

Abstract
The paralyzed zebrafish strain relaxed carries a null mutation for the skeletal muscle dihydropyridine receptor (DHPR) β1a subunit. Lack of β1a results in (i) reduced membrane expression of the pore forming DHPR α1S subunit, (ii) elimination of α1S charge movement, and (iii) impediment of arrangement of the DHPRs in groups of four (tetrads) opposing the ryanodine receptor (RyR1), a structural prerequisite for skeletal muscle-type excitation-contraction (EC) coupling. In this study we used relaxed larvae and isolated myotubes as expression systems to discriminate specific functions of β1a from rather general functions of β isoforms. Zebrafish and mammalian β1a subunits quantitatively restored α1S triad targeting and charge movement as well as intracellular Ca2+ release, allowed arrangement of DHPRs in tetrads, and most strikingly recovered a fully motile phenotype in relaxed larvae. Interestingly, the cardiac/neuronal β2a as the phylogenetically closest, and the ancestral housefly βM as the most distant isoform to β1a also completely recovered α1S triad expression and charge movement. However, both revealed drastically impaired intracellular Ca2+ transients and very limited tetrad formation compared with β1a. Consequently, larval motility was either only partially restored (β2a-injected larvae) or not restored at all (βM). Thus, our results indicate that triad expression and facilitation of 1,4-dihydropyridine receptor (DHPR) charge movement are common features of all tested β subunits, whereas the efficient arrangement of DHPRs in tetrads and thus intact DHPR-RyR1 coupling is only promoted by the β1a isoform. Consequently, we postulate a model that presents β1a as an allosteric modifier of α1S conformation enabling skeletal muscle-type EC coupling.
Excitation-contraction (EC)3 coupling in skeletal muscle is critically dependent on the close interaction of two distinct Ca2+ channels. Membrane depolarizations of the myotube are sensed by the voltage-dependent 1,4-dihydropyridine receptor (DHPR) in the sarcolemma, leading to a rearrangement of charged amino acids (charge movement) in the transmembrane segments S4 of the pore-forming DHPR α1S subunit (1, 2). This conformational change induces via protein-protein interaction (3, 4) the opening of the sarcoplasmic type-1 ryanodine receptor (RyR1) without need of Ca2+ influx through the DHPR (5). The release of Ca2+ from the sarcoplasmic reticulum via RyR1 consequently induces muscle contraction. The protein-protein interaction mechanism between DHPR and RyR1 requires correct ultrastructural targeting of both channels. In Ca2+ release units (triads and peripheral couplings) of the skeletal muscle, groups of four DHPRs (tetrads) are coupled to every other RyR1 and hence are geometrically arranged following the RyR-specific orthogonal arrays (6).
The skeletal muscle DHPR is a heteromultimeric protein complex, composed of the voltage-sensing and pore-forming α1S subunit and auxiliary subunits β1a, α2δ-1, and γ1 (7). While gene knock-out of the DHPR γ1 subunit (8, 9) and small interfering RNA knockdown of the DHPR α2δ-1 subunit (10-12) have indicated that neither subunit is essential for coupling of the DHPR with RyR1, the lack of the α1S or of the intracellular β1a subunit is incompatible with EC coupling and accordingly null model mice die perinatally due to asphyxia (13, 14). β subunits of voltage-gated Ca2+ channels were repeatedly shown to be responsible for the facilitation of α1 membrane insertion and to be potent modulators of α1 current kinetics and voltage dependence (15, 16). Whether the loss of EC coupling in β1-null mice was caused by decreased DHPR membrane expression or by the lack of a putative specific contribution of the β subunit to the skeletal muscle EC coupling apparatus (17, 18) was not clearly resolved. Recently, other β-functions were identified in skeletal muscle using the β1-null mutant zebrafish relaxed (19, 20). Like the β1-knock-out mouse (14) zebrafish relaxed is characterized by complete paralysis of skeletal muscle (21, 22). While β1-knock-out mouse pups die immediately after birth due to respiratory paralysis (14), larvae of relaxed are able to survive for several days because of oxygen and metabolite diffusion via the skin (23). Using highly differentiated myotubes that are easy to isolate from these larvae, the lack of EC coupling could be described by quantitative immunocytochemistry as a moderate ∼50% reduction of α1S membrane expression although α1S charge movement was nearly absent, and, most strikingly, as the complete lack of the arrangement of DHPRs in tetrads (19). Thus, in skeletal muscle the β subunit enables EC coupling by (i) enhancing α1S membrane targeting, (ii) facilitating α1S charge movement, and (iii) enabling the ultrastructural arrangement of DHPRs in tetrads.
The question arises, which of these functions are specific for the skeletal muscle β1a and which ones are rather general properties of Ca2+ channel β subunits. Previous reconstitution studies made in the β1-null mouse system (24, 25) using different β subunit constructs (26) did not allow differentiation between β-induced enhancement of non-functional α1S membrane expression and the facilitation of α1S charge movement, due to the lack of information on α1S triad expression levels. Furthermore, the β-induced arrangement of DHPRs in tetrads was not detected as no ultrastructural information was obtained.
In the present study, we established zebrafish mutant relaxed as an expression system to test different β subunits for their ability to restore skeletal muscle EC coupling. Using isolated myotubes for in vitro experiments (19, 27) and complete larvae for in vivo expression studies (28-31) and freeze-fracture electron microscopy, a clear differentiation between the major functional roles of β subunits was feasible in the zebrafish system. The cloned zebrafish β1a and a mammalian (rabbit) β1a were shown to completely restore all parameters of EC coupling when expressed in relaxed myotubes and larvae. However, the phylogenetically closest β subunit to β1a, the cardiac/neuronal isoform β2a from rat, as well as the ancestral βM isoform from the housefly (Musca domestica), could recover functional α1S membrane insertion, but led to very restricted tetrad formation when compared with β1a, and thus to impaired DHPR-RyR1 coupling. This impairment caused drastic changes in skeletal muscle function.
The present study shows that the enhancement of functional α1S membrane expression is a common function of all the tested β subunits, from β1a to even the most distant βM, whereas the effective formation of tetrads and thus proper skeletal muscle EC coupling is an exclusive function of the skeletal muscle β1a subunit. In context with previous studies, our results suggest a model according to which β1a acts as an allosteric modifier of α1S conformation. Only in the presence of β1a, the α1S subunit is properly folded to allow RyR1 anchoring and thus skeletal muscle-type EC coupling.
EXPERIMENTAL PROCEDURES
Zebrafish Embryos—Adult zebrafish, heterozygous for the β1-null redts25 (relaxed) mutation were maintained and bred under standard aquarium conditions (28, 29). Freshly spawned eggs were directly used for zygote RNA microinjection (see below) and/or raised until 25-32 h post-fertilization (hpf) at 28 °C to be used for experiments.
