

Abstract
A new stereoselective synthesis of the antifungal and antitumor agents Preussin and 3-epi-Preussin via a Pd-catalyzed carboamination of a protected amino alcohol is described. The key transformation leads to simultaneous formation of the N—C2 bond and the C1’-aryl bond, and allows installation of the aryl group one step from the end of the sequence. This strategy permits the facile construction of a variety of preussin analogs bearing different aromatic groups.
The natural product preussin (1) was first isolated in 1988 by Schwartz and coworkers from the fermentation extracts of Preussia sp. and Aspergillus ochraceus.1 Initial screens revealed that this compound had significant antifungal activity,1, 2 and more recent work has demonstrated that preussin induces apoptosis in a number of human cancer cell lines and is a potent (IC50 = 500 nm) inhibitor of cyclin-E kinase.3 Preussin has also shown antiviral activity, and is believed to inhibit —1 ribosomal frameshifting of RNA-based viruses.4 Interestingly, all eight stereoisomers of preussin exhibit biological activity.5
Owing to its interesting biological properties, preussin has been a popular target for total synthesis, and has been prepared via 22 different routes ranging from five steps to over 23 steps.6,7,8,9 However, the large majority of these syntheses employ phenylalanine as the source of the C1’-phenyl group, and most other routes also install this group early in the synthetic sequence.6,10 Thus, the previously described syntheses of preussin are generally not well suited to the rapid generation of preussin analogs that differ in the nature of the aryl substituent. A concise approach to this molecule that involves the installation of the aryl group near the end of the synthetic route would be of value for the straightforward construction of preussin derivatives, particularly if the arene could be incorporated in a manner that would permit synthesis of functionalized and/or heteroaryl analogs from readily available precursors.
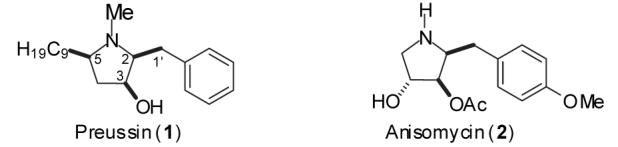
Due to the limitations of existing synthetic routes, very little work has been conducted on the synthesis and study of preussin analogs.11 However, limited studies on the effect of arene substitution on the activity of the related alkaloid anisomycin (2) have been performed that demonstrate the nature of the C1’-aryl group has a profound effect on biological activity.12,13 For example, an anisomycin analog bearing a phenyl group in place of the p-methoxyphenyl moiety showed 40-fold less cytotoxic potency than 2 against a human KB cell line.13 Thus, a synthetic route to preussin that allows facile modification of the arene moiety may be of significant biological interest.
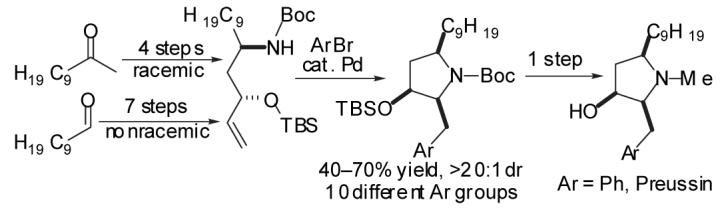
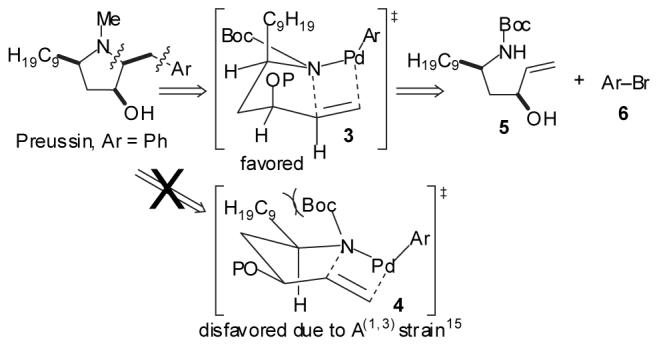
In this Letter we describe a new strategy for the stereoselective synthesis of (±)-preussin via the Pdcatalyzed carboamination14 of a protected amino alcohol. The key disconnection in this six-step route is the retrosynthetic cleavage of both the N—C2 bond and C1’—aryl bond to yield starting amino alcohol 5 and aryl bromide 6 (Scheme 1). This strategy has two significant implications for the construction of the molecule. It allows the installation of a variety of different functionalized arenes one step from the end of the sequence, which permits the facile construction of a number of preussin analogs. It also allows control of the relative stereochemistry at C2 through use of A(1,3)-strain15 in conjunction with a favorable eclipsed orientation of the Pd-N and alkene C-C bonds during the stereochemistry determining step of the carboamination,14 which promotes cyclization through transition state 3 in which the C-3 oxygen substituent is oriented in a pseudoaxial position.
