the former, heme a3 is present as (1−)OH–Fe3+ state and in the latter a Fe2+–OH2 state.
iii) His-Tyr/Farnesyl-OH interaction: K-channel gating
Our K-Gate model for the regulation of proton access from the K-channel into the BNC is based on two critical observations. Firstly, the universal presence of the very strong hydrogen bond between the farnesyl hydroxide and the phenol group of Y288 precludes the entry of H+(SK) into the BNC and represents the K-gate in the normal, ground state, closed position. Secondly, it has been shown by EXAF examinations of the oxidase that CuB may sometimes become three coordinate, bound only to two histidines and one aqueous ligand (Fann et al. 1995; Powers et al. 1994).
We postulate that reduction of CuB results in the release of the H284-Y288 ligand, and its movement, resulting in breaking the strong hydrogen bond between the farnesyl hydroxide and Y288, opening the K-gate. This opening allows a proton to move from H+-K362 into the BNC, protonating the cuprus hydroxide. Figure 2A shows the proposed positions of His-Tyr, CuB and Farn-OH during the K-gate opening cycle.
The interaction of the H284–Y288 pair with Farn-OH is a central feature of the metal reduction phase of our proposed model. However, Y288 and Farn-OH are both absent in the b3 class of oxidases and may not be universally present in oxidases with unusual hemes in their BNCs. The apparent paradox is resolved by comparative sequence analysis, modeling studies and more recently by site-directed mutagenesis studies on a b3-type oxidase. In the non-classical b3 type oxidases, the roles we postulate for Y288 and Farn-OH are undertaken by two different and fully conserved alcohol groups. In the b3 oxidases, Y327 (helix 7) replaces Farn-OH and Y395 (helix 9) replaces Y288. Our modeling studies indicate the functionality of the Y327/Y395 K-gate in b3 oxidases.
iv) The third and fourth electrons
Soon after it binds to the fully reduced BNC, molecular oxygen is reduced and split, with two electrons taken from heme a3 (generating a ferryl), one electron taken from CuB, and a H+/e− is abstracted from His-Tyr, to generate a ferryl/radical state which is generally called PM, Fig. 2B. There are two major differences in proton pumping in the oxygen reduction phase, when compared to the metal reduction phase: H+(S) are supplied from HE286 at the top of the D-channel and not from H+-K362 of the K-channel; and the radical form of the H284-Y288 crosslinked pair, His-•Tyr, is the initial acceptor of both electrons and H+(S).
Transfer of the 3rd electron into the BNC reduces His-•Tyr, forming a protonatable anionic CuB 2+ ligand, (1−)His-Tyr. Electron entry into the BNC is charge compensated by H+(S) uptake, gating ensures that (1−)H334 is protonated via the H+–PWC. There now follows a slower internal movement of protons: HE286, of the D-channel, passes a H+(S) to (1−)His-Tyr, consequentially CuB 2+ requires an additional ionic bond and so HH334 is forced to deprotonate, ejecting a H+(P) out of the BNC.
We propose that prior to the arrival for the 4th electron into the BNC there is a second regeneration reaction. The F-state decays oxidizing His-Tyr and regenerating the proton pumping acceptor, His-•Tyr (Blomberg et al. 2000a, b; Shi et al. 2000; Proshlyakov et al. 1998, 2000). Thus His-•Tyr is again the acceptor of the 4th electron and 4th substrate proton from HD286.
v) The overall movements of charges and counter charges through oxidase.
Figure 3 provides an overview of our stepwise, sequential model of H+(P) pumping. The center section shows a cutaway section of oxidase that is color coded to show some of the key amino acids, cofactors and water molecules that are important for H+(P) pumping.
Fig. 3.

Is a color coded, diagrammatic representation of the electrostatically coupled movements of substrate electrons, substrate protons from the K-channel (H+(SK)), substrate protons from the D-channel (H+(SD)) and pumped protons (H+(P)), into, through and out of the oxidase. The figure is divided into three parts: The central panel is a cutaway diagram of the R.s. oxidase and shows the route taken by protons, during both phases of the catalytic cycle, and key amino acids, metal centers and water molecules. The panel on the left shows the counter charge movements of H+(P), in numbered sequence, that occur during the consumption of each of the four electrons in the overall metal/oxygen reduction phases. The panel on the right shows how discrete, electrostatic potentials are generated by the movement of substrate electrons and protons, in both the metal (MR) and oxygen (OR) reduction phases (first/second and third/fourth electrons respectively)
Electrogenic movements of substrate electrons and H+(S), are shown on the right-hand-side.