Expression Plasmids—All β subunit cDNAs were N-terminally fused in-frame to GFP cDNA and cloned into expression vector pCI-neo (Promega) that allows both, in vitro RNA synthesis for zygote injection as well as transient expression in cultured relaxed myotubes. Constructs were designed as follows, with nucleotide numbers (nt) given in parentheses and asterisks indicating restriction enzyme (RE) sites introduced by the PCR technique using proofreading Pfu Turbo DNA polymerase (Stratagene). The integrity of cDNA sequences generated by PCR was confirmed by sequence analysis (Eurofins MWG Operon, Martinsried, Germany).
zf-β1a—Total RNA from adult wild type (WT) zebrafish muscle was isolated using the RNeasy Mini kit (Qiagen) and reverse transcribed using the Ready-To-Go T-primed first-strand kit (Amersham Biosciences). From the first-strand cDNA, the zf-β1a open reading frame (GenBank™ AY952462) was PCR-generated in three fragments: HindIII*-XhoI (nt -5-502), XhoI-HindIII (nt 502-1352), and HindIII-BamHI* (nt 1352-1577). A subclone was created by co-ligating fragments XhoI-HindIII (nt 502-1352) and HindIII-BamHI* (nt 1352-1577) into the XhoI/BamHI polylinker RE sites of pBlue-script SK+ (pBS) (Stratagene). For N-terminal GFP tagging, fragment HindIII*-XhoI (nt -5-502) was in-frame ligated together with the excised fragment XhoI-BamHI* (nt 502-1577) into the HindIII/BamHI polylinker RE sites of the proprietary expression plasmid pGFP37 (32). From this subclone GFP-zf-β1a cDNA was excised with PstI-XhoI (nt -734-502) and XhoI-BamHI* (nt 502-1577) and ligated into the PstI/BamHI cut pBS. For the final construct zf-β1a, the SalI-BamHI* (nt -771-1577) insert was co-ligated with the 226-bp poly(A) tail excised with BamHI-NotI from the proprietary transcription plasmid pNKS2 (a gift of O. Pongs) into the XhoI/NotI cut polylinker of pCI-neo.
rb-β1a—The open reading frame of rabbit β1a cDNA (Gen-Bank NM_001082279) was isolated from plasmid pcDNA3 (33) as the HindIII-BstXI fragment (nt -20-834) and as the BstXI-BamHI (nt 834-1575) PCR fragment, reintroducing its original stop codon at nt 1572. For N-terminal GFP tagging, both fragments were co-ligated into the HindIII/BamHI polylinker RE sites of pGFP37. From this subclone GFP-rb-β1a cDNA was excised with PstI-KpnI (nt -734 to -10) and KpnI-BamHI* (nt -10-1575) and ligated into the PstI/BamHI cut pBS. To gain the final construct for rb-β1a, the SalI-BamHI* (nt -771-1575) insert was co-ligated with the BamHI-NotI-excised poly(A) tail (see above) into the XhoI/NotI cut polylinker of pCI-neo.
β2a—The open reading frame of rat β2a cDNA (GenBank M80545) was isolated from plasmid p91023(B) (34) as the HindIII-XhoI fragment (nt -11-1064) and the XhoI-BamHI* (nt 1064-1816) PCR fragment. For GFP tagging, fragments were co-ligated into the HindIII/BamHI polylinker RE sites of pGFP37. From this subclone GFP-β2a cDNA was excised with PstI-BamHI* (nt -740-1816) and ligated into the PstI/BamHI opened pBS. As a final step, the SalI-BamHI* (nt -777-1816) insert was co-ligated with the BamHI-NotI cut poly(A) tail into the XhoI/NotI opened polylinker of pCI-neo.
βM—Musca β (βM) cDNA (GenBank X78561) was isolated from plasmid βM-pNKS2 (35) as HindIII-DraIII (nt 8-2369) and DraIII-XbaI (nt 2369-3506) fragments and was co-ligated in-frame with GFP cDNA into the HindIII/XbaI polylinker of pGFP37. GFP-βM cDNA was isolated as PstI-DraIII (nt -721-2369) and DraIII-XbaI (nt 2369-3506) fragments and ligated into the PstI/XbaI opened pBS. To generate the final βM construct, fragments SalI-DraIII (nt -758-2369) and DraIII-XbaI (nt 2369-3506) were ligated into the XhoI/XbaI cut polylinker of pCI-neo.
GFP—For standardizing experimental conditions, GFP alone was cloned into expression vector pCI-neo in the following way: the GFP cDNA was excised EcoRI-HindIII (nt -24-716) from subclone GFP-zf-β1a in pBS (see above; GFP-open reading frame numbering) and was co-ligated with the HindIII-NotI cut poly(A) tail into the EcoRI/NotI-cleaved polylinker of pCI-neo.
Primary Culture of Zebrafish Myotubes—For the isolation of myoblasts, 25-28 hpf chorionated embryos derived from heterozygous relaxed parental fish were surface-sterilized using 0.5% sodium hypochlorite for 2 min and then enzymatically dechorionated using 2 mg/ml Pronase (Protease, Type XIV, Sigma) (28) for 20 min at 28 °C and collected in 0.5× Hanks' buffered salt saline (Sigma). Homozygous relaxed larvae were identified by their inability to move despite tactile stimulation. Motile “normal” siblings (i.e. heterozygous and WT) were used for control experiments. 100-150 larvae were anesthetized with 0.02% tricaine (MS-222; Sigma), decapitated, and the tails digested for 1 h in 200 units/ml collagenase type I in Hanks' buffered salt saline (Sigma) at 28 °C in a thermomixer with continuous trituration. Collagenase digestion was stopped by adding 7 ml of zebrafish culture medium containing 60% L-15 medium (Sigma) with 3% fetal calf serum, 3% horse serum (both Invitrogen), and 4 mm l-glutamine (Sigma). After centrifugation for 5 min at 200 × g cells were resuspended and transfected with 2 μg of expression plasmid cDNA using the AMAXA™ rat neonatal cardiomyocyte nucleofector kit (AMAXA Biosystems, Köln, Germany) according to the manufacturer's manual. Myocytes were resuspended in 200 μl of zebrafish medium supplemented with 4 units/ml penicillin/streptomycin (Invitrogen) (“full zebrafish medium”) and plated on carbon, gelatin, and collagen-coated glass coverslips (for immunocytochemical experiments) or as droplets in the center of collagen-coated plastic dishes (for electrophysiological experiments). After 20 min, 1.5 ml of full zebrafish medium was added and cells were cultured at 28 °C for 4 to 6 days.
Immunocytochemistry—Myotubes cultured on glass coverslips were washed in phosphate-buffered saline supplemented with 100 μm N-benzyl-p-toluene sulfonamide. Cells were fixed with 4% paraformaldehyde in 0.1 m sodium phosphate buffer for 20 min, permeabilized, and blocked by incubating with 5% normal goat serum in phosphate-buffered saline supplemented with 0.2% bovine serum albumin and 0.2% Triton X-100 (PBT) for 30 min, followed by incubation with primary antibodies in PBT overnight at 4 °C. Primary antibodies used were monoclonal antibody 1A against α1S (Affinity Bioreagents) at 1:2,000 (36, 37) and rb-anti-GFP (Invitrogen) at 1:5,000 dilutions. After several washes with PBT, secondary antibodies, goat anti-mouse Alexa Fluor 594, and goat anti-rabbit Alexa Fluor 488 (Invitrogen) at a concentration of 1:4,000 in PBT were applied for 1 h at room temperature. Specimens were mounted in 90% glycerol, 0.1 m Tris with 5 mg/ml p-phenylendiamine to retard photobleaching (38). Images were taken with a cooled CCD camera (Diagnostic Instruments) mounted on a Zeiss Axiophot microscope equipped with a ×63, 1.4 NA objective lens, using MetaVue image-processing software (Universal Imaging, West Chester, PA). For quantification of α1S triad expression, images were acquired with identical exposure times, followed by background subtraction and shading correction. Transfected cells were identified by positive anti-GFP staining.
Quantification of α1S triad expression was determined by measuring the average fluorescence intensity of Alexa Fluor 595 along a line across a row of α1S clusters (triadic junctions; see Fig. 1A) in 5 measurements on each myotube, which were obtained from at least 2 different cultures. Myotubes that barely expressed GFP-β (and as a consequence also α1S) and were only visible because of signal amplification by anti-GFP/Alexa Fluor 488 staining were excluded from the α1S quantification to allow a quantitative link to our patch-clamp data. To this aim we determined the percentage of expressing myotubes, either identified by direct GFP fluorescence (patch-clamp approach) or by GFP-antibody enhancement (immunocytochemical approach) from the total number of myotubes. Calculations were done from 2 different preparations for both approaches. Fractions of expressing cells were 7 ± 1%, n = 460; and 21 ± 6%, n = 325, for the patch-clamp and immunocytochemical approach, respectively. Thus, to enable a link between both approaches, only the values of the highest1/3 of expressing myotubes were considered for α1S fluorescence quantification.
FIGURE 1.

