

Abstract
The cholinergic system is crucial for cognitive processes and the deficient acetylcholine (ACh) function has been implicated in Alzheimer’s disease (AD). Inhibitors of acetylcholinesterase (AChE), which act to enhance cholinergic function by prolonging the action of endogenously released ACh, have been used as the major therapy of AD. To understand the functional roles of cholinergic enhancement in prefrontal cortex (PFC), a key brain region for cognition, we examined the impact of AChE inhibitors in PFC neurons on synaptic responses mediated by the NMDA receptor (NMDAR), an important player in learning and memory. We found that AChE inhibitors produced a strong and persistent reduction of the amplitude of NMDA receptor-mediated excitatory postsynaptic current (NMDAR-EPSC). This effect was mainly mediated by nicotinic ACh receptors, and through a Ca2+-dependent mechanism. Inhibition of extracellular signal-regulated kinases (ERK) abolished the regulation of NMDAR function by AChE inhibitors, suggesting the involvement of ERK. In the transgenic mouse model of AD overexpressing mutant β-amyloid precursor protein (APP), the effect of AChE inhibitors on NMDAR-EPSC was significantly impaired, which was associated with their diminished effect on ERK activation. Taken together, these results suggest that one of the key targets of endogenous ACh involved in cognition is the NMDAR-mediated transmission. Loss of the regulation of synaptic NMDAR responses by endogenous ACh may contribute to the cognitive deficiency in AD.
1. Introduction
Multiple lines of evidence suggest that the central cholinergic system plays a key role in cognitive processes [56] and deterioration of the cholinergic system contributes to memory failure and cognitive decline associated with aging [3] and AD [34]. Drugs that potentiate central cholinergic function, such as the AChE inhibitor physostigmine, have been found to significantly enhance storage of information into long-term memory and improve retrieval of information from long-term memory [11]. Physostigmine also improves working memory efficiency [12] by augmenting the selectivity of perceptual processing during encoding [13]. A prominent feature consistently found in AD patients is the severe degeneration of basal forebrain cholinergic systems [54, 55, 8]. Corresponding to the degeneration, there is a significant loss of nicotinic ACh receptors and certain types of muscarinic ACh receptors in AD brains [ 15, 17, 37]. So far, the mostly used therapeutic strategy in AD treatment is to enhance cholinergic transmission with AChE inhibitors [47, 53]. Despite the clinical effectiveness of these drugs in alleviating memory and cognitive problems of many AD patients [4], their molecular targets and cellular mechanisms remain largely unknown. Since a key player implicated in the regulation of learning and memory is the NMDA receptor [38], and the NMDAR-mediated transmission in PFC has been proposed to be crucial for “working memory” that is maintained by neuronal activity [30], we speculate that one of the targets of AChE inhibitors involved in cognitive process is the NMDAR-mediated synaptic response in PFC neurons.
In addition to cholinergic deficiency, another prominent feature of AD is the accumulation of β-amyloid peptides (Aβ) in cholinergic target areas, such as cortex and hippocampus [32, 44]. Aβ peptides are produced by proteolytic cleavage of the β-amyloid precursor protein (APP) [43]. Mutations in the APP gene around the cleavage sites result in the increased rate of cleavage and increased generation of Aβ [42, 52]. Transgenic mice overexpressing mutant APP genes exhibit behavioral and histopathological abnormalities resembling AD, including increased Aβ deposits and deficits in learning and memory, and therefore are widely used as an AD model [14, 21, 6]. Recent advances in AD research have pointed Aβ blockade as the major new therapy directly targeting the mechanisms underlying AD [39, 16, 28], which potentially challenges the cholinergic hypothesis. However, converging lines of evidence indicate that cholinergic system actually has a close functional relationship with amyloids [2, 48]. Aβ peptides are able to reduce choline uptake, inhibit acetylcholine releases [5], bind to α7 nAChR and blunt the receptor function [31]. On the other hand, activation of nAChR results in the enhanced release of a secreted form of APP that has a wide range of trophic and protective functions [26], and this effect is reversed by cholinergic antagonists [27]. Thus, the interaction between cholinergic system and the abnormal protein aggregates in AD needs to be further investigated.
In this study, we examined the functional role of AChE inhibitors and its potential impairment in the APP transgenic mouse model of AD.
2. Materials and Methods
2.1. Slice preparation
Prefrontal cortex (PFC) slices from Sprague Dawley rats (3–4 weeks old), wild-type and APP transgenic mice (6 weeks to 16 months old) were prepared as previously described [7, 59]. APP transgenic mice carry the human APP 695 with the double mutation (Swedish mutation) of K670N and M671L [21]. All experiments were performed with the approval of “Institutional Animal Care and Use Committee” (IACUC) of the State University of New York at Buffalo, and our animal care procedures were in accordance with the IACUC guidelines under the Animal Welfare Act. In brief, animals were anesthetized with halothane vapour before decapitation. Slices (300µM) were cut with a Leica VP1000S Vibratome, and were incubated in oxygenated ACSF (in mM, 130 NaCl, 26 NaHCO3, 2 CaCl2, 5 MgCl2, 3 KCl, 10 glucose, 1.25 NaH2PO4) at room temperature.
2.2 Recording of synaptic NMDAR responses in slices
To evaluate the regulation of NMDAR-EPSC in pyramidal neurons located in deep layers (V–VI) of PFC slices, the whole-cell voltage-clamp recording technique was used [59, 58]. A patch electrode (3–5 MΩ) was filled with an internal solution containing (in mM):130 Cs-methanesulfonate, 10 HEPES, 10 CsCl ,4 NaCl, 1 MgCl2, 1 EGTA, 5 N-methyl-d-glucamine, 12 phosphocreatine, 5 MgATP, and 0.5 Na2GTP (pH 7.2, 275–290 mOsm). Slices were bathed in ACSF containing CNQX (20 µM) and bicuculline (10 µM) to block AMPA/kainate receptors and GABAA receptors. Evoked currents were generated with a 50 µs pulse (intensity: 20–28 mA) from a stimulation isolation unit controlled by a S48 pulse generator (Astro-Med). A bipolar stimulating electrode (FHC) was positioned in the same layer as the recording electrode, and was ~50 µm from the neuron under recording. Before stimulation, cells (voltage-clamped at −70 mV) were depolarized to +60 mV for 3 seconds to fully relieve the voltage-dependent Mg2+ block of NMDAR channels. Clampfit Program (Axon Instrument) was used to analyze evoked synaptic activity. Mann-Whitney U test was used to compare the current amplitudes before and after drug application. Student t-test was performed to compare the differential degrees of current modulation between groups subjected to different treatment.
2.3. Western Blot analysis in brain slices
Procedures were similar to what we described previously [18]. Primary antibodies used for blotting include: anti-phospho-p44/42 MAP kinase (Thr202/Tyr 204) and anti-p44/42 MAPK from Cell Signaling.
2.4. Primary culture and gene transfection
Frontal cortical cultures from rats were prepared as described before [58]. At 8–11 DIV, neurons were cotransfected with a plasmid encoding the enhanced green fluorescent protein (EGFP, Clontech) and a plasmid encoding either wild type MEK1 or dominant negative MEK1 (carrying M substitution at K97). Transfection was conducted with the Lipofectamine 2000 method following manufacturer’s instruction (Invitrogene, San Diego, CA). Two to three days after transfection, GFP-positive neurons were selected for electrophysiological recoding.