Scheme 1.
Key Disconnection and Stereocontrol
In order to probe the feasibility of the key Pd-catalyzed carboamination reaction, our initial studies focused on the development of a synthesis of (±)-preussin. As outlined in Scheme 2, aldol reaction between commercially available 2-undecanone and acrolein provided keto-alcohol 7 in 78% yield. Conversion of 7 to the O-benzyl oxime 8 proceeded smoothly, and was followed by a one-pot sequence of LiAlHL4 reduction and Boc-protection to provide an 86% yield of a 1:1.2 mixture of readily separable amino alcohol diastereomers 9 and 10. Although stereoselective reductions of β-hydroxy oxime ethers have been previously described,16we elected to employ non-selective conditions to allow access to both syn- and anti-amino alcohol substrates for the Pd-catalyzed cyclization reactions, which ultimately provide two different biologically active pyrrolidine derivatives (1 and 14).
Scheme 2.
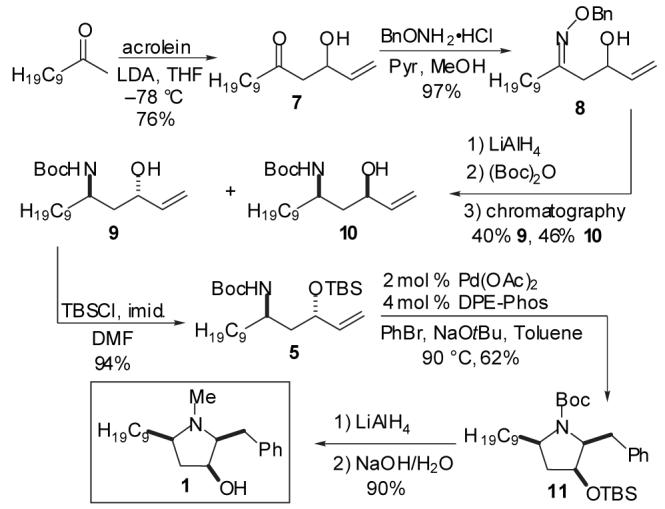
Synthesis of (±)-Preussin
TBS-protection of anti-amino alcohol 9 provided 5, the substrate for the key Pd-catalyzed carboamination reaction. In the event, treatment of 5 with bromobenzene and NaOtBu in the presence of catalytic Pd(OAc)2/dpe-phos17provided pyrrolidine 11 in 62% isolated yield with >20:1 diastereoselectivity. As noted above, the desired stereoisomer most likely arises from transition state 3 (Scheme 1) in which the C9-chain is oriented in a pseudoaxial position in order to minimize A(1,3)-strain between this substituent and the N-Boc group.15 Although the C3-OTBS group is also pseudoaxial, the energetic cost of this conformation is lower than that of the A(1,3)-strain in transition state 4 in which both the C9-chain and the OTBS group are equatorial. One-pot reduction and deprotection of 11 afforded (±) — preussin (1) in 90% yield as a single diastereomer. This six-step sequence proceeded with an overall yield of 15% from 2-undecanone.