Qualitative and quantitative restoration of α1S triad expression in relaxed myotubes with all tested β subunit isoforms. A, representative images of double immunofluorescence labeling of the DHPR α1S subunit (anti-α1S) and GFP-tagged β subunits (anti-GFP). Normal (1st row) and relaxed (2nd row) myotubes mock-transfected with pure GFP revealed diffuse GFP staining throughout the cell (center images). Relaxed myotubes transfected with zf-β1a, rb-β1a, β2a, or βM showed correct targeting of the β subunits into triadic clusters (center images) and co-localization (merge) with clusters of the endogenous α1S subunit (left). B, quantification of α1S triad expression by measuring average fluorescence intensity along a line across a row of α1S triadic clusters (exemplified 1st row, left; indicated by a red arrowhead) showed that both, skeletal and non-skeletal muscle β subunit isoforms were able to completely restore α1S triad expression in relaxed myotubes (**, p < 0.01; ***, p < 0.001).
Whole-cell Patch Clamp Analysis—Immobilization-resistant intramembrane charge movement, as a measure of functional α1S expression (39), as well as intracellular Ca2+ transients were recorded from myotubes cultured for 4-6 days after transfection. GFP fluorescing myotubes were patch clamp analyzed on an Olympus IX70 inverted fluorescence microscope equipped with Hoffmann modulation contrast. Patch pipettes were pulled from borosilicate glass (Harvard Instruments), fire-polished (Microforge MF-830, Narishige), and had resistances of 3.5-5 MΩ after back-filling with pipette solution containing 100 mm Cs-aspartate, 10 mm HEPES, 0.5 mm CsEGTA, 3 mm MgATP, and 0.2 mm Fluo-4 (pH 7.4 with CsOH). The bath solution consisted of 10 mm Ca(OH)2, 100 mm l-aspartate, and 10 mm HEPES (pH 7.4 with tetraethylammonium hydroxide). Contractions of myotubes were blocked by adding 100 μm of the myosin-II blocker N-benzyl-p-toluene sulfonamide (Sigma) to the bath solution (40). Recordings were performed with an Axopatch 200B amplifier controlled by pClamp software (version 7.0; Axon Instruments Inc., Foster City, CA) and leak currents were subtracted by a P/4 prepulse protocol. To inactivate endogenous T-type currents all test pulses were preceded by a 1-s prepulse to -30 mV (39). Recordings were low-pass Bessel-filtered at 1 kHz and sampled at 5 kHz. DHPR charge movement was measured in 20-ms depolarizing test pulses starting from a test potential of +70 down to -60 mV in 10-mV increments. Total charge movement was calculated by integrating the ON-component of gating currents. 0.2 mm Fluo-4 was added to the patch pipette solution to measure intracellular Ca2+ release. Fluo-4 fluorescence was recorded using a PTI 814 photomultiplier system (PTI, S. Brunswick, NJ). Average fluorescence intensity (F) of a rectangular region on the patched myotube was recorded in 200-ms depolarizing test pulses from +80 to -50 mV in 10-mV increments with a holding potential of -80 mV. The average fluorescence was normalized to the resting fluorescence and expressed as ΔF/F0. The voltage dependence of charge movement (Q) and maximum intracellular Ca2+ release for each test potential were fitted according to the following Boltzmann distribution,
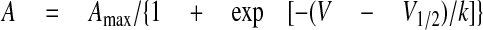 |
(Eq.1) |
where A is Q or ΔF/F0, V½ is the potential at which A = Amax/2, and k is a slope factor. Data were analyzed using ClampFit 9.0 and 10.0 (Axon Instruments) and SigmaPlot 9.0 and 10.0 (SPSS Science, Chicago, IL) software.
Zygote Injection of in Vitro Synthesized RNA—For in vitro RNA synthesis 50 μg of all β subunit cDNAs and GFP cDNA were linearized with restriction enzymes XbaI and NotI, respectively, purified with phenol/chloroform and precipitated with 3 m NH4Ac in 70% EtOH and the pellet redissolved in RNase-free water. Linearized DNA templates were fidelity checked on an agarose gel. In vitro transcription was performed in a volume of 100 μl containing: 5 μg of linearized DNA template, 10 mm NTPs, with GTP supplemented by m7(5′)ppp(5′)G-cap (Roche Diagnostics), 100 units of T7-RNA Polymerase (Roche), and 200 units of RNase inhibitor (RNAsin; Roche), and incubated at 37 °C for 1 h. Template DNA was digested with 100 units of RNase-free DNase (Roche) for 15 min at 37 °C. After phenol/chloroform extraction and ethanol precipitation, the RNA pellet was redissolved in RNase-free water and aliquots frozen at -80 °C. RNA fidelity and concentration were checked on a 7% formaldehyde-agarose gel in MOPS running buffer.
For RNA injection, eggs from heterozygous parental zebrafish in the one-cell stage were collected immediately after spawning and positioned in a 0.9-mm groove of an agarose tray to be microinjected within 20 min. Injection needles were pulled from heat-sterilized borosilicate glass capillaries (Harvard Instruments) and front-filled with RNA solution (0.2 μg/μl), containing 0.1% phenol red as an injection volume tracer (29). Injection volume of RNA solution was ∼1/5 of total zygote volume (calculated 13 nl) and was injected using a motorized micromanipulator DC3001 and the pneumatic PicoPump PV830 (both WPI, Germany). Eight hours after injection, GFP fluorescence of healthy embryos was quantified using a PTI 814 photomultiplier system. Only proper developing injected embryos with a mean fluorescence signal exceeding 40% above uninjected control embryos were considered for freeze-fracture electron microscopy or digital motion analysis.
Identification of Rescued Relaxed Larvae—Discrimination of the 25% of motility restored homozygous relaxed larvae used in motion analysis experiments from the injected normal siblings was done by keeping all injected larvae separated and thus identifiable for up to 5 days and by observing a gradual fallback to the paralyzed phenotype due to degradation of the injected β-RNAs and translated proteins. Only in the case where larval tail muscle tissue was used for freeze-fracture electron microscopy, motility restored relaxed larvae had to be identified by a restriction fragment length polymorphism test on the larval heads. For this, larvae were anesthetized, decapitated, and the tails fixed as identifiable specimen as described below. Genomic DNA was extracted by incubating the larval heads in DNA extraction buffer containing: 10 mm Tris (pH 8.2), 10 mm EDTA, 200 mm NaCl, 0.5% SDS, and 200 μg/ml proteinase K for 1 h at 55 °C with intermittent vortexing, followed by ethanol precipitation. After washing in 70% ethanol, the pellet was redissolved in water (29) and the DNA was used as PCR template to amplify a 459-bp fragment containing the relaxed mutation. The relaxed genotype was identified by restriction enzyme digest of the PCR product with BsrI. The PCR product was cleaved into 279- and 180-bp fragments only in the presence of WT alleles.
Freeze-fracture Electron Microscopy—Immediately after decapitating, tails of the injected motile larvae at 27-30 hpf were fixed with 6% glutaraldehyde in 0.1 m cacodylate buffer at neutral pH (both Sigma) and incubated for 30 min at room temperature. Tails were mechanically skinned in 3% glutaraldehyde in 0.1 m cacodylate buffer and stored at 4 °C until processing for freeze-fracture. Tails from motility restored relaxed larvae were cryoprotected in 30% glycerol, mounted between two copper holders covered with a thin layer of 20% polyvinyl alcohol in 30% glycerol, and frozen in liquid nitrogen-cooled propane. Finally, tails were freeze fractured by separating the two holders under vacuum, shadowed with platinum at 45 °C, and replicated with carbon in a freeze-fracture unit (model BFA 400; Balzers SpA). Replicas were analyzed in an electron microscope (model 410; Philips) and searched for myotomes that contained myotubes at the developmental stage when peripheral couplings between the sarcoplasmic reticulum and the surface membrane were present. At the age used, these comprise a group of 3-4 myotomes in the middle of the tail. Sites of peripheral couplings were identified by the clusters of unique large particles representing the position of DHPRs.