2.5. Recording of NMDAR-mediated ionic current in neuronal cultures
Standard voltage-clamp techniques [7, 58] were used to record the whole-cell NMDA-evoked current. The internal solution consisted (in mM): 180 N-methyl-D-glucamine, 40 HEPES, 4 MgCl2, 0.5 BAPTA, 12 phosphocreatine, 3 Na2ATP, 0.5 Na2GTP, 0.1 leupeptin, pH 7.2, 265–270 mOsm. The external solution contained (in mM): 127 NaCl, 20 CsCl, 10 HEPES, 1 CaCl2, 5 BaCl2, 12 glucose, 0.001 TTX, 0.02 glycine, pH 7.3–7.4, 300–305 mOsm. Drugs were applied with a gravity-fed 'sewer pipe' system. The array of application capillaries (ca. 150 µm i.d.) was positioned a few hundred microns from the cell under study. Solution changes were effected by the SF-77B fast-step solution stimulus delivery device (Warner Instrument). Cells were held at −60mV and exposed to NMDA (100 µM) for 2 sec every 30 sec.
3. Results
3.1. AChE inhibitors reduce NMDAR-mediated synaptic transmission in cortical pyramidal neurons
To understand the potential impact of endogenous acetylcholine on NMDAR-mediated synaptic transmission in frontal cortex, we assessed the effect of physostigmine, a commonly used AChE inhibitor, on NMDA receptor-mediated excitatory postsynaptic current (NMDAR-EPSC) in PFC pyramidal neurons. As shown in Figure 1A and 1B, under the control condition, NMDAR-EPSC amplitudes were stable throughout the recording (up to 60 min). Application of physostigmine (40 µM) caused a significant reduction of NMDAR-EPSC amplitudes (39.4 ± 2.1%, n = 19, p < 0.001, Mann-Whitney U test). The physostigmine-induced reduction was persistent, without complete recovery after 20 min of washing. The dose response of physostigmine effects on NMDAR-EPSC is shown in Figure 1C (1 µM: 13.3 ± 1.4%; 10 µM: 30.7 ± 2.5%; 40 µM: 39.4 ± 2.1%; 100 µM: 49.4 ± 3.1%; n = 8). Another AChE inhibitor, methomidophos (80 µM), gave similar results, reducing NMDAR-EPSC amplitudes by 42.1 ± 4.5% (n = 5, Figure 1D). In contrast, physostigmine or methomidophos had no significant effect on AMPAR-EPSC (5.1 ± 0.4%, n = 6).
Figure 1. AChE inhibitors cause a reduction of NMDAR-EPSC amplitudes in cortical pyramidal neurons.
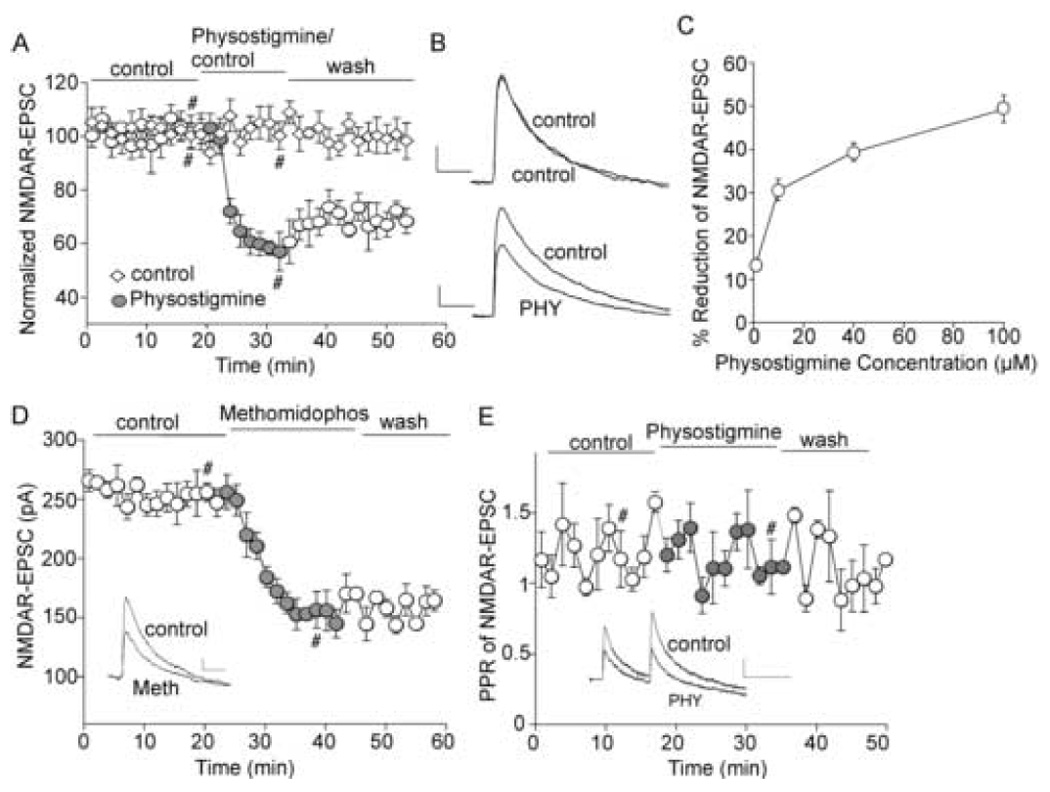
A, Plot of normalized peak NMDAR-EPSC amplitudes as a function of time and drug (the AChE inhibitor physostigmine, 40 µM, or control ACSF) application in two representative PFC pyramidal neurons. B. Representative traces of NMDAR-EPSC from the records used to construct A (at time points denoted by #). Scale bars: 20pA/100ms. C. Dose response curve showing the effect of physostigmine on NMDAR-EPSC. D. Plot of peak NMDAR-EPSC amplitudes as a function of time and methomidophos (50 µM, another AChE inhibitor) application in a representative PFC pyramidal neuron. E. Plot of paired pulse ratio (PPR) of NMDAR-EPSC as a function of time and drug application. Inset (D, E), representative traces of NMDAR-EPSC (at time points denoted by #). Scale bars: 20pA/100ms.
We next sought to determine whether the effect of physostigmine on NMDAR-EPSC is mediated by a presynaptic mechanism on neurotransmitter (glutamate) release or a postsynaptic mechanism on NMDA receptors. To do so, we assessed the effect of physostigmine on the ratio of paired-pulse facilitation (PPR) of NMDAR-EPSC evoked by double pulses (100 ms intervals), an index reflecting presynaptic processes. As shown in Figure 1E, PPR remained largely unaltered after physostigmine application (105 ± 6.1% of control, n = 12), suggesting that AChE inhibitors regulate NMDAR-EPSC by acting on postsynaptic cholinergic receptors in cortical neurons.