The syn-amino alcohol 10 was converted to (±)-3-epi-preussin (14)8a,c using a sequence of reactions analogous to that described above (Scheme 3). The stereoselectivity of the Pd-catalyzed cyclization of 12 was high, although the chemical yield obtained in the reaction of the syn-diastereomer 12 (54%) was slightly lower than that achieved for the reaction of the anti-isomer 5 (62%). This route provided the desired pyrrolidine 14 as a single isomer in an overall yield of 15%. Thus, the strategy described herein provides straightforward access to both racemic preussin diastereomers in comparable yields and stereoselectivities.18
Scheme 3.
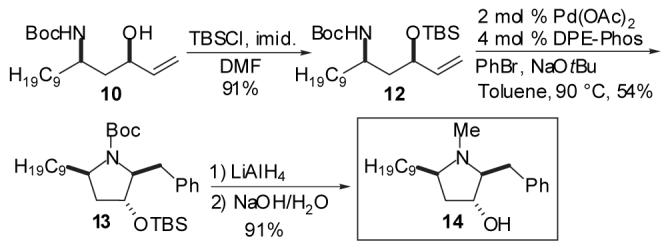
Synthesis of (±)-3-epi-Preussin.
Having demonstrated the feasibility of Pd-catalyzed carboamination for the construction of Preussin, we turned our attention towards the development of an asymmetric synthesis of the naturally occurring (+)-enantiomer. As shown in Scheme 4, addition of allylmagnesium bromide to enantiopure sulfinylimine 15 under Ellman’s conditions19 afforded 16, which was isolated in 77% yield as a single diastereomer upon purification.20 Cleavage of the chiral auxiliary and protection of the resulting primary amine afforded 17 in 85% yield. Ozonolysis of 17 (63%) followed by vinylcuprate addition21 to the resulting β-amino aldehyde 18 generated (+)-9 with 3:1 diastereoselectivity; the desired pure anti-diastereomer was obtained in 54% yield after chromatographic separation. The remaining three steps proceeded with similar yields and selectivities to those obtained from the racemic route, and provided (+)-preussin {[α]23D +21.2° (c 1.0, CHCl3) [lit.2 [α}25D +22.0° (c 1.0, CHCl3)]} with an overall yield of 12% in nine steps from commercially available decanal.
Scheme 4.
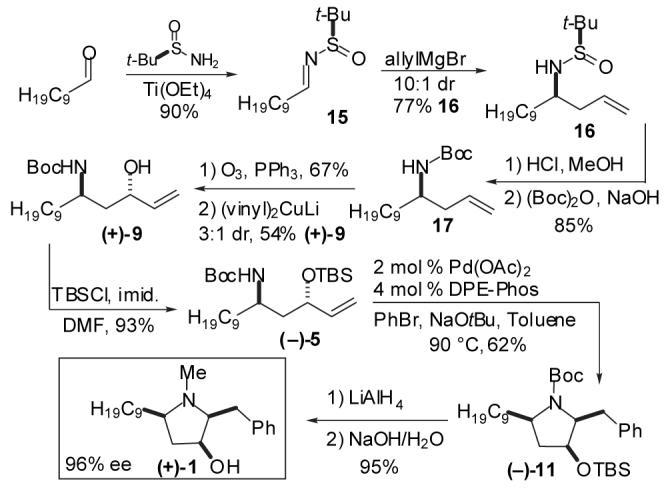
Synthesis of (+)-Preussin
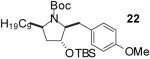
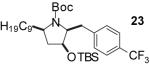
In order to demonstrate the amenability of this strategy toward the synthesis of differentially arylated preussin analogs, Pd-catalyzed reactions of 5 and 12 were conducted with a variety of different aryl bromide coupling partners. To illustrate the utility of this method for the rapid generation of useful quantities of analogs the Pd/dpe-phos catalyst was employed for all substrate combinations. However, previous studies indicate that other ligands may provide superior results for certain aryl bromide coupling partners.14c Thus, the yields obtained in these transformations are not optimized on a case-by-case basis. As shown in Table 1, the cyclization reactions proceed in moderate to good yields for a variety of different aryl bromides including strates that are electron-rich (entries 3-4),22 electron-poor (entries 1-2, 5-6, 9, and 12), or sterically hindered (entry 8). Several functional groups are tolerated, and heterocyclic aryl bromides can also be employed (entries 10-11). In most cases examined, the cyclizations of anti-amino alcohol derivative 5 proceeded in higher yield than the analogous reactions of syn-amino alcohol derivative 12. In all cases the reactions proceeded with >20:1 diastereoselectivity as judged by 1H and 13C NMR analysis.