The route and storage sites of H+(P) are shown on the left-hand-side.
Summary
Pumped protons move through cytochrome oxidase from the inner to the outer bulk phases along the same route, in a stepwise fashion utilizing six acceptor/donor sites. Only two of the acceptor/donor sites are amino acids. The other four acceptor/donor sites consist of water molecules or clusters of water molecules (Fig. 3): a protonateable water cluster (PWC), near the top of the D-channel; W1a3 between the heme a3 propionates; W1Mg associated with the Mg ion; and W1E254II associated with CuA/Mg2+. Storage of a proton as part of a water cluster, near the top of the D-channel, has previously been suggested on the basis of computational analysis (Xu et al. 2007; Xu and Voth 2006). The model proposed for proton pumping is dependent on the coupling of electron movements to proton movements outside the BNC as well as the chemistry that occurs within it. The chemistry that occurs in the BNC is only responsible for the movement of a H+(P) from the H+–PWC to H+–W1a3, at the BNC interface, which is ≈ 30% of the total distance between H+(P) input and output sites. Recycling in the BNC is a key component of this pumping model. The BNC undergoes a rearrangement following the entry of the first and third electrons and their associated substrate protons, to allow the regeneration of CuB to its common pumping state. Generally, in common with many proposed pumping mechanisms for cytochrome oxidase, protons move through the protein along water chains, utilizing Grotthuss-type mechanisms, but these movements are constrained by gates, which open and close at specific parts of the cycle.
Contributor Information
Martyn A. Sharpe, M. A. Sharpe, Department of Neurosurgery, The Methodist Hospital, 6565 Fannin Street, Houston, TX 77030, USA, e-mail: MASharpe@tmhs.org
Shelagh Ferguson-Miller, S. Ferguson-Miller, Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48824-1319, USA.
References
- Antonini E, Brunori M, Colosimo A, Greenwood C, Wilson MT. Oxygen pulsed cytochrome c oxidase—functional properties and catalytic relevance. Proc Natl Acad Sci USA. 1977;74:3128–3132. doi: 10.1073/pnas.74.8.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama H, Muramoto K, Hirata K, Suga M, Yamashita E, Shinzawa-Itoh K, Ogura T, Tsukihara T, Yoshikawa S. A peroxide bridge between the two metals in the dinuclear center of the fully oxidized cytochrome c oxidase. Biochim Biophys Acta (BBA)—Bioenerg. 2008;1777:S69–S69. [Google Scholar]
- Artzatbanov VY, Konstantinov AA, Skulachev VP. Involvement of intra-mitochondrial protons in redox reactions of cytochrome a. FEBS Lett. 1978;87:180–185. doi: 10.1016/0014-5793(78)80327-5. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Tomson FL, Mecklenburg SL, MacDonald GM, Katsonouri A, Puustinen A, Gennis RB, Woodruff WH, Dyer RB. Time-resolved step-scan Fourier transform infrared spectroscopy of the CO adducts of bovine cytochrome c oxidase and of cytochrome bo(3) from Escherichia coli. Biochemistry. 2002;41:2675–2683. doi: 10.1021/bi010823g. [DOI] [PubMed] [Google Scholar]
- Belevich I, Verkhovsky MI. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxid Redox Signal. 2008;10:1–29. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI, Wikstrom M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci USA. 2004;101:529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg MRA, Siegbahn PEM, Babcock GT, Wikstrom M. Modeling cytochrome oxidase: a quantum chemical study of the O–O bond cleavage mechanism. J Am Chem Soc. 2000a;122:12848–12858. [Google Scholar]
- Blomberg MRA, Siegbahn PEM, Babcock GT, Wikstrom M. O–O bond splitting mechanism in cytochrome oxidase. J Inorg Biochem. 2000b;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- Brand SE, Rajagukguk S, Ganesan K, Geren L, Fabian M, Han D, Gennis RB, Durham B, Millett F. A new ruthenium complex to study single-electron reduction of the pulsed OH state of detergent-solubilized cytochrome oxidase. Biochemistry. 2007;46:14610–14618. doi: 10.1021/bi701424d. [DOI] [PubMed] [Google Scholar]