3.2. The effect of AChE inhibitor on NMDAR-EPSC is primarily mediated by nicotinic ACh receptors
Both muscarinic (mAChR) and nicotinic (nAChR) acetylcholine receptors are widely expressed in frontal cortex. To identify the major receptor subtypes that mediate the physostigmine-induced reduction of NMDAR-EPSC amplitudes, we examined several acetylcholine receptor antagonists. Slices were pretreated with antagonists for at least 2 hours and further exposed to antagonists throughout the recording. As shown in Figure 2A and 2B, treatment with mAChR antagonist atropine (50 µM) failed to show a significant blockade on the inhibitory effect of physostigmine. Atropine itself did not have a significant effect on NMDAR-EPSC (1.5% ± 0.6%, n = 7). Incubating slices with an allosteric activator of nAChR, Benzoquinonium (BZQ, 50 µM), to occupy the binding site of physostigmine on nAChR [36] significantly attenuated the effect of physostigmine on NMDAR-EPSC. Direct application of BZQ itself caused a significant reduction of NMDAR-EPSC (35.0 ± 7.5%, n = 4), similar to the inhibitory effect of physostigmine. As summarized in Figure 2C, the percentage reduction of NMDAR-EPSC by physostigmine was 39.7 ± 2.0% in control cells (n = 7), 30.9 ± 4.0% in atropine-treated cells (n = 9), 14.5 ± 1.5% in BZQ-treated cells (n = 10, p < 0.001, t-test), and 8.6 ± 2.9% in BZQ + atropine-treated cells (n = 6, p < 0.001, t-test), respectively.
Figure 2. The reduction of NMDAR-EPSC by physostigmine is mainly mediated by nicotinic acetylcholine receptors.
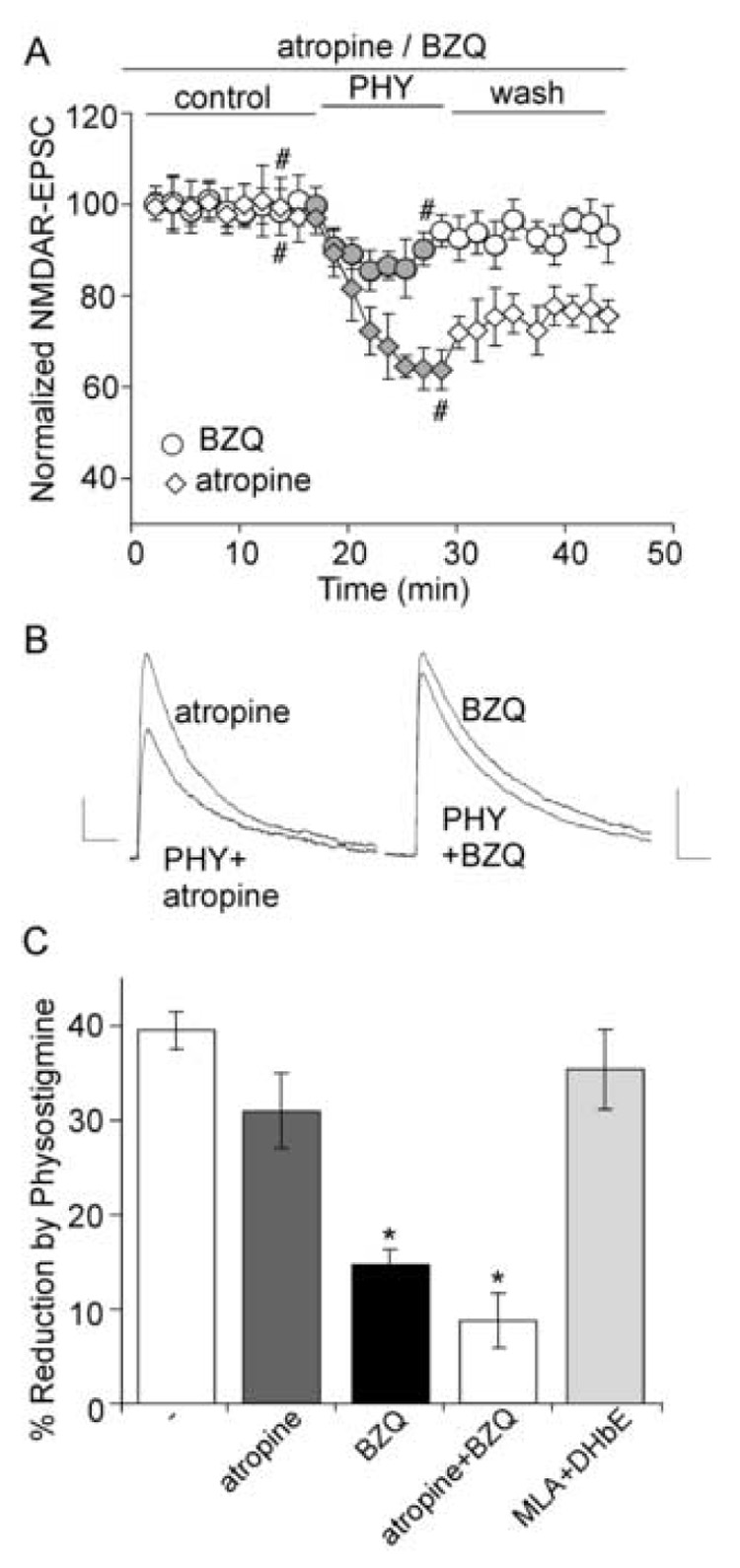
A. Plot of normalized NMDAR-EPSC amplitudes as a function of time and physostigmine (40 µM) application in neurons treated with either atropine (50 µM) or benzoquinonium (BZQ, 50 µM). B. Current traces taken from the records used to construct A (at time points denoted by #). Scale bars: 10pA/100ms. C. Bar plot summary showing the percentage reduction of NMDAR-EPSC by physostigmine in the presence of atropine, BZQ, atropine + BZQ, or MLA (10 µM) + DHβE (0.1 µM). *: p < 0.001, t-test.
In the CNS, α4β2 heteromeric receptors and α7 homomeric receptors are believed to be the major nAChR subtypes [33]. However, combined application of α7 selective antagonist MLA (methyllycaconitine, 10 µM) and α4β2 selective antagonist DHβE (dihydro-β-erythroidine, 0.1 µM) did not prevent the inhibition of NMDAR-EPSC by physostigmine (36.4 ± 3.5%, n = 9, Figure 2C). These results suggest that physostigmine regulates NMDAR-EPSC by acting on nAChR at the novel allosteric binding site, but not the classical binding sites for established nAChR agonists or antagonists [41].
3.3. The regulation of synaptic NMDAR responses by AChE inhibitor is through a calcium-dependent mechanism
We next examined the molecular mechanism that may underlie physostigmine regulation of NMDAR-EPSC. It is known that many nAChRs have a high permeability for calcium [10]. The role of Ca2+ was first examined by dialyzing with a calcium chelator, BAPTA. As shown in Figure 3A, the inhibition of NMDAR-EPSC by physostigmine was significantly reduced in the presence of intracellular BAPTA (10 mM). To further verify the role of calcium, we used the calcium pump inhibitor thapsigargin. Perfusion of thapsigargin (10 µM) caused a reduction of NMDAR-EPSC and largely occluded the effect of subsequently applied physostigmine (Figure 3B).
Figure 3. The effect of physostigmine on NMDAR-EPSC is through a mechanism depending on Ca2+.