Table 1.
Synthesis of N-Boc-O-TBS-Preussin Analogs
| entry | substrate | ArBr | product | yield (%) |
|---|---|---|---|---|
| 1 | 5 |  |
 |
70 |
| 2 | 12 | 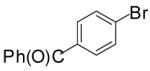 |
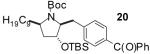 |
65 |
| 3 | 5 | 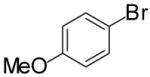 |
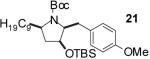 |
52 |
| 4 | 12 | 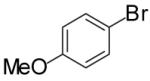 |
 |
49 |
| 5 | 5 | 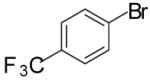 |
 |
69 |
| 6 | 12 | 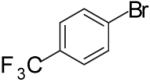 |
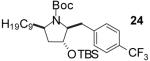 |
52 |
| 7 | 5 | 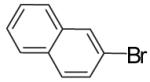 |
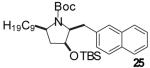 |
69 |
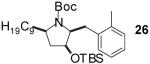
| 8 | 5 | 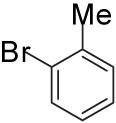 |
 |
57 |
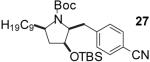
| 9 | 5 | 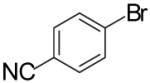 |
 |
62 |
| 10 | 5 | 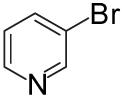 |
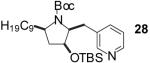 |
54 |
| 11 | 5 | 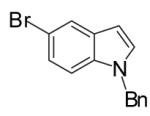 |
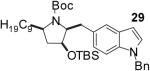 |
40 |
| 12 | 5 | 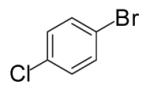 |
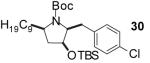 |
70 |
Conditions: 1.0 equiv substrate, 1.2 equiv ArBr, 2.3 equiv NaOtBu, 2 mol% Pd(OAc)2, 4 mol % dpe-phos, toluene, 90 °C.
To illustrate that the N-Boc-O-TBS-preussin analogs could be converted to N-methyl-3-hydroxy-2-(arylmethyl)pyrrolidines that are closely related to the natural product, a subset of the products shown in Table 1 were deprotected in a 1-2 step sequence. As shown in Table 2, deprotection was effected using LiAlH4 followed by treatment with aqueous base or TBAF for products bearing unreactive aromatic functionality. Alternatively, deprotection was also achieved through one-pot Boc-cleavage and reductive amination with formic acid/formaldehyde followed by treatment with TBAF. The latter conditions tolerate functional groups (e.g., ketones, trifluoromethyl substituents) that would be reduced by LiAlH4. Both deprotection protocols provided the desired products in excellent yields.
Table 2.