Digital Motion Analysis—Motion analysis was performed on custom made computer programs using different image and data analysis platforms: Optimas 6.5, Image Pro 6, and LabView 8.5. For quantification of larval motility, larvae were dechorionated (see above) and transferred to 24-well plates to keep individuals identifiable for several days. 2-Min video sequences with 25 frames/s were acquired with a Sony CCD AVC-D7CE b/w camera and stored as multipaged TIFF stacks. The TIFF stacks were converted into stacks of differential images by subtracting subsequent images (41). To eliminate pixel noise, all differential images were 3 × 3 median filtered. The mean luminance of every image reflecting larval movement was quantified by automated counting of total dynamic pixels per image and plotting against time. On this processed signal, peak detection was performed. Peak detection is based on an algorithm that fits a quadratic polynomial to sequential groups of data points. The number of data points used in the fit was specified by the width of typical peaks found in the acquired signals. For each peak, the quadratic fit was tested against the threshold level that in turn was determined for each individual larva separately and depended on small differences in illumination and larval orientation. Peak amplitudes below the threshold level (e.g. small peaks induced by passive movements of the larva) were ignored. Peaks were detected only after the procession of approximately width/2 data points beyond the location of the peak (42). The cumulative dynamic pixels per peak were calculated and the mean value for all larval movements of each experimental group was determined.
Statistics—Statistical significance from experimental approaches was determined by unpaired Student's t test and data are reported as mean ± S.E., unless noted otherwise.
RESULTS
A prerequisite to be able to use muscle cells and the entire larvae of the β1-null zebrafish mutant relaxed as expression systems is the complete phenotype rescue with the homologous zebrafish β1a. Therefore, we cloned β1a cDNA from WT zebrafish skeletal muscle (19) and expressed it in relaxed myotubes and larvae. To test if the zebrafish expression system permits species-independent conclusions we also used a mammalian (rabbit) skeletal muscle β1a subunit (43). These two β1a subunits, which share 76% all-over amino acid identity, were tested against two non-skeletal muscle isoforms to dissect specific functions of β1a from general β functions. For this aim, we tested the cardiac/neuronal β2a subunit, as the phylogenetically closest isoform to β1a and a β subunit that is phylogenetically basal to all four mammalian β subunit isoforms (35), namely the neuronal βM from the housefly (M. domestica) (77 and 60% amino acid identity of the core region (35) to rabbit β1a, respectively).
Triad Targeting of α1S: A Common Feature of β Subunits—Expression and targeting of the heterologously expressed β subunits and their influence on the targeting of the endogenous α1S subunit was investigated with immunocytochemistry on transfected, primary cultures of relaxed myotubes. Normal and relaxed myotubes transfected with pure GFP showed a diffuse pattern upon anti-GFP staining and a punctuate pattern of foci aligned along transverse stripes with the sarcomeric spacing upon anti-α1S staining (Fig. 1A, 1st and 2nd rows). As previously shown (19) the foci correspond to the location of triads. By quantifying α1S immunofluorescence we could determine α1S membrane expression in relaxed as 44 ± 2% (n = 90) compared with GFP mock-transfected normal myotubes (100 ± 1%, n = 359, p < 0.001) (Fig. 1B), and thus confirm previous results (19). Relaxed myotubes transfected with either GFP-tagged zf-β1a or rb-β1a showed co-clustering of β1a, detected by anti-GFP antibody, with the endogenous α1S (Fig. 1A, 3rd and 4th rows) in a pattern indistinguishable from normal myotubes (not shown, but see Ref. 19). Quantification of α1S immunofluorescence revealed a complete rescue of α1S triad expression comparable or slightly above normal myotubes or of relaxed myotubes transfected with zf-β1a (98 ± 3%; n = 17; p > 0.05) or rb-β1a (112 ± 2%; n = 47; p < 0.01) (Fig. 1B). To test whether this facilitation of α1S triad targeting, as a muscle-specific targeting feature, is an exclusive function of the skeletal muscle β1a we also tested the cardiac/neuronal β2a and βM subunits. Interestingly, both heterologous β subunits correctly colocalized with α1S in triadic clusters (Fig. 1A, 5th and 6th rows). Importantly, α1S triad targeting was restored by both, β2a and βM to levels (88 ± 11%, n = 16 and 97 ± 5%, n = 17, respectively; p > 0.05) comparable with normal myotubes (Fig. 1B).
Facilitation of α1S Charge Movement: A Common Feature of β Subunits—Relaxed myotubes showed almost complete lack of immobilization-resistant intramembrane (α1S) charge movement, despite the fact that α1S immunofluorescence was still ∼44% of normal myotubes (19) (Figs. 1B and 2). This divergence in immunocytochemical and electrophysiological data strongly suggests a role of β1a in the voltage-dependent conformational change of α1S that triggers skeletal muscle-type EC coupling. To test if this can be similarly achieved by homologous and heterologous β subunits, we recorded charge movement from relaxed myotubes transfected with all four β constructs. Both β1a isoforms were able to recover robust α1S intramembrane charge movements (Fig. 2A) with Qmax values of 11.13 ± 1.72 nC/μF, n = 12, for zf-β1a and 11.07 ± 0.77 nC/μF, n = 22, for rb-β1a (Fig. 2B). Interestingly, β2a and the phylogenetically even more distant βM were also able to recover α1S intramembrane charge movements to a level comparable with that of the homologous β1a subunits (9.94 ± 2.06, n = 26, for β2a and 11.09 ± 1.04, n = 9 for βM) (Fig. 2C). Qmax values recorded from all expression experiments were somewhat higher than those recorded from normal myotubes mock-transfected with GFP (7.76 ± 0.48 nC/μF, n = 31), pointing to a moderate β overexpression. The voltage dependence of charge movement was similar to that of normal myotubes (p > 0.05) for all constructs (half-maximal activation in mV: normal + GFP, -4.99 ± 0.94; zf-β1a, -3.97 ± 2.19; rb-β1a, -4.49 ± 0.85; β2a, -5.16 ± 1.04; βM, -3.47 ± 2.75).
FIGURE 2.

Complete restoration of intramembrane α1S charge movement in relaxed myotubes with all tested β subunit isoforms. A, representative recordings of intramembrane α1S charge movement at test potentials of -50, 0, and +50 mV from a holding potential of -80 mV of normal myotubes mock-transfected with GFP, untransfected relaxed myotubes, and relaxed myotubes transfected with zf-β1a, rb-β1a, β2a, or βM. B, voltage dependence of the integrated ON-component of intramembrane α1S charge movement (Qon) were comparable for zf-β1a-, rb-β1a-transfected relaxed myotubes and normal myotubes mock-transfected with GFP. Maximal charge movement (Qmax) values were somewhat higher (p < 0.05) for zf-β1a (♦) and rb-β1a (⋄) compared with normal myotubes (○). No charge movement at any potential could be recorded from untransfected relaxed myotubes (•). C, heterologous β isoforms, β2a (▴) and βM (▿) were likewise able to fully recover intramembrane α1S charge movement comparable with the two homologous β1a isoforms.