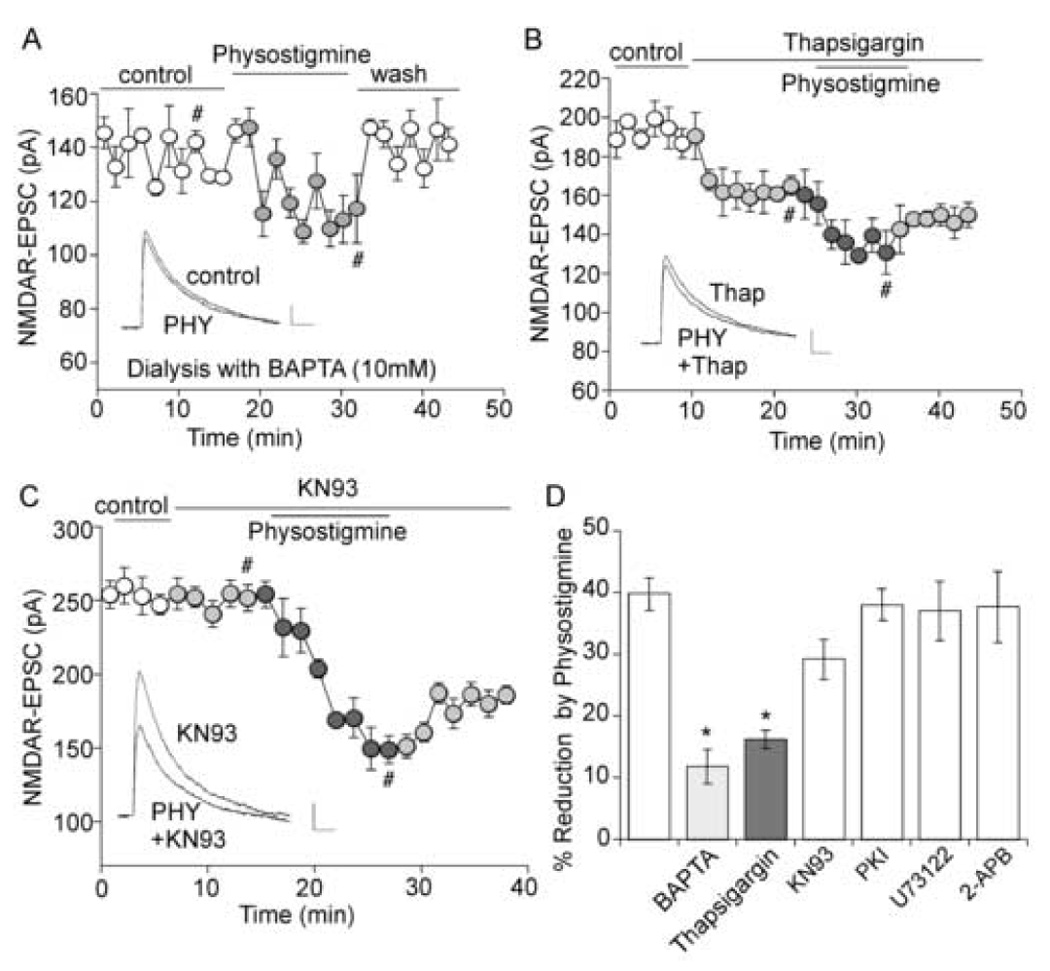
A. Plot of NMDAR-EPSC as a function of time and physostigmine (40 µM) application in a neuron loaded with BAPTA (10 mM). Inset, representative current traces (at time points denoted by #). Scale bars: 20pA/100ms. B, C. Plot of NMDAR-EPSC as a function of time and drug application. Note that thapsigargin (10 µM, B), but not KN93 (10 µM, C), prevented the effect of physostigmine. Inset (B, C), representative current traces (at time points denoted by #). Scale bars: 10pA/100ms. D. Bar plot summary showing the percentage reduction of NMDAR-EPSC by physostigmine in neurons treated with different agents. *: p < 0.005, t-test.
Elevated intracellular Ca2+ can activate many Ca2+-dependent signaling molecules, which in turn modulate NMDA responses [46]. Thus, we further examined the potential role of some of these molecules, including Ca2+/calmodulin-dependent kinase II (CaMKII), phospholipase C (PLC) and IP3. As shown in Figure 3C, bath application of the CaMKII inhibitor KN93 (10 µM) failed to block the effect of physostigmine on NMDAR-EPSC. Summarized in Figure 3D is the percentage reduction of NMDAR-EPSC by physostigmine in the presence of different agents. Compared to control cells (39.8 ± 2.6%, n = 12), the effect of physostigmine was significantly (p < 0.005, t-test) smaller in BAPTA-injected cells (11.8 ± 2.7%, n = 11) or thapsigargin-treated cells (16.2 ± 1.4%, n = 9), but was unchanged in cells treated with KN93 (29.2 ± 3.2%, n = 9), protein kinase A inhibitor PKI6–22 (2 µM, 38.5 ± 1.6%, n = 4), PLC inhibitor U73122 (5 µM, 37.0 ± 4.8%, n = 6), or IP3 receptor antagonist 2-APB (20 µM, 37.7 ± 5.7%, n = 4).
3.4. ERK is involved in the AChE regulation of NMDAR-mediated current
Our previous studies have found that ERK is involved in the regulation of NMDA receptors by neuromodulators via mechanisms depending on microtubule dynamics or actin cytoskeleton [58, 19]. ERK can be activated as a result of nAChR-mediated calcium entry. To test whether ERK also plays a role in AChE modulation of NMDAR-mediated synaptic transmission, we treated neurons with inhibitors of MEK (the upstream kinase of ERK). As shown in Figure 4A–C, the reduction of NMDAR-EPSC by physostigmine was significantly (p < 0.001, t-test) smaller in neurons treated with the MEK inhibitor U0126 (10 µM, 10.2 ± 1.9%, n = 9), compared to non-treated control neurons (39.2 ± 1.8%, n = 6). Another MEK inhibitor PD98059 (25 µM) gave similar results, significantly (p < 0.001, t-test) attenuating the effect of physostigmine on NMDAR-EPSC (14.6 ± 3.1% with internal application, n = 8; 13.3 ± 3.3% with external application, n = 9).
Figure 4. ERK is involved in the regulation of NMDAR current by physostigmine.