Deprotection of N-Boc-O-TBS-Preussin Analogs
| entry | substrate | method | product | yield (%) |
|---|---|---|---|---|
| 1 | 19 | B | 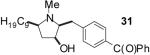 |
83 |
| 2 | 20 | B | 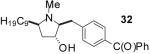 |
84 |
| 3 | 21 | A | 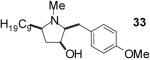 |
85 |
| 4 | 22 | A | 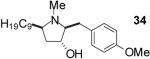 |
89 |
| 5 | 23 | B | 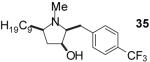 |
80 |
| 6 | 24 | B | 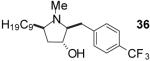 |
81 |
Conditions: Method A: i) LiAlH4, THF, 60 °C; ii) H2O or TBAF. Method
B: i) HCO2H, HCHO; ii) TBAF, THF.
In conclusion, we have developed concise, stereoselective routes to (+)-preussin, (±)-preussin and (±)-3-epi-preussin. These routes feature a new strategy for the synthesis of 2-benzylpyrrolidine alkaloids that allows the construction of both the C2-N and the C1’ -Ar bonds in a single step near the end of the synthetic sequence. This strategy allows the facile synthesis of differentially arylated preussin analogs, and provides straightforward access to derivatives that could not be easily and rapidly prepared using previously developed routes. This strategy is potentially applicable to other members of this class of natural products; further studies in this area are currently underway.
Supplementary Material
Acknowledgment
This research was supported by the NIH-NIGMS (GM 071650) and the University of Michigan JPW thanks the Camille and Henry Dreyfus Foundation for a New Faculty Award, Research Corporation for an Innovation Award, and Eli Lilly for a Grantee Award. Additional unrestricted support was provided by Amgen, Eli Lilly, and 3M.
Footnotes
Supporting Information Available. Experimental procedures, spectroscopic data, and copies of 1H and 13C spectra for all compounds reported in the text (89 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schwartz RE, Liesch J, Hensens O, Zitano L, Honeycutt S, Garrity G, Fromtling RA, Onishi J, Monaghan R. J. Antibiot. 1988;41:1774. doi: 10.7164/antibiotics.41.1774. [DOI] [PubMed] [Google Scholar]
- 2(a).Johnson JH, Phillipson DW, Kahle AD. J. Antibiot. 1989;42:1184. doi: 10.7164/antibiotics.42.1184. [DOI] [PubMed] [Google Scholar]; (b) Kasahara K, Yoshida M, Eishima J, Takesako K, Beppu T, Horinouchi S. J. Antibiot. 1997;50:267. [PubMed] [Google Scholar]
- 3.Achenbach TV, Slater PE, Brummerhop H, Bach T, Müller R. Antimicrob. Agents Chemother. 2000;44:2794. doi: 10.1128/aac.44.10.2794-2801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinzy TG, Harger JW, Carr-Schmid A, Kwon J, Shastry M, Justice M, Dinman JD. Virology. 2002;300:60. doi: 10.1006/viro.2002.1567. [DOI] [PubMed] [Google Scholar]
- 5.Okue M, Watanabe H, Kasahara K, Yoshida M, Horinouchi S, Kitahara T. Biosci. Biotechnol. Biochem. 2002;66:1093. doi: 10.1271/bbb.66.1093. [DOI] [PubMed] [Google Scholar]
- 6.Kitahara has described a non-stereoselective route that affords all eight stereoisomers of preussin in a two-step sequence. The isomers were separated by preparative chiral HPLC. See reference 5.
- 7.For a recent review, see:Basler B, Brandes S, Spiegel A, Bach T. Top. Curr. Chem. 2005;243:1.
- 8(a).For early synthetic studies see:Shimazaki M, Okazaki F, Nakajima F, Ishikawa T, Ohta A. Heterocycles. 1993;36:1823.McGrane PL, Livinghouse T. J. Am. Chem. Soc. 1993;115:11485.Overhand M, Hecht SM. J. Org. Chem. 1994;59:4721.Deng W, Overman LE. J. Am. Chem. Soc. 1994;116:11241.