Skeletal-type EC Coupling: A Specific Feature of β1a—After triad targeting and functional expression of α1S were shown to be non-exclusive features of β1a, we raised the question if this nonspecificity also holds true for the restoration of proper DHPR-RyR1 coupling. Unlike skeletal muscle of other vertebrates, zebrafish skeletal muscle shows no DHPR inward Ca2+ current (19)4 and thus a possible contamination of the measurements of intracellular RyR1 Ca2+ release by influx of extracellular Ca2+ and thus a cardiac-type EC coupling component (44-46) is not present. Both skeletal muscle isoforms, zf-β1a and rb-β1a, were able to restore intracellular Ca2+ transients (Fig. 3A) with similar voltage dependence and with maximum ΔF/F0 values of 1.91 ± 0.39 for zf-β1a (n = 9) and 2.07 ± 0.17 for rb-β1a (n = 23), indistinguishable (p > 0.05) from normal transients with a maximal ΔF/F0 value of 1.87 ± 0.2 (n = 30) (Fig. 3B). However, in relaxed myotubes transfected with the non-skeletal muscle isoform β2a, intracellular Ca2+ transients were dramatically different in kinetic and voltage dependence (Fig. 3, A and C). First, contrary to normal or β1a expressing myotubes, β2a expressing myotubes were unable to maintain stable Ca2+ release over the entire pulse duration of 200 ms (Fig. 3A). Following an initial upstroke of intracellular Ca2+, the transient rapidly decayed. Second, the voltage dependence of transients was shifted toward more positive potentials with a half-maximal activation at 22.02 ± 3.59 mV compared with -0.66 ± 2.52 mV in normal myotubes (p < 0.001). Third, the maximum intracellular Ca2+ release had a ΔF/F0 value of 1.49 ± 0.17 (n = 23; p = 0.18) that is 80 ± 9% of normal myotubes (Fig. 3C). In the case of the ancestral βM the aberrant shape of Ca2+ transients and the voltage shift (half-maximal activation: 31.41 ± 2.52 mV) were even most pronounced. Maximum ΔF/F0 reached only 0.93 ± 0.16 (n = 6; p = 0.05) that is 50 ± 9% of normal myotubes. These substantial differences in voltage-dependent intracellular Ca2+ release point to a less efficient DHPR-RyR1 coupling upon expression of the non-skeletal muscle β subunits compared with the β1a subunits.
FIGURE 3.

Differential rescue of voltage-dependent intracellular Ca2+ release in relaxed myotubes with skeletal muscle and non-skeletal muscle β subunit isoforms. A, representative recordings of intracellular Ca2+ release in response to 200-ms depolarizing step pulses to -50, 0, and +60 mV. Intracellular Ca2+ transients recorded from relaxed myotubes transfected with the skeletal muscle isoforms zf-β1a or rb-β1a were identical to those recorded from normal myotubes mock-transfected with GFP and during the 200 ms of depolarization displayed a rapid upstroke that was followed by a constant plateau of intracellular Ca2+ release that finally declined due to Ca2+ re-uptake into the sarcoplasmic reticulum. In contrast, Ca2+ transients recorded from relaxed myotubes transfected with the heterologous β2a or βM subunits were not able to sustain a plateau but showed a decline in intracellular Ca2+ immediately after initiation of the pulse. B, size and voltage-dependence of intracellular Ca2+ transients were indistinguishable (p > 0.05) between normal myotubes mock-transfected with GFP (○) and relaxed myotubes transfected with zf-β1a (♦, note, both graphs are superimposed) or rb-β1a (⋄). Dashed lines indicate half-maximal activation potentials of all three groups. C, voltage dependence of intracellular Ca2+ transients obtained from myotubes transfected with β2a (▴) or βM (▿) were significantly (p < 0.001) shifted toward more positive potentials compared with GFP-transfected normal myotubes (dashed lines, half-maximal activation). Furthermore, β2a or βM were unable to restore maximum ΔF/F0 values like normal myotubes.
DHPR Tetrad Formation: A Specific Feature of β1a—To assess whether the impaired functional DHPR-RyR1 coupling in relaxed larvae expressing non-skeletal β subunits could be explained by failure to restore the appropriate spatial DHPR-RyR1 association we performed freeze-fracture electron microscopy of tail myotomes from injected larvae. This guarantees that our studied β subunits are expressed in every muscle cell. In normal larvae and zf-β1a-injected larvae the DHPRs were normally arranged into groups of four (tetrads, marked by a central red dot in Fig. 4, upper row) and the centers of tetrads in turn were disposed in an orthogonal array related to the array of underlying RyRs. Note that tetrads may be incomplete, i.e. they may lack one or more DHPR particles, but the majority of DHPRs were located in the appropriate position relative to the predicted centers of tetrad within an array. The percentages of total particles in a cluster that constitute complete or almost complete tetrads (3 or 4 clearly visible particles) are essentially the same (p > 0.05) in normal (68 ± 19%, mean ± S.D.; n, number of clusters = 38) and zf-β1a-injected relaxed larvae (67 ± 26%, n = 17). The position of 88 ± 11 and 82 ± 12% of the particles pertained to orthogonal arrays related to those of RyRs. In contrast, in myotubes from β2a- and βM-injected relaxed larvae DHPRs were arranged in clusters (38, 47, 48), but a small and very variable portion of the particles formed tetrads (β2a, 24 ± 23% n = 34; βM, 28 ± 30% n = 21; p < 0.001 compared with normal) (red dots, Fig. 4, center and bottom rows). Although none of the particles in the β2a expressing tails showed any indication of organization into arrays, a small and variable portion of the βM particles (48 ± 35%) seemed to form limited arrays.
FIGURE 4.

Impaired tetrad formation with non-skeletal muscleβ isoforms in relaxed myotubes. Freeze-fracture electron microscopy on tail muscle tissue of 30-32 hpf relaxed larvae, zygote-injected with in vitro synthesized RNA coding for different β isoforms, revealed assembly of DHPRs in triadic clusters, indicated by yellow ellipses. In control experiments on normal larvae (upper row, left) DHPR particles were predominantly found in tetrad-like groups of 3 or 4 (indicated by red dots), indistinguishable (p > 0.05) from relaxed larvae injected with zf-β1a (upper row, right). No particles could be found between the tetrads. In contrast, arrangement of DHPRs in β2a-(center row) or βM-(bottom row) injected larvae was less organized. Arrangement of DHPR particles in tetrads was lacking in many of the DHPR clusters (β2a and βM, left images) or was very limited (right images).
Full Restoration of Larval Motility: A Specific Feature of β1a—Expression of the heterologous β subunits, β2a and βM, restored aberrant intracellular Ca2+ release with a pronounced right shift of its voltage dependence, and almost completely failed to rescue DHPR tetrad formation (Figs. 3 and 4). The next question addressed was, what are the effects, if any, of these in vitro observed changes on skeletal muscle function in an intact in vivo muscle expression system? Thus, we analyzed the motility of β subunit-injected relaxed larvae at 30-32 hpf. These larvae spontaneously and repeatedly twitch their tails by bending them in a tight arch, holding the bend position for a very brief period of time and then relaxing to the straight position. The movements involve the simultaneous activity of the myotomes on one side of the tail. For analysis of larval movements 2-min videos of single larvae were recorded and converted into sequences of differential images (Fig. 5A, see “Experimental Procedures”). The total number of dynamic pixels per frame, i.e. the pixels that showed a displacement relative to the previous image in the sequence, was plotted against time (Fig. 5, B and C). Single larval twitches display double-peaks representing larval muscle contraction and relaxation (Fig. 5B). The mean value of cumulative dynamic pixels per movement for each experimental group was calculated and standardized to that of normal larvae (100 ± 3%, n = 160). The movement extent of relaxed larvae injected with zf-β1a or rb-β1a was 94 ± 4 (n = 103) and 91 ± 3% (n = 37), and was indistinguishable (p > 0.05) from that of normal larvae (Fig. 5D). Relaxed larvae, injected with the cardiac β2a subunit displayed movements with a comparable profile to that of normal or β1a-injected relaxed larvae (data not shown), but movement extent was significantly reduced to 26 ± 2% (n = 65, p < 0.001) compared with normal larvae (Fig. 5, C and D). An even more severe failure to restore larval motility was found with the most heterologous βM. This β subunit was completely unable to recover a motile phenotype in relaxed larvae (Fig. 5D).
FIGURE 5.