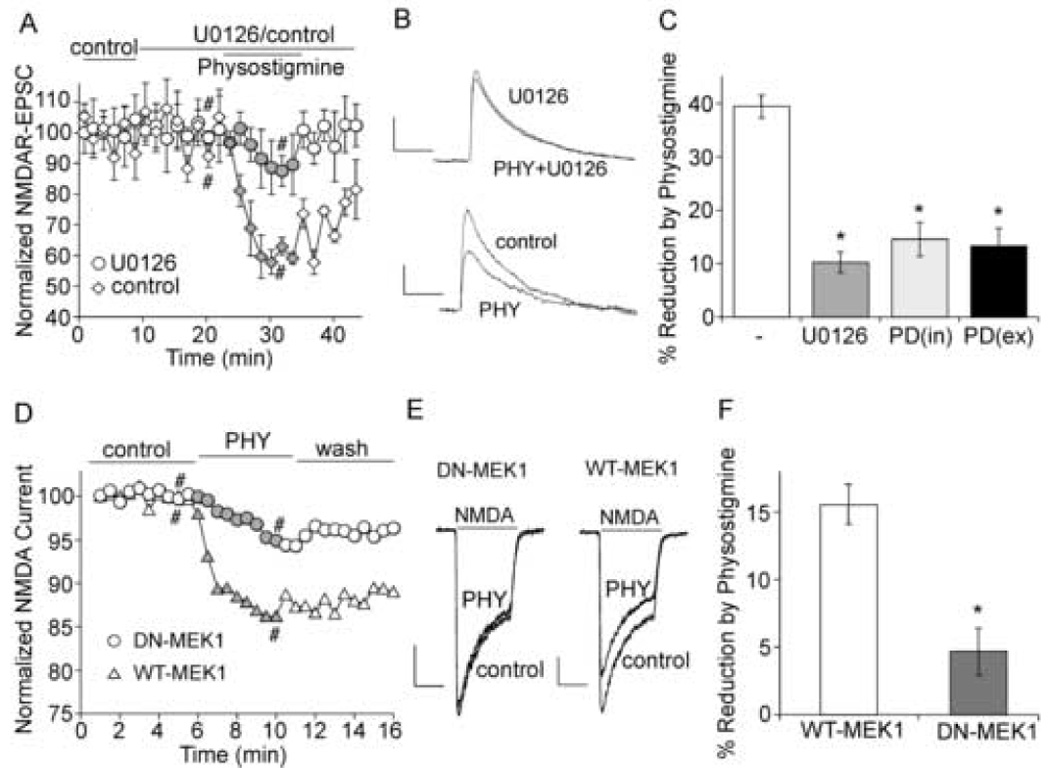
A. Plot of normalized NMDAR-EPSC as a function of time and physostigmine (40 µM) application in the absence (control) or presence of U0126 (20 µM). B. Representative current traces taken from records used to construct A (at time points denoted by #). Scale bars: 20pA/100ms. C. Bar plot summary of the percentage reduction of NMDAR-EPSC by physostigmine in neurons treated without or with U0126 or PD98059 (25 µM internal or external application). *: p < 0.001, t-test. D. Plot of normalized NMDA (100 µM)-evoked currents as a function of time and physostigmine (40 µM) application in cultured cortical neurons transfected with either wild type MEK1 (WT-MEK1) or dominant negative MEK1 (DN-MEK1). E. Representative current traces taken from records used to construct D (at time points denoted by #). Scale bars: 50pA/0.5sec. F. Bar plot summary of the percentage reduction of NMDAR current by physostigmine in neurons transfected with WT-MEK1 or DN-MEK1. *: p < 0.001, t-test.
We further examined the involvement of ERK using cultured cortical neurons transfected with a dominant-negative MEK1 construct that is catalytically inactive [58, 19] to block the ERK signaling. As shown in Figure 4D–F, in neurons transfected with wild-type MEK1, physostigmine caused a reduction of NMDA-elicited current (15.2 ± 1.5%, n = 5). This effect was significantly (p < 0.001, t-test) smaller in neurons transfected with dominant-negative MEK1 (4.7 ± 1.7%, n = 5). These results suggest that ERK is involved in the regulation of NMDAR-mediated current by AChE inhibitors.
3.5. The effect of AChE inhibitor on NMDAR-EPSC is impaired in APP transgenic mice
Compared to wild-type mice, APP transgenic mice exhibit significantly higher (~ 20–30 fold) levels of Aβ peptides at two months of age, even though no amyloid plaques, neuronal death or cognitive deficit are observed at the early presymptomatic stage [21], and amyloid deposits are found in frontal cortex, along with other brain regions, in aged APP transgenic mice [21]. Previous studies have suggested that cholinergic signaling could be diminished by the elevated level of Aβ in Alzheimer’s disease [2, 57], thus we examined whether the effect of AChE inhibitors on NMDAR-EPSC is altered in the APP transgenic mouse model of AD. As shown in Figure 5A and 5B, application of physostigmine caused a significantly smaller effect on NMDAR-EPSC in cortical pyramidal neurons from an APP transgenic mouse (2-month-old) than an age-matched WT mouse. The percentage reduction of NMDAR-EPSC by physostigmine in three age-matched groups (6 weeks, 2 months and 16 months) were summarized in Figure 5C. The effect of physostigmine was unchanged in the 6-week group (WT: 48.5 ± 2.7%, n = 6; APP: 46.0 ± 4.2%, n = 7). However, it was significantly smaller in the 2-month group (WT: 44.6 ± 2.2%, n =14; APP: 27.8 ± 3.0%, n =14, p <0.001, t-test), and the 16-month group (WT: 40.8 ± 2.2%, n = 7; APP: 27.2 ± 3.0%, n = 7, p <0.005, t-test). We further compared the dose responses of physostigmine-induced reduction of NMDAR-EPSC in wild-type and APP transgenic mice at the 4-month group. As shown in Figure 5D, physostigmine produced significantly smaller effects at different doses in APP transgenic mice (10 µM: WT: 41.1 ± 3.5%, n = 5; APP: 31.1 ± 2.6%, n = 7; 40 µM: WT: 54.4 ± 3.4%, n = 6; APP: 36.2 ± 1.5%, n = 6; 100 µM: WT: 64.6 ± 4.6%, n = 6; APP: 50.7 ± 4.2%, n = 6). The impairment of endogenous ACh effects on NMDAR-EPSC occurs at 2 months of age, supporting the notion that synaptic dysfunction is an early event in AD [45].
Figure 5. The regulation of NMDAR-EPSC by physostigmine is impaired in APP transgenic mice.