- Branden G, Pawate AS, Gennis RB, Brzezinski P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunori M, Colosimo A, Rainoni G, Wilson MT, Antonini E. Functional intermediates of cytochrome oxidase—role of pulsed oxidase in the pre-steady state and steady-state reactions of the beef enzyme. J Biol Chem. 1979;254:769–775. [PubMed] [Google Scholar]
- Brunori M, Colosimo A, Sarti P, Antonini E, Wilson MT. Pulsed cytochrome-oxidase may be produced without the advent of dioxygen. FEBS Lett. 1981;126:195–198. doi: 10.1016/0014-5793(81)80240-2. [DOI] [PubMed] [Google Scholar]
- Brzezinski P, Adelroth P. Pathways of proton transfer in cytochrome c oxidase. J Bioenerg Biomembranes. 1998;30:99–107. doi: 10.1023/a:1020567729941. [DOI] [PubMed] [Google Scholar]
- Bu YX, Cukier RI. Structural character and energetics of tyrosyl radical formation by electron/proton transfers of a covalently linked histidine-tyrosine: a model for cytochrome c oxidase. J Phys Chem B. 2005;109:22013–22026. doi: 10.1021/jp053046t. [DOI] [PubMed] [Google Scholar]
- Buse G, Soulimane T, Dewor M, Meyer HE, Bluggel M. Evidence for a copper-coordinated histidine-tyrosine cross-link in the active site of cytochrome oxidase. Protein Sci. 1999;8:985–990. doi: 10.1110/ps.8.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busenlehner LS, Branden G, Namslauer I, Brzezinski P, Armstrong RN. Structural elements involved in proton translocation by cytochrome c oxidase as revealed by backbone amide hydrogen-deuterium exchange of the E286H mutant. Biochemistry. 2008;47:73–83. doi: 10.1021/bi701643a. [DOI] [PubMed] [Google Scholar]
- Cappuccio JA, Ayala I, Elliott GI, Szundi I, Lewis J, Konopelski JP, Barry BA, Einarsdottir O. Modeling the active site of cytochrome oxidase: synthesis and characterization of a cross-linked histidine-phenol. J Am Chem Soc. 2002;124:1750–1760. doi: 10.1021/ja011852h. [DOI] [PubMed] [Google Scholar]
- Fadda E, Chakrabarti N, Pomes R. Acidity of a Cu-bound histidine in the binuclear center of cytochrome c oxidase. J Phys Chem B. 2005;109:22629–22640. doi: 10.1021/jp052734+. [DOI] [PubMed] [Google Scholar]
- Fadda E, Yu CH, Pomes R. Electrostatic control of proton pumping in cytochrome c oxidase. Biochim Biophys Acta Bioenerg. 2008;1777:277–284. doi: 10.1016/j.bbabio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Fann YC, Ahmed I, Blackburn NJ, Boswell JS, Verkhovskaya ML, Hoffman BM, Wikstrom M. Structure of CuB in the binuclear heme–copper center of the cytochrome aa3-type quinol oxidase from Bacillus subtilis—an ENDOR and EXAFS study. Biochemistry. 1995;34:10245–10255. doi: 10.1021/bi00032a019. [DOI] [PubMed] [Google Scholar]
- Fetter J, Sharpe M, Qian J, Mills D, FergusonMiller S, Nicholls P. Fatty acids stimulate activity and restore respiratory control in a proton channel mutant of cytochrome c oxidase. FEBS Lett. 1996;393:155–160. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- Gennis RB. Coupled proton and electron transfer reactions in cytochrome oxidase. Front Biosci. 2004;9:581–591. doi: 10.2741/1237. [DOI] [PubMed] [Google Scholar]
- Heberle J, Nyquist RM, Heitbrink D, Bolwien C, Gennis RB. Direct observation of protonation reactions during the catalytic cycle of cytochrome c oxidase: controversies on the role of E286. Biochim Biophys Acta Bioenerg. 2004;1658:25–25. [Google Scholar]
- Hosler JP, Shapleigh JP, Mitchell DH, Kim Y, Pressler MA, Georgiou C, Babcock GT, Alben JO, FergusonMiller S, Gennis RB. Polar residues in helix VIII of subunit I of cytochrome c oxidase influence the activity and the structure of the active site. Biochemistry. 1996;35:10776–10783. doi: 10.1021/bi9606511. [DOI] [PubMed] [Google Scholar]
- Jancura D, Berka V, Antalik M, Bagelova J, Gennis RB, Palmer G, Fabian M. Spectral and kinetic equivalence of oxidized cytochrome c oxidase as isolated and “activated” by reoxidation. J Biol Chem. 2006;281:30319–30325. doi: 10.1074/jbc.M605955200. [DOI] [PubMed] [Google Scholar]
- Ji H, Rousseau DL, Yeh SR. Heme–heme communication during the alkaline-induced structural transition in cytochrome c oxidase. J Inorg Biochem. 2008;102:414–426. doi: 10.1016/j.jinorgbio.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills DA, Ferguson-Miller S. Understanding the mechanism of proton movement linked to oxygen reduction in cytochrome c oxidase: lessons from other proteins. FEBS Lett. 2003;545:47–51. doi: 10.1016/s0014-5793(03)00392-2. [DOI] [PubMed] [Google Scholar]