Full restoration of larval motility in relaxed larvae injected with β1a RNA but only very weak or no motility with β2a and βM, respectively. To analyze larval motility, 2-min video sequences of normal larvae and relaxed larvae zygote-injected with β subunit RNAs were recorded and converted into sequences of differential images. A, representative sequences of differential images (only every 3rd frame is shown) of a single spontaneous larval movement, and B, the resulting plot of the total number of dynamic pixels per frame as described under “Experimental Procedures.” The first peak represents muscle contraction, the trough is tension maintenance, and the second peak is relaxation. C, representative recordings of relaxed larvae zygote-injected with zf-β1a (left) or β2a (right). Larvae injected with β2a displayed fewer and weaker movements, than larvae injected with zf-β1a. D, to quantify larval movement extent, the mean value of cumulative dynamic pixels per movement for each experimental group was calculated and standardized to normal larvae. Full recovery of larval movement extent was obtained in relaxed larvae when zygote-injected with either zf-β1a or rb-β1a. However, movement extent of β2a-injected relaxed larvae was rescued to only 26 ± 2% of normal larvae, whereas βM was unable to recover any motility in relaxed larvae (***, p < 0.001).
DISCUSSION
In the present study we established the β1-null mutant zebrafish relaxed as an expression system to investigate the specific role of the DHPR β1a subunit for skeletal muscle-type EC coupling. We could show that both, the homologous zf-β1a as well as the mammalian rb-β1a were equally able to completely restore all parameters of skeletal muscle-type EC coupling in in vitro and in vivo approaches, thus demonstrating a species independence of the relaxed expression system. Exogenously expressed β1a subunits led to triad expression of the DHPR α1S subunit qualitatively and quantitatively indistinguishable from normal myotubes. Immobilization-resistant intramembrane charge movement of the α1S, as the first step in the EC coupling signaling pathway, was as well properly restored as it was for the downstream intracellular sarcoplasmic reticulum Ca2+ release. Thus, together with the correct targeting of the DHPRs into tetrads opposite to the RyR1, the structural and functional prerequisites were fulfilled to allow the complete transient restoration of motility in β1a-RNA-injected relaxed zebrafish larvae.
Expression of the phylogenetically nearest isoform to β1a, the cardiac/neuronal β2a subunit or the ancestral β subunit, βM in relaxed myotubes and larvae, was likewise able to completely restore functional α1S membrane insertion and charge movement. However, myotubes expressing β2a and βM, in contrast to β1a, revealed drastic impairments in intracellular Ca2+ release. Only a minor fraction of DHPRs were grouped into tetrads that are essential for direct EC coupling. Therefore, the very weak motility of β2a-expressing relaxed larvae, and the complete absence of motility in βM-expressing relaxed larvae did not come unexpectedly. Thus, the newly established zebrafish relaxed expression system allowed us to clearly differentiate between functions of β subunits that seem common to all of them (α1S triad targeting, charge movement restoration) and functions that are essentially β1a-specific, like supporting proper intracellular Ca2+ release and effective tetrad targeting.
Previous results from β1-knock-out mice demonstrated a complete lack of intracellular Ca2+ release, strongly reduced DHPR currents, charge movements, and isradipine membrane binding (17) but the exact reason for the loss of EC coupling capability remained enigmatic. The β1-null zebrafish mutant relaxed system (19, 20) allows a higher differentiated view on isolated functions of β1a, as an elaborated set of appropriate methodological approaches is practicable with this model system. In contrast to mouse myotubes, zebrafish myotubes show a higher degree of differentiation in culture and thus allow quantification of α1S membrane expression in the absence of β1a or upon expression of different β subunits. Because quantification of the α1S protein expression in the mouse myotube-typical peripheral couplings was never performed, a clear differentiation between non-functional α1S expression and functional α1S expression in the membrane (charge movement) was not feasible. Thus, the β1a-induced facilitation of α1S charge movement was not detected in the mouse system and consequently experimental attempts on the ultrastructural level were not pushed forward (14, 17, 18). Due to the lack of these essential informations, the data of a large series of β-expression experiments (24, 25, 49-52) were in general interpreted in a way that domains of the DHPR β1a subunit, similar to elements present in the α1S subunit, might be directly involved in activation of RyR1 channels (26).
The β1a Subunit as a Signal Transducer in EC Coupling?—However, previous observations in studies with chimeric α1 subunits (53) do not support a model with β1a as a signal-transducing DHPR element, e.g. the α1S II-III loop (44, 54-58). Substitution of the α1S II-III loop by the heterologous II-III loop of a housefly (M. domestica) α1 subunit completely erased EC coupling in heterologous expression experiments in dysgenic (α1S-null) mouse myotubes (57, 58). Surprisingly, this II-III loop chimera (SkLM) was perfectly targeted into tetrads opposite the RyR1 (53) and fully restored charge movement (57). Thus, except the deletion of the II-III loop RyR1-interaction domain (critical domain) (57), chimera SkLM fulfilled all basic requirements for proper skeletal muscle-type EC coupling. To our judgment, if the β1a subunit has any intrinsic signal transducing function in EC coupling, this should have been revealed in the above experiments in which all other factors were optimal.
Theβ1a Subunit as a Scaffold for DHPRα1S Tetrad Targeting?—Now the question arises, if the β1a subunit could act as a scaffold to anchor the DHPR α1 subunits into tetrads opposite the RyR1? Again, earlier chimeric studies on α1 subunits (53) rather disagree with such a hypothesis. In experiments, where the cardiac α1C subunit was expressed in dysgenic mouse myotubes, the endogenous β1a subunit interacted with α1C and supported functional membrane expression (59), but it was not capable to promote DHPR tetrad formation (53). Thus, it seems rather obvious that in combination with the β1a subunit intracellular components of α1S are also essential for proper tetrad formation (53).
The β1a Subunit as an Additional RyR1 Binding Protein or as an Allosteric Modifier of α1S Conformation?—To sum up previous and recent results, two alternative models for the role of the β1a subunit for tetrad formation and subsequent EC coupling are possible. (i) The β1a subunit might be understood as a RyR1-anchoring/binding protein acting in addition to the anchoring functions of the II-III loop and other α1S regions, like e.g. the III-IV loop (60) or the C terminus (61). Consequently, the α1S subunit alone, despite its active binding domains, cannot bind sufficiently firm to the RyR1. It needs additional β1a binding site(s) to complete RyR1 binding of the α1S-β1a couple and thus allow tetrad formation. This model would be in accordance with a previous study (62) that described a β1a-binding domain on RyR1. Because in dysgenic myotubes no β1a/RyR1 co-localization was observed (63) we have to assume also that β1a alone would not be able to target to RyR1. (ii) An attractive alternative model that would more completely subsume our recent data is that the α1S subunit lacking β1a as a partner protein is in a state of massive conformational distortion. In this state α1S is unable to bind to RyR1 with its anchoring sites sterically hindered to appropriately interact with RyR1 (Fig. 6A). With this model also the lack of charge movement and thus of any EC coupling signal generation can be well explained by a possible misfolding of the hydrophobic core region of the voltage sensor (symbolized by the tilted cylinders in Fig. 6A). The α1S-β1a interaction would lead to a conformational correction of α1S subunit protein folding, which can now perform charge movement and also sterically orientate and therefore activate its binding domains to enable tetrad formation by accurate RyR1 anchoring. Resetting both functions would now allow proper skeletal muscle-type EC coupling (Fig. 6B). Hence, in this model β1a would function not primarily as an additional binding entity (though this has not to be excluded) but as an allosteric modifier to restore functional α1S conformation.
FIGURE 6.