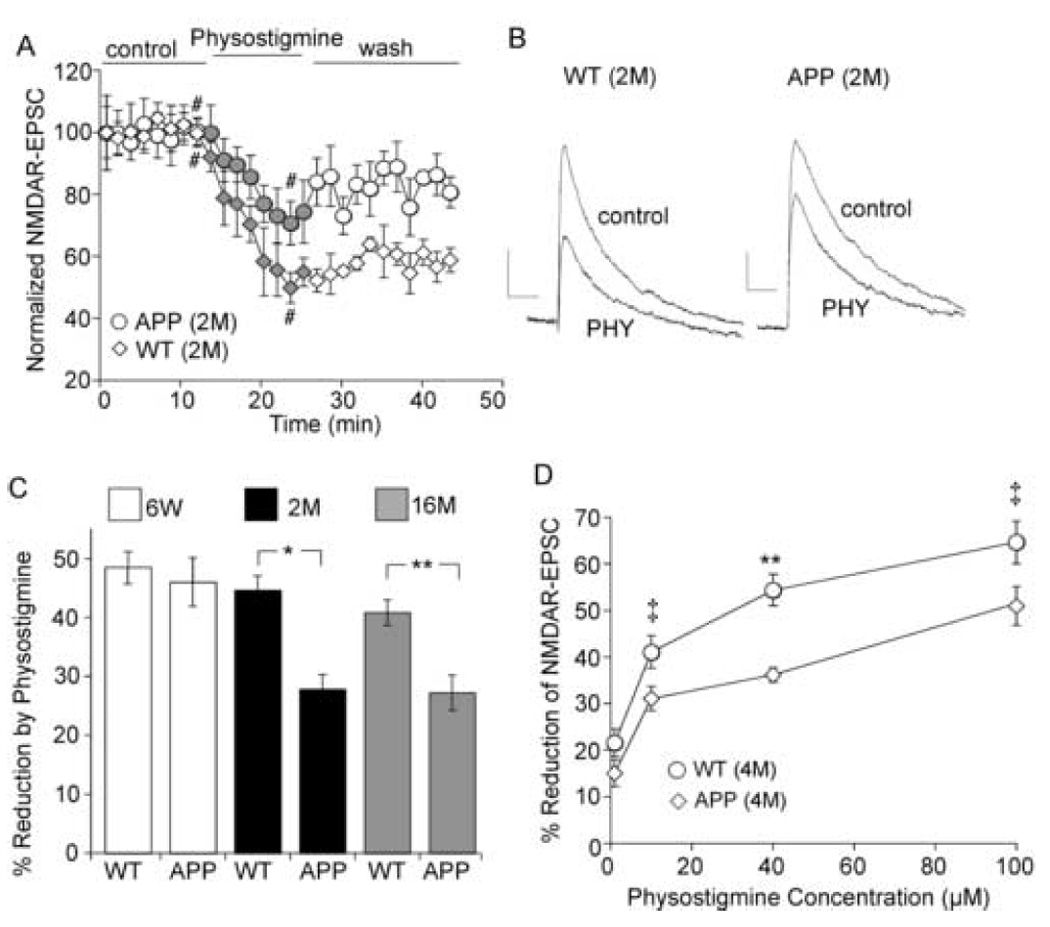
A. Plot of normalized NMDAR-EPSC as a function of time and physostigmine (40 µM) application in cortical pyramidal neurons from a 2-month-old wild type mouse (WT) and a 2-month-old APP transgenic mouse (APP). B. Representative current traces taken from records used to construct A (at time points denoted by #). Scale bars: 20pA/100ms. C. Bar plot summary of the percentage reduction of NMDAR-EPSC by physostigmine in wild-type and APP transgenic mice at different ages (6 weeks, 2 months, and 16 months). *: p < 0.001; **: p < 0.005, t-test. D. Dose response curves showing the effect of physostigmine on NMDAR-EPSC in 4-month-old WT and APP transgenic mice. ‡: p < 0.01; **: p < 0.005, t-test.
3.6. The activation of ERK by AChE inhibitors is diminished in APP transgenic mice
We next sought to determine mechanisms underlying the impaired regulation of NMDA responses by AChE inhibitors in the AD model. Given the role of ERK in this regulatory event, we speculated that the activation of ERK by AChE inhibitors might be impaired in APP transgenic mice. Consistent with this, both physostigmine and methomidophos (10 min treatment) significantly induced the activation of ERK (as indicated by phosphorylated ERK) in cortical slices from WT mice (4-month-old); however, this effect was significantly attenuated in APP mice (4-month-old, Figure 6A). The total level of ERK was not changed by AChE inhibitor treatment. As summarized in Figure 6B, ERK activation in response to AChE inhibitors was largely abolished in APP transgenic mice (increase of p-ERK in WT mice: physostigmine: 2.00 ± 0.12 fold; methomidophos: 2.00 ± 0.50 fold; p < 0.005, t-test; increase of p-ERK in APP mice: physostigmine: 0.33 ± 0.33 fold; methomidophos: 0.13 ± 0.19 fold; n = 6; p > 0.05, t-test).
Figure 6. The ERK activation by AChE inhibitors is diminished in APP transgenic mice.
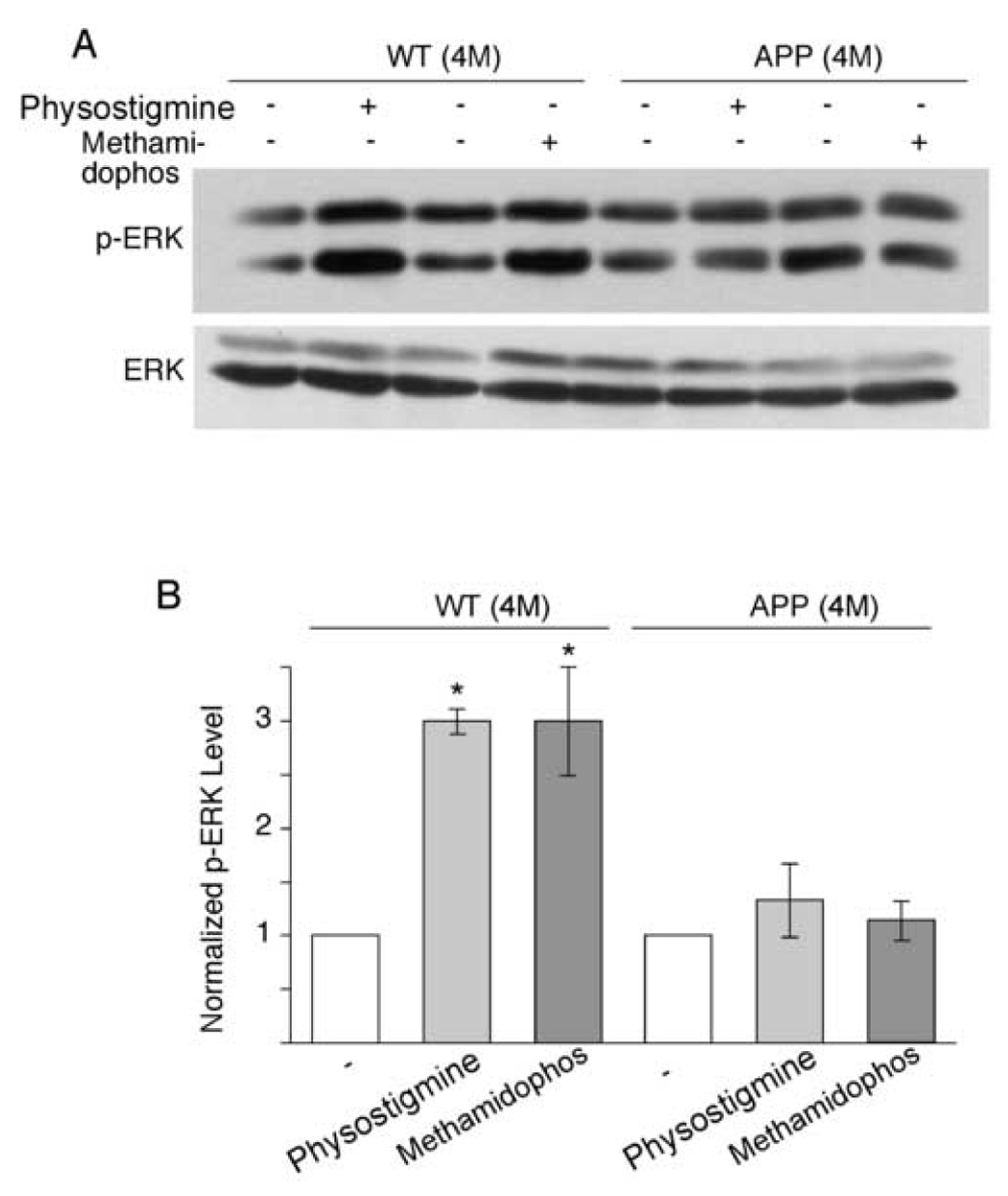
A. Representative Western blots of phospho-ERK and total ERK in cortical slices treated without or with AChE inhibitors (physostigmine: 40 µM; methomidophos: 40 µM) from WT and APP transgenic mice (4-month-old). B. Bar plot summary showing the phospho-ERK levels in cortical slices from WT and APP mice under different treatments. *: p < 0.005, t-test.