- Mills DA, Hosler JP. Slow proton transfer through the pathways for pumped protons in cytochrome c oxidase induces suicide inactivation of the enzyme. Biochemistry. 2005;44:4656–4666. doi: 10.1021/bi0475774. [DOI] [PubMed] [Google Scholar]
- Mills DA, Florens L, Hiser C, Qian J, Ferguson-Miller S. Where is ‘outside’ in cytochrome c oxidase and how and when do protons get there? Biochim Biophys Acta Bioenerg. 2000;1458:180–187. doi: 10.1016/s0005-2728(00)00067-0. [DOI] [PubMed] [Google Scholar]
- Mochizuki M, Aoyama H, Shinzawa-Itoh K, Usui T, Tsukihara T, Yoshikawa S. Quantitative reevaluation of the redox active sites of crystalline bovine heart cytochrome c oxidase. J Biol Chem. 1999;274:33403–33411. doi: 10.1074/jbc.274.47.33403. [DOI] [PubMed] [Google Scholar]
- Muramoto K, Hirata K, Shinzawa-Itoh K, Yoko-O S, Yamashita E, Aoyama H, Tsukihara T, Yoshikawa S. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc Natl Acad Sci USA. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyquist RM, Heitbrink D, Bolwien C, Gennis RB, Heberle J. Direct observation of protonation reactions during the catalytic cycle of cytochrome c oxidase. Proc Natl Acad Sci USA. 2003;100:8715–8720. doi: 10.1073/pnas.1530408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson MHM, Warshel A. Monte Carlo simulations of proton pumps: on the working principles of the biological valve that controls proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:6500–6505. doi: 10.1073/pnas.0510860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisliakov AV, Sharma PK, Chu ZT, Haranczyk M, Warshel A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105:7726–7731. doi: 10.1073/pnas.0800580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: coulomb pump model with kinetic gating. FEBS Lett. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Popovic DM, Quenneville J, Stuchebrukhov AA. DFT/ electrostatic calculations of pK(a) values in cytochrome c oxidase. J Phys Chem B. 2005;109:3616–3626. doi: 10.1021/jp046535m. [DOI] [PubMed] [Google Scholar]
- Powers L, Lauraeus M, Reddy KS, Chance B, Wikstrom M. Structure of the binuclear heme iron-copper site in the quinol-oxidizing cytochrome aa(3), from Bacillus subtilis. Biochim Biophys Acta Bioenerg. 1994;1183:504–512. doi: 10.1016/0005-2728(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Proshlyakov DA, Pressler MA, Babcock GT. Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc Natl Acad Sci USA. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Oxygen activation and reduction in respiration: involvement of redox-active tyrosine 244. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- Qin L, Mills DA, Hiser C, Murphree A, Garavito RM, Ferguson-Miller S, Hosler J. Crystallographic location and mutational analysis of Zn and Cd inhibitory sites and role of lipidic carboxylates in rescuing proton path mutants in cytochrome c oxidase. Biochemistry. 2007;46:6239–6248. doi: 10.1021/bi700173w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhamaki V, Baumann M, Soliymani R, Puustinen A, Wikstrom M. Identification of a histidine-tyrosine cross-link in the active site of the cbb(3)-type cytochrome c oxidase from Rhodobacter sphaeroides. Proc Natl Acad Sci USA. 2006;103:16135–16140. doi: 10.1073/pnas.0606254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR. Towards an understanding of the chemistry of oxygen reduction and proton translocation in the iron–copper respiratory oxidases. Aust J Plant Physiol. 1995;22:479–486. [Google Scholar]
- Seibold SA, Mills DA, Ferguson-Miller S, Cukier RI. Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: a molecular dynamics comparison of the wild type and R481K mutant. Biochemistry. 2005;44:10475–10485. doi: 10.1021/bi0502902. [DOI] [PubMed] [Google Scholar]
- Sharpe MA, Qin L, Ferguson-Miller S. Proton entry, exit and pathways in cytochrome oxidase: insight from ‘conserved’ water. In: Wilkstrom M, editor. Biophysical and Structural Aspects of Bioenergetics. RSC Biomolecular Series, RSC Publishing; Cambridge UK: 2005. [Google Scholar]