Model of β-induced DHPR α1S-RyR1 interactions, incorporating results of the present and previous studies. A, hypothetical situation of the lack of any DHPR α1S-RyR1 interaction due to the lack of the β1a subunit in the relaxed zebrafish triad. Without β the conformation of the α1S subunit is severely distorted, which hampers α1S charge movement (Q) and also inactivates presumptive α1S-RyR1 interaction sites (indicated by arrows). Bold arrow represents the primary interaction site in the α1S II-III loop (53, 57), and small arrows represent an unspecific number of additional α1S-specific interaction sites as previously postulated (53). The deficiency of direct α1S-RyR1 interaction correlates with the lack of tetrad formation (19) (Tetrads). Consequently, by the lack of charge movement and tetrad formation skeletal-type EC coupling (sk-ECC) is completely hampered. In B, the situation in normal muscle or muscle from the mutant relaxed reconstituted with β1a is depicted. The β1a subunit leads to full and correct restoration of α1S conformation, allowing charge movement and appropriate targeting of the α1S into tetrads opposite the RyR1. If the β subunit adds interaction sites with RyR1 (red arrow) (62), or not, is irrelevant for our model according to which β1a acts as an allosteric modifier of α1S conformation and thus function. C, if heterologousβ subunits like the cardiac/neuronalβ2a or the ancestral, neuronal Musca βM are expressed in relaxed muscle, a partial restoration of α1S conformation takes place. Charge movement (Q) is now fully possible, but tetrad formation and thus proper α1S-RyR1 protein-protein interaction is still impaired. This “fuzzy targeting” of α1S opposite the RyR1 leads to unstable Ca2+ release and thus to very weak (in the case of β2a) or even no (for βM) muscle motility.
Non-skeletal muscle β isoforms in this model are able to endorse only a partial conformational restoration (Fig. 6C). These isoforms would fully reinstate the voltage-sensing hydrophobic α1S core region because charge movement is completely restored (Fig. 2C), but the intracellular anchoring domains seem to undergo only a very limited conformational correction. This would lead to an only partial (fuzzy) targeting of the α1S-β pair to the RyR1 without allowing accurate tetrad formation and thus only weak EC coupling interaction is possible (Figs. 3C, 4, 5D, and 6C).
For future studies it will be of high interest which molecular regions of β1a do promote this specific influence on the α1S conformation and thus are responsible for proper skeletal muscle-type EC coupling? Previous loss-of-function expression studies in the β1-null mouse system revealed no effect on intracellular Ca2+ transients, upon deletion of the β1a hook region, but a drastic reduction when either the C or N terminus of β1a were deleted (49). However, follow up truncation studies were done solely on the C terminus and the gradual loss of EC coupling was interpreted that the C terminus of β1a is the critical determinant of skeletal muscle-type EC coupling (52). However, loss-of-function studies always raise concerns that the observed effects are solely due to a more general loss of function due to induced global protein misfolding. The ancestral βM subunit is able to perform all basic β-functions like triad targeting and charge movement restoration but not the specific β1a functions. Because βM lacks major parts of the variable C- and N-terminal, and hook regions, it will serve as a valuable tool for gain-of-function approaches using the elaborate set of methods practicable with the newly established zebrafish relaxed system. Thus the next aim will be to identify in detail the crucial regions of the β1a subunit responsible for correct α1S subunit protein folding as it is required for proper skeletal muscle EC coupling.
Acknowledgments
We thank Sandra Schleret for excellent technical assistance, Dr. Gerald Obermair for expert advice with immunocytochemistry, and Dr. Hartmut Glossmann for continuous support.
This work was supported, in whole or in part, by National Institutes of Health Grant HL-48093 (to C. F.-A.). This work was also supported by Austrian Fonds zur Förderung der wissenschaftlichen Forschung Research Grant FWF-16098-B04 (to M. G.) and Medizinische Forschungsförderung Innsbruck Grant MFI-6180 (to J. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: EC, excitation-contraction; DHPR, 1,4-dihydropyridine receptor; RyR1, ryanodine receptor type-1; hpf, hours post fertilization; GFP, green fluorescent protein; nt, nucleotide(s); RE, restriction enzyme; WT, wild type; rb-β1a, rabbit β1a; zf-β1a, zebrafish β1a; MOPS, 4-morpholinepropanesulfonic acid.
J. Schredelseker, M. Shrivastav, A. Dayal, and M. Grabner, manuscript in preparation.
References
- 1.Liman, E. R., Hess, P., Weaver, F., and Koren, G. (1991) Nature 353 752-756 [DOI] [PubMed] [Google Scholar]
- 2.Papazian, D. M., Timpe, L. C., Jan, Y. N., and Jan, L. Y. (1991) Nature 349 305-310 [DOI] [PubMed] [Google Scholar]
- 3.Schneider, M. F., and Chandler, W. K. (1973) Nature 242 244-246 [DOI] [PubMed] [Google Scholar]
- 4.Rios, E., and Brum, G. (1987) Nature 325 717-720 [DOI] [PubMed] [Google Scholar]
- 5.Armstrong, C. M., Bezanilla, F. M., and Horowicz, P. (1972) Biochim. Biophys. Acta 267 605-608 [DOI] [PubMed] [Google Scholar]
- 6.Block, B. A., Imagawa, T., Campbell, K. P., and Franzini-Armstrong, C. (1988) J. Cell Biol. 107 2587-2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arikkath, J., and Campbell, K. P. (2003) Curr. Opin. Neurobiol. 13 298-307 [DOI] [PubMed] [Google Scholar]
- 8.Freise, D., Held, B., Wissenbach, U., Pfeifer, A., Trost, C., Himmerkus, N., Schweig, U., Freichel, M., Biel, M., Hofmann, F., Hoth, M., and Flockerzi, V. (2000) J. Biol. Chem. 275 14476-14481 [DOI] [PubMed] [Google Scholar]
- 9.Ursu, D., Sebille, S., Dietze, B., Freise, D., Flockerzi, V., and Melzer, W. (2001) J. Physiol. (Lond.) 533 367-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obermair, G. J., Kugler, G., Baumgartner, S., Tuluc, P., Grabner, M., and Flucher, B. E. (2005) J. Biol. Chem. 280 2229-2237 [DOI] [PubMed] [Google Scholar]
- 11.Gach, M. P., Cherednichenko, G., Haarmann, C., Lopez, J. R., Beam, K. G., Pessah, I. N., Franzini-Armstrong, C., and Allen, P. D. (2008) Biophys. J. 94 3023-3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.García, K., Nabhani, T., and García, J. (2008) J. Physiol. (Lond.) 586 727-738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beam, K. G., Knudson, C. M., and Powell, J. A. (1986) Nature 320 168-170 [DOI] [PubMed] [Google Scholar]
- 14.Gregg, R. G., Messing, A., Strube, C., Beurg, M., Moss, R., Behan, M., Sukhareva, M., Haynes, S., Powell, J. A., Coronado, R., and Powers, P. A. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 13961-13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birnbaumer, L., Qin, N., Olcese, R., Tareilus, E., Platano, D., Costantin, J., and Stefani, E. (1998) J. Bioenerg. Biomembr. 30 357-375 [DOI] [PubMed] [Google Scholar]
- 16.Dolphin, A. C. (2003) J. Bioenerg. Biomembr. 35 599-620 [DOI] [PubMed] [Google Scholar]