4. Discussion
AChE inhibitors are currently used as the major therapy of AD. However, as indicated in [16], inhibitors of AChE symptomatically enhance cognitive state to some degree but are not disease modifying. One of the consensuses about the new therapy directly targeting the mechanisms underlying AD is to block pathogenic Aβ peptides [39]. However, cognitive impairment and behavioral abnormalities are not directly caused by the pathological hallmarks, such as amyloid plaques and neurofibrillary tangles, but by the loss of synaptic connections [49]. Thus, it has been perceived that variations in the activity of neural networks underlie the fluctuations in neurological functions of AD patients [35]. The currently approved AChE inhibitor drugs are more likely to alter the synaptic processes without affecting the underlying disease pathways [48]. In this study, we have shown a functional role of physostigmine in PFC pyramidal neurons, i.e. to suppress the NMDAR-mediated synaptic current. It provides a mechanism for the AChE inhibitor to alter synaptic processes and neural network activity. Moreover, it provides a potential explanation for the neuroprotective role of AChE inhibitors against glutamate (or NMDA) toxicity [51,1], which seems to be partially mediated by inhibition of the increase of [Ca2+]i [1]. Interestingly, the newly approved drug for moderate to severe AD, memantine, is an uncompetitive, low-affinity NMDAR antagonist [40, 50], suggesting that downregulation of NMDA responses may be one of the beneficial effects of AChE inhibitors in AD treatment. It supports the idea that modulation of glutamatergic function may represent a viable therapeutic strategy for AD [29]. Interestingly, the reduction of NMDA-EPSCs by physostigmine survived the washout for at least 30 min, suggesting that the AChE inhibitor may have caused long-lasting changes on synaptic NMDA receptors.
AChE inhibitors act to enhance cholinergic function by prolonging the action of endogenously released ACh, which could exert their functions via either mAChRs or nAChRs. Physostigmine and several other AChE inhibitors have also been shown to act as noncompetitive nAChR agonists by binding to sites on the alpha-polypeptide that are distinct from those for the natural transmitter ACh [36, 41]. The physostigmine-induced suppression of synaptic NMDAR responses is largely prevented by the allosteric activator of nAChR, Benzoquinonium, which occupies the binding site of physostigmine on nAChR [36], suggesting that the effect of AChE inhibitors on NMDARs is mainly mediated by its direct binding to nicotinic receptors. Although activation of presynaptic nAChRs can cause an increase in transmitter release, postsynaptic effects of nAChRs have also been suggested [23]. The lack of changes in paired-pulse ratio of NMDAR-EPSC by AChE inhibitors and the blockade of their effect by intracellular application of various agents suggest that AChE inhibitors regulate synaptic NMDAR responses by acting on postsynaptic nAChRs.
Using selective inhibitors and dominant negative constructs, we have shown that ERK is involved in the regulation of NMDAR current by AChE inhibitors. Calcium entry through nAChRs could activate calcium-dependent RasGEF (Ras Guanine Nucleotide Exchange Factor) [9], leading to activation of ERK. ERK in turn could affect NMDAR function by phosphorylating NMDAR subunits and/or related proteins. Alternatively, ERK could affect NMDAR trafficking through mechanisms dependent of cytoskeleton [58, 19].
To find out whether the regulation of NMDAR-EPSC by AChE inhibitors is potentially implicated in AD, we examined APP transgenic mice. Interestingly, the effect of physostigmine on NMDAR-EPSC is significantly attenuated in this AD model, which seems to be associated with the diminished ERK activation by AChE inhibitors. It suggests that the elevated level of Aβ in APP transgenic mice (older than 6 weeks) may have altered nAChR expression or signaling. Similarly, our previous study has demonstrated that the muscarinic regulation of GABAergic transmission in PFC pyramidal neurons is impaired in APP transgenic mice, which is due to the loss of PKC activation by mAChRs [59]. These results are consistent with the notion that Aβ can directly induce cholinergic hypofunction without apparent neurotoxicity [2]. Aβ has been found to have pleiotropic actions on the cholinergic system, including the suppression of acetylcholine synthesis in primary cultures of basal forebrain neurons [20], the inhibition of acetylcholine release from hippocampal slices [24], and the disruption of muscarinic receptor – G proteins coupling in cortical cultures [25]. Our present study suggests that under pathological conditions, Aβ acts as a cholinergic neuromodulator to disturb the cholinergic enhancer-mediated inhibition of synaptic NMDAR responses, which could contribute to the perturbed cellular calcium homeostasis that plays an important role in the progressive neuronal loss underlying the evolving dementia [22, 23].
Acknowledgments
This work was supported by grants from National Institutes of Health (AG21923, MH63128) and National Science Foundation in China (NSFC 30528009, 30370311) to Z.Y and F.D. We would like to thank Xiaoqing Chen for her technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
All authors have no actual or potential conflicts of interest with other people or organizations within three years of beginning the work presented here.
References
- 1.Akasofu S, Kimura M, Kosasa T, Ogura H, Sawada K. Protective effect of donepezil in primary-cultured rat cortical neurons exposed to N-methyl-d-aspartate (NMDA) toxicity. Eur J Pharmacol. 2006;530(3):215–222. doi: 10.1016/j.ejphar.2005.11.057. [DOI] [PubMed] [Google Scholar]
- 2.Auld DS, Kar S, Quirion R. Beta-amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci. 1998;21(1):43–49. doi: 10.1016/s0166-2236(97)01144-2. [DOI] [PubMed] [Google Scholar]
- 3.Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 4.Benzi G, Moretti A. Is there a rationale for the use of acetylcholinesterase inhibitors in the therapy of Alzheimer's disease? Eur J Pharmacol. 1998;346(1):1–13. doi: 10.1016/s0014-2999(98)00093-4. [DOI] [PubMed] [Google Scholar]
- 5.Blusztajn JK, Berse B. The cholinergic neuronal phenotype in Alzheimer's disease. Metab Brain Dis. 2000;15(1):45–64. doi: 10.1007/BF02680013. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2(3):271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 7.Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101(8):2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 9.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3(5):339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 10.Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25(6):317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Davis KL, Mohs RC, Tinklenberg JR, Pfefferbaum A, Hollister LE, Kopell BS. Physostigmine: improvement of long-term memory processes in normal humans. Science. 1978;201(4352):272–274. doi: 10.1126/science.351807. [DOI] [PubMed] [Google Scholar]
- 12.Furey ML, Pietrini P, Haxby JV, Alexander GE, Lee HC, VanMeter J, Grady CL, Shetty U, Rapoport SI, Schapiro MB, Freo U. Cholinergic stimulation alters performance and task-specific regional cerebral blood flow during working memory. Proc Natl Acad Sci U S A. 1997;94(12):6512–6516. doi: 10.1073/pnas.94.12.6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furey ML, Pietrini P, Haxby JV. Cholinergic enhancement and increased selectivity of perceptual processing during working memory. Science. 2000;290(5500):2315–2319. doi: 10.1126/science.290.5500.2315. [DOI] [PubMed] [Google Scholar]