- 9(a).For recent syntheses, see:Canova S, Bellosta V, Cossy J. Synlett. 2004:1811.Davis FA, Deng J. Tetrahedron. 2004;60:5111.Raghavan S, Rasheed MA. Tetrahedron. 2003;59:10307.Huang P-Q, Wu T-J, Ruan Y-P. Org. Lett. 2003;5:4341. doi: 10.1021/ol035617a.Dikshit DK, Goswami LN, Singh VS. Synlett. 2003:1737.
- 10 b).Two strategies allow installation of the aryl moiety within 1–3 steps of the final target. Davis generated the C-2 benzyl group as the final step via reaction of lithium diphenyl cuprate with a pyrrolidinylmethyl iodide (40% yield, single diastereomer; 10 steps total, 9% overall yield). Bach employed a Paternò-Büchi reaction of benzaldehyde with a dihydropyrrole (4:1 dr, 53% yield after separation of diastereomers) followed by a two-step deprotection sequence to generate the benzyl stituent (39% over 3 steps; 9 steps total, 11% overall yield). See: a) reference 9b.Bach T, Brummerhop H. Angew. Chem., Int. Ed. 1998;37:3400. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3400::AID-ANIE3400>3.0.CO;2-3.
- 11 a).To date, no analogs of preussin have been prepared that are modified on the aromatic ring. Bach has reported the synthesis of two analogs that differ in the nature of the C5 alkyl chain, and four analogs that differ in the nature of the C-3 stituent have been described in the patent literature. See:Bach T, Brummerhop H, Harms K. Chem. Eur. J. 2000;6:3838. doi: 10.1002/1521-3765(20001016)6:20<3838::aid-chem3838>3.0.co;2-1.Suzuki K, Miike N, Kawamoto E. 2003.
- 12.Hall SS, Loebenberg D, Schumacher DP. J. Med. Chem. 1983;26:469. doi: 10.1021/jm00358a003. [DOI] [PubMed] [Google Scholar]
- 13.Schwardt O, Veith U, Gaspard C, Jäger V. Synthesis. 1999:1473. [Google Scholar]
- 14 a).Ney JE, Wolfe JP. Angew. Chem., Int. Ed. 2004;43:3605. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; b) Ney JE, Wolfe JP. J. Am. Chem. Soc. 2005;127:8644. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. [Google Scholar]; d) Yang Q, Ney JE, Wolfe JP. Org. Lett. 2005;7:2575. doi: 10.1021/ol050647u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15 a).For a discussion of allylic strain related to the partial double bond character between the nitrogen atom and the carbonyl carbon in α-stituted amides and carbamates see:Hoffmann RW. Chem. Rev. 1989;89:1841.Hart DJ. J. Am. Chem. Soc. 1980;102:397.Williams RM, Sinclair PJ, Zhai D, Chen D. J. Am. Chem. Soc. 1988;110:1547.Kano S, Yokomatsu T, Iwasawa H, Shibuya S. Heterocycles. 1987;26:2805.
- 16.Narasaka K, Ukaji Y, Yamazaki S. Bull. Chem. Soc. Jpn. 1986;59:525. [Google Scholar]
- 17.Dpe-phos = bis(2-diphenylphosphinophenyl)ether
- 18.The (—)-B-chlorodiisopinocampheylborane-mediated asymmetric aldol reaction between 2-undecanone and acrolein provided 7 in 78% yield and 48% ee using Paterson’s conditions. See:Paterson I, Goodman JM, Lister MA, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663.
- 19.Cogan DA, Liu G, Ellman J. Tetrahedron. 1999;55:8883. [Google Scholar]
- 20.The crude reaction mixture was judged to be a 10:1 mixture of diastereomers by 1H NMR analysis.
- 21.Toujas J-L, Toupet L, Vaultier M. Tetrahedron. 2000;56:2665. [Google Scholar]
- 22.Use of high purity starting material (> 97% by NMR and GC) was essential to obtain high yields in reactions of electron-rich aryl bromides. Use of starting material with trace (ca 5%) impurities led to catalyst deactivation and formation of complex mixtures of products with these strate combinations.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.