- 17.Strube, C., Beurg, M., Powers, P. A., Gregg, R. G., and Coronado, R. (1996) Biophys. J. 71 2531-2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strube, C., Beurg, M., Sukhareva, M., Ahern, C. A., Powell, J. A., Powers, P. A., Gregg, R. G., and Coronado, R. (1998) Biophys. J. 75 207-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schredelseker, J., Di Biase, V., Obermair, G. J., Felder, E. T., Flucher, B. E., Franzini-Armstrong, C., and Grabner, M. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 17219-17224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou, W., Saint-Amant, L., Hirata, H., Cui, W. W., Sprague, S. M., and Kuwada, J. Y. (2006) Cell Calcium 39 227-236 [DOI] [PubMed] [Google Scholar]
- 21.Granato, M., van Eeden, F. J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C. P., Jiang, Y. J., Kane, D. A., Kelsh, R. N., Mullins, M. C., Odenthal, J., and Nusslein-Volhard, C. (1996) Development 123 399-413 [DOI] [PubMed] [Google Scholar]
- 22.Haffter, P., Granato, M., Brand, M., Mullins, M. C., Hammerschmidt, M., Kane, D. A., Odenthal, J., van Eeden, F. J., Jiang, Y. J., Heisenberg, C. P., Kelsh, R. N., Furutani-Seiki, M., Vogelsang, E., Beuchle, D., Schach, U., Fabian, C., and Nusslein-Volhard, C. (1996) Development 123 1-36 [DOI] [PubMed] [Google Scholar]
- 23.Pelster, B., and Burggren, W. W. (1996) Circ. Res. 79 358-362 [DOI] [PubMed] [Google Scholar]
- 24.Beurg, M., Sukhareva, M., Strube, C., Powers, P. A., Gregg, R. G., and Coronado, R. (1997) Biophys. J. 73 807-818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beurg, M., Sukhareva, M., Ahern, C. A., Conklin, M. W., Perez-Reyes, E., Powers, P. A., Gregg, R. G., and Coronado, R. (1999) Biophys. J. 76 1744-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coronado, R., Ahern, C. A., Sheridan, D. C., Cheng, W., Carbonneau, L., and Bhattacharya, D. (2004) Biol. Res. 37 565-575 [DOI] [PubMed] [Google Scholar]
- 27.Ono, F., Higashijima, S., Shcherbatko, A., Fetcho, J. R., and Brehm, P. (2001) J. Neurosci. 21 5439-5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westerfield, M. (2000) The Zebrafish Book, University of Oregon Press, Eugene
- 29.Nüsslein-Volhard, C., and Dahm, R. (2002) in Zebrafish (Nüsslein-Volhard, C., and Dahm, R., eds) Oxford University Press, Oxford
- 30.Hirata, H., Saint-Amant, L., Waterbury, J., Cui, W., Zhou, W., Li, Q., Goldman, D., Granato, M., and Kuwada, J. Y. (2004) Development 131 5457-5468 [DOI] [PubMed] [Google Scholar]
- 31.Hirata, H., Watanabe, T., Hatakeyama, J., Sprague, S. M., Saint-Amant, L., Nagashima, A., Cui, W. W., Zhou, W., and Kuwada, J. Y. (2007) Development 134 2771-2781 [DOI] [PubMed] [Google Scholar]
- 32.Grabner, M., Dirksen, R. T., and Beam, K. G. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1903-1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neuhuber, B., Gerster, U., Mitterdorfer, J., Glossmann, H., and Flucher, B. E. (1998) J. Biol. Chem. 273 9110-9118 [DOI] [PubMed] [Google Scholar]
- 34.Perez-Reyes, E., Castellano, A., Kim, H. S., Bertrand, P., Baggstrom, E., Lacerda, A. E., Wei, X. Y., and Birnbaumer, L. (1992) J. Biol. Chem. 267 1792-1797 [PubMed] [Google Scholar]
- 35.Grabner, M., Wang, Z., Mitterdorfer, J., Rosenthal, F., Charnet, P., Savchenko, A., Hering, S., Ren, D., Hall, L. M., and Glossmann, H. (1994) J. Biol. Chem. 269 23668-23674 [PubMed] [Google Scholar]
- 36.Morton, M. E., and Froehner, S. C. (1987) J. Biol. Chem. 262 11904-11907 [PubMed] [Google Scholar]
- 37.Kugler, G., Grabner, M., Platzer, J., Striessnig, J., and Flucher, B. E. (2004) Arch. Biochem. Biophys. 427 91-100 [DOI] [PubMed] [Google Scholar]
- 38.Flucher, B. E., Andrews, S. B., and Daniels, M. P. (1994) Mol. Biol. Cell 5 1105-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams, B. A., Tanabe, T., Mikami, A., Numa, S., and Beam, K. G. (1990) Nature 346 569-572 [DOI] [PubMed] [Google Scholar]
- 40.Cheung, A., Dantzig, J. A., Hollingworth, S., Baylor, S. M., Goldman, Y. E., Mitchison, T. J., and Straight, A. F. (2002) Nat. Cell Biol. 4 83-88 [DOI] [PubMed] [Google Scholar]
- 41.Schwerte, T., and Pelster, B. (2000) J. Exp. Biol. 203 1659-1669 [DOI] [PubMed] [Google Scholar]
- 42.Schwerte, T., Prem, C., Mairösl, A., and Pelster, B. (2006) J. Exp. Biol. 209 1093-1100 [DOI] [PubMed] [Google Scholar]
- 43.Ruth, P., Rohrkasten, A., Biel, M., Bosse, E., Regulla, S., Meyer, H. E., Flockerzi, V., and Hofmann, F. (1989) Science 245 1115-1118 [DOI] [PubMed] [Google Scholar]
- 44.García, J., Tanabe, T., and Beam, K. G. (1994) J. Gen. Physiol. 103 125-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ríos, E., and Stern, M. D. (1997) Annu. Rev. Biophys. Biomol. Struct. 26 47-82 [DOI] [PubMed] [Google Scholar]
- 46.Cheng, H., and Wang, S. (2002) Front. Biosci. 7 d1867-78 [DOI] [PubMed] [Google Scholar]
- 47.Flucher, B. E., Andrews, S. B., Fleischer, S., Marks, A. R., Caswell, A., and Powell, J. A. (1993) J. Cell Biol. 123 1161-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun, X. H., Protasi, F., Takahashi, M., Takeshima, H., Ferguson, D. G., and Franzini-Armstrong, C. (1995) J. Cell Biol. 129 659-671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beurg, M., Ahern, C. A., Vallejo, P., Conklin, M. W., Powers, P. A., Gregg, R. G., and Coronado, R. (1999) Biophys. J. 77 2953-2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahern, C. A., Sheridan, D. C., Cheng, W., Mortenson, L., Nataraj, P., Allen, P., De Waard, M., and Coronado, R. (2003) Biophys. J. 84 942-959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheridan, D. C., Carbonneau, L., Ahern, C. A., Nataraj, P., and Coronado, R. (2003) Biophys. J. 85 3739-3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheridan, D. C., Cheng, W., Ahern, C. A., Mortenson, L., Alsammarae, D., Vallejo, P., and Coronado, R. (2003) Biophys. J. 84 220-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takekura, H., Paolini, C., Franzini-Armstrong, C., Kugler, G., Grabner, M., and Flucher, B. E. (2004) Mol. Biol. Cell 15 5408-5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanabe, T., Beam, K. G., Adams, B. A., Niidome, T., and Numa, S. (1990) Nature 346 567-569 [DOI] [PubMed] [Google Scholar]
- 55.Nakai, J., Tanabe, T., Konno, T., Adams, B., and Beam, K. G. (1998) J. Biol. Chem. 273 24983-24986 [DOI] [PubMed] [Google Scholar]
- 56.Grabner, M., Dirksen, R. T., Suda, N., and Beam, K. G. (1999) J. Biol. Chem. 274 21913-21919 [DOI] [PubMed] [Google Scholar]
- 57.Wilkens, C. M., Kasielke, N., Flucher, B. E., Beam, K. G., and Grabner, M. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 5892-5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kugler, G., Weiss, R. G., Flucher, B. E., and Grabner, M. (2004) J. Biol. Chem. 279 4721-4728 [DOI] [PubMed] [Google Scholar]
- 59.Tanabe, T., Mikami, A., Numa, S., and Beam, K. G. (1990) Nature 344 451-453 [DOI] [PubMed] [Google Scholar]
- 60.Leong, P., and MacLennan, D. H. (1998) J. Biol. Chem. 273 29958-29964 [DOI] [PubMed] [Google Scholar]
- 61.Slavik, K. J., Wang, J. P., Aghdasi, B., Zhang, J. Z., Mandel, F., Malouf, N., and Hamilton, S. L. (1997) Am. J. Physiol. 272 C1475—C1481 [DOI] [PubMed] [Google Scholar]
- 62.Cheng, W., Altafaj, X., Ronjat, M., and Coronado, R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 19225-19230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neuhuber, B., Gerster, U., Döring, F., Glossmann, H., Tanabe, T., and Flucher, B. E. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 5015-5020 [DOI] [PMC free article] [PubMed] [Google Scholar]