- Sharpe MA, McCracken J, Xu S, Krzyaniak M, Ferguson-Miller S. EPR evidence of cyanide and azide binding to the Mn (Mg) center of cytochrome c oxidase: support for CuA-Mg involvement in proton pumping. Biochemistry. 2008 doi: 10.1021/bi801391r. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi WJ, Hoganson CW, Espe M, Bender CJ, Babcock GT, Palmer G, Kulmacz RJ, Tsai AL. Electron paramagnetic resonance and electron nuclear double resonance spectroscopic identification and characterization of the tyrosyl radicals in prostaglandin H synthase 1. Biochemistry. 2000;39:4112–4121. doi: 10.1021/bi992561c. [DOI] [PubMed] [Google Scholar]
- Smirnova IA, Adelroth P, Gennis RB, Brzezinski P. Aspartate-132 in cytochrome c oxidase from Rhodobacter sphaeroides is involved in a two-step proton transfer during oxo-ferryl formation. Biochemistry. 1999;38:6826–6833. doi: 10.1021/bi982865j. [DOI] [PubMed] [Google Scholar]
- Vakkasoglu AS, Morgan JE, Han D, Pawate AS, Gennis RB. Mutations which decouple the proton pump of the cytochrome c oxidase from Rhodobacter sphaeroides perturb the environment of glutamate 286. FEBS Lett. 2006;580:4613–4617. doi: 10.1016/j.febslet.2006.07.036. [DOI] [PubMed] [Google Scholar]
- Verkhovskaya ML, GarciaHorsman A, Puustinen A, Rigaud JL, Morgan JE, Verkhovsky MI, Wikstrom M. Glutamic acid 286 in subunit I of cytochrome bo(3) is involved in proton translocation. Proc Natl Acad Sci USA. 1997;94:10128–10131. doi: 10.1073/pnas.94.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky MI, Jasaitis A, Verkhovskaya ML, Morgan JE, Wikstrom M. Proton translocation by cytochrome c oxidase. Nature. 1999;400:480–483. doi: 10.1038/22813. [DOI] [PubMed] [Google Scholar]
- Vygodina TV, Pecoraro C, Mitchell D, Gennis R, Konstantinov AA. Mechanism of inhibition of electron transfer by amino acid replacement K362M in a proton channel of Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 1998;37:3053–3061. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- Wikstrom MKF. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- Wikstrom M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta Bioenerg. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Wikstrom M, Krab K. Proton-pumping cytochrome c oxidase. Biochim Biophys Acta. 1979;549:177–222. doi: 10.1016/0304-4173(79)90014-4. [DOI] [PubMed] [Google Scholar]
- Wikstrom M, Verkhovsky MI. Mechanism and energetics of proton translocation by the respiratory heme–copper oxidases. Biochim Biophys Acta Bioenerg. 2007;1767:1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Wikstrom M, Bogachev A, Finel M, Morgan JE, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky MI. Mechanism of proton translocation by the respiratory oxidases—the histidine cycle. Biochim Biophys Acta Bioenerg. 1994;1187:106–111. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Wrigglesworth JM, Elsden J, Chapman A, Vanderwater N, Grahn MF. Activation by reduction of the resting form of cytochrome c oxidase—tests of different models and evidence for the involvement of cub. Biochim Biophys Acta. 1988;936:452–464. doi: 10.1016/0005-2728(88)90023-0. [DOI] [PubMed] [Google Scholar]
- Xu JC, Voth GA. Free energy profiles for H+ conduction in the D-pathway of cytochrome c oxidase: a study of the wild type and N98D mutant enzymes. Biochim Biophys Acta Bioenerg. 2006;1757:852–859. doi: 10.1016/j.bbabio.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Xu JC, Sharpe MA, Qin L, Ferguson-Miller S, Voth GA. Storage of an excess proton in the hydrogen-bonded network of the D-pathway of cytochrome c oxidase: identification of a protonated water cluster. J Am Chem Soc. 2007;129:2910–2913. doi: 10.1021/ja067360s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- Zaslavsky D, Gennis RB. Substitution of lysine-362 in a putative proton-conducting channel in the cytochrome c oxidase from Rhodobacter sphaeroides blocks turnover with O2 but not with H2O2. Biochemistry. 1998;37:3062–3067. doi: 10.1021/bi971877m. [DOI] [PubMed] [Google Scholar]