- 14.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373(6514):523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 15.Giacobini E. Cholinergic receptors in human brain: effects of aging and Alzheimer disease. J Neurosci Res. 1990;27(4):548–560. doi: 10.1002/jnr.490270416. [DOI] [PubMed] [Google Scholar]
- 16.Golde TE. Disease modifying therapy for AD? J Neurochem. 2006;99(3):689–707. doi: 10.1111/j.1471-4159.2006.04211.x. [DOI] [PubMed] [Google Scholar]
- 17.Greenamyre JT, Maragos WF. Neurotransmitter receptors in Alzheimer disease. Cerebrovasc Brain Metab Rev. 1993;5(2):61–94. [PubMed] [Google Scholar]
- 18.Gu Z, Yan Z. Bidirectional regulation of Ca2+/calmodulin-dependent protein kinase II activity by dopamine D4 receptors in prefrontal cortex. Mol Pharmacol. 2004;66(4):948–955. doi: 10.1124/mol.104.001404. [DOI] [PubMed] [Google Scholar]
- 19.Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25(20):4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshi M, Takashima A, Murayama M, Yasutake K, Yoshida N, Ishiguro K, Hoshino T, Imahori K. Nontoxic amyloid beta peptide 1–42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer's disease. J Biol Chem. 1997;272(4):2038–2041. doi: 10.1074/jbc.272.4.2038. [DOI] [PubMed] [Google Scholar]
- 21.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 22.Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45(5):583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22(12):555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 24.Kar S, Seto D, Gaudreau P, Quirion R. Beta-amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal slices. J Neurosci. 1996;16(3):1034–1040. doi: 10.1523/JNEUROSCI.16-03-01034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly JF, Furukawa K, Barger SW, Rengen MR, Mark RJ, Blanc EM, Roth GS, Mattson MP. Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U S A. 1996;93(13):6753–6758. doi: 10.1073/pnas.93.13.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SH, Kim YK, Jeong SJ, Haass C, Kim YH, Suh YH. Enhanced release of secreted form of Alzheimer's amyloid precursor protein from PC12 cells by nicotine. Mol Pharmacol. 1997;52(3):430–436. doi: 10.1124/mol.52.3.430. [DOI] [PubMed] [Google Scholar]
- 27.Lahiri DK, Utsuki T, Chen D, Farlow MR, Shoaib M, Ingram DK, Greig NH. Nicotine reduces the secretion of Alzheimer's beta-amyloid precursor protein containing beta-amyloid peptide in the rat without altering synaptic proteins. Ann N Y Acad Sci. 2002;965:364–372. doi: 10.1111/j.1749-6632.2002.tb04178.x. [DOI] [PubMed] [Google Scholar]
- 28.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443(7113):774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 29.Lawlor BA, Davis KL. Does modulation of glutamatergic function represent a viable therapeutic strategy in Alzheimer's disease? Biol Psychiatry. 1992;31(4):337–350. doi: 10.1016/0006-3223(92)90227-q. [DOI] [PubMed] [Google Scholar]
- 30.Lisman JE, Fellous JM, Wang XJ. A role for NMDA-receptor channels in working memory. Nat Neurosci. 1998;1(4):273–275. doi: 10.1038/1086. [DOI] [PubMed] [Google Scholar]
- 31.Liu Q, Kawai H, Berg DK. beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98(8):4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 34.Mesulam MM. The systems-level organization of cholinergic innervation in the human cerebral cortex and its alterations in Alzheimer's disease. Prog Brain Res. 1996;109:285–297. doi: 10.1016/s0079-6123(08)62112-3. [DOI] [PubMed] [Google Scholar]
- 35.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443(7113):768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 36.Pereira EF, Reinhardt-Maelicke S, Schrattenholz A, Maelicke A, Albuquerque EX. Identification and functional characterization of a new agonist site on nicotinic acetylcholine receptors of cultured hippocampal neurons. J Pharmacol Exp Ther. 1993;265(3):1474–1491. [PubMed] [Google Scholar]
- 37.Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience. 1995;64(2):385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 38.Robbins TW, Murphy ER. Behavioural pharmacology: 40+ years of progress, with a focus on glutamate receptors and cognition. Trends Pharmacol Sci. 2006;27(3):141–148. doi: 10.1016/j.tips.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314(5800):781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003;9(3):275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schroder B, Reinhardt-Maelicke S, Schrattenholz A, McLane KE, Kretschmer A, Conti-Tronconi BM, Maelicke A. Monoclonal antibodies FK1 and WF6 define two neighboring ligand binding sites on Torpedo acetylcholine receptor alpha-polypeptide. J Biol Chem. 1994;269(14):10407–10416. [PubMed] [Google Scholar]
- 42.Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271(31):18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 43.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8(11):447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 44.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 46.Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298(5594):776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- 47.Sitaram N, Weingartner H, Gillin JC. Human serial learning: enhancement with arecholine and choline impairment with scopolamine. Science. 1978;201(4352):274–276. doi: 10.1126/science.351808. [DOI] [PubMed] [Google Scholar]
- 48.Sivaprakasam K. Towards a unifying hypothesis of Alzheimer's disease: cholinergic system linked to plaques, tangles and neuroinflammation. Curr Med Chem. 2006;13(18):2179–2188. doi: 10.2174/092986706777935203. [DOI] [PubMed] [Google Scholar]
- 49.Smith AD. Imaging the progression of Alzheimer pathology through the brain. Proc Natl Acad Sci U S A. 2002;99(7):4135–4137. doi: 10.1073/pnas.082107399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonkusare SK, Kaul CL, Ramarao P. Dementia of Alzheimer's disease and other neurodegenerative disorders--memantine, a new hope. Pharmacol Res. 2005;51(1):1–17. doi: 10.1016/j.phrs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Takada-Takatori Y, Kume T, Sugimoto M, Katsuki H, Sugimoto H, Akaike A. Acetylcholinesterase inhibitors used in treatment of Alzheimer's disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology. 2006;51(3):474–486. doi: 10.1016/j.neuropharm.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Tanzi RE, Bertram L. New frontiers in Alzheimer's disease genetics. Neuron. 2001;32(2):181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- 53.Weinstock M. The pharmacotherapy of Alzheimer's disease based on the cholinergic hypothesis: an update. Neurodegeneration. 1995;4(4):349–356. doi: 10.1006/neur.1995.0042. [DOI] [PubMed] [Google Scholar]
- 54.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10(2):122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 55.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 56.Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. Essential role of neocortical acetylcholine in spatial memory. Nature. 1995;375(6531):484–487. doi: 10.1038/375484a0. [DOI] [PubMed] [Google Scholar]
- 57.Yan Z, Feng J. Alzheimer's disease: interactions between cholinergic functions and beta-amyloid. Curr Alzheimer Res. 2004;1(4):241–248. doi: 10.2174/1567205043331992. [DOI] [PubMed] [Google Scholar]
- 58.Yuen EY, Jiang Q, Chen P, Gu Z, Feng J, Yan Z. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J Neurosci. 2005;25(23):5488–5501. doi: 10.1523/JNEUROSCI.1187-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong P, Gu Z, Wang X, Jiang H, Feng J, Yan Z. Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer's disease. J Biol Chem. 2003;278(29):26888–26896. doi: 10.1074/jbc.M302789200. [DOI] [PubMed] [Google Scholar]