

Abstract
This paper discusses the suitability of manganese for its function in catalysing the formation of molecular oxygen from water. Manganese is an abundant element. In terms of its inherent properties, Mn has a particularly rich redox chemistry compared with other d-block elements, with several oxidizing states accessible. The most stable-state Mn2+ behaves like a Group 2 element—it is mobile, weakly complexing, easily taken up by cells and redox-inactive in simple aqueous media. Only in the presence of suitable ligands does Mn2+ become oxidized, so it provides an uncomplicated building unit for the oxygen-evolving centre (OEC). The intermediate oxidation states Mn(III) and Mn(IV) are strongly complexed by O2− and form robust mixed-valence poly-oxo clusters in which the Mn(IV)/Mn(III) ratio can be elevated, one electron at a time, accumulating oxidizing potential and capacity. The OEC is a Mn4CaOx cluster that undergoes sequential oxidations by P680+ at potentials above 1 V, ultimately to a super-oxidized level that includes one Mn(V) or a Mn(IV)-oxyl radical. The latter is powerfully oxidizing and provides the crucial ‘power stroke’ necessary to generate an O–O bond. This leaves a centre still rich in Mn(IV), ensuring a rapid follow-through to O2.
Keywords: manganese, oxygen, catalyst, evolution, water oxidation
1. Background and goals
The 21% oxygen (O2) in our atmosphere results from the action of a single catalyst, the O2-evolving complex of photosystem II, for which important structural details are now emerging (Ferreira et al. 2004; McEvoy & Brudvig 2006; Yano et al. 2006; Pushkar et al. 2007). Oxidation of H2O in the so-called S-cycle occurs at a unique active site, the ‘oxygen-evolving complex’ (OEC) that contains 4Mn and 1Ca, and this process has continued, very likely with little alteration, since the advent of cyanobacteria more than 2.5 Gyr ago. This uniqueness and the fact that O2 is essential to all higher life forms mean that the OEC is the most important catalyst on Earth. Furthermore, outside biology there are intense efforts to find efficient photo- and electro-catalysts for H2O oxidation, not because we need more O2 but because it is highly desirable to produce hydrogen (H2) by artificial water splitting through photolysis or electrolysis, and a half an equivalent of O2 must be produced simultaneously with H2. However we choose to use the H2 that is produced, either directly as a fuel or as a reduced raw material for hydrocarbon production, water splitting is an obvious and sensible way to capture the energy from the Sun (Alstrum-Acevedo et al. 2005; Lewis & Nocera 2006; Barber 2007).
So why was Mn chosen for this task? The presence of Ca is not quite so specific as it can be replaced by Sr, but Mn is somehow indispensable. This paper summarizes the properties of Mn that make it special. We start by considering the reactions involved and derive what we can from ‘textbook knowledge’ that is available to any undergraduate.
2. H2O to O2
The energetics of O2 formation from H2O have been considered by Krishtalik (1986) and I will outline his conclusions. The half-cell reaction equation (2.1), involves transfer of four electrons and four protons, and the reduction potential has a pH dependence of approximately −0.06 V per pH unit.
| (2.1) |
At pH 7, formation of a free molecule of O2 requires an energy input equivalent to 0.82 V per electron, that is 3.68 eV or approximately 350 kJ mol−1. In photosystem II this compares with an available energy of approximately 1.25 V per electron, the driving force provided by P680+ (Grabolle & Dau 2005). Outside biology, in laboratory experiments, it is relevant to note that no reversible electrochemistry of the four-electron O2/H2O couple has been reported. Reduction of O2 to H2O requires a sizeable overpotential, even at a Pt electrode, and the sluggish kinetics of this reaction are a major concern in the performance of fuel cells (contrast with 2H+/H2 which is reversible at Pt and some enzyme electrodes; Anderson & Albu 2000). Electrochemically, the reverse reaction, electrochemical oxidation of H2O to O2, is at least as difficult.
Krishtalik's analysis of the O2 evolution problem was more sophisticated than I outline here, as he corrected reduction potentials for transfer of species between the catalytic active site and the environment. I will present only the simple acid–base and electrochemistry considerations that are most familiar to all chemists. The substrate H2O is present at approximately 55 M and although the products of deprotonation, OH− and O2−, are much easier to oxidize, they are present only at tiny or immeasurable amounts. One function of the catalyst is therefore to generate these minority species in the active site by abstracting H+ and stabilizing OH−/O2− by coordination to a metal ion M. Since OH− and particularly O2− are strong π-donor ligands, they coordinate preferentially to transition metal ions having a high availability of suitable acceptor orbitals, most notably metals to the left side of the d-block and increasingly upon oxidation, since removing electrons from a metal ion releases H+ from coordinated H2O. Taking simple Mn species as examples, the pK of a coordinated H2O molecule is approximately 10.5 for Mn(II), approximately 0 for Mn(III) and even the remaining proton is lost upon oxidation to Mn(IV) which therefore coordinates (and stabilizes) O2− (Wiberg 2001). Transition metals coordinated by O2− ligands are commonly written as [M=O] units, although the ‘double bond’ is just a notation to indicate additional π-bonding that can vary in its extent. As the formal oxidation state of M increases, so does the likelihood that the electron is removed instead from the O2− ligand so it becomes an oxyl radical O·−.
The aim of this article is not to discuss detailed mechanism; several recent articles have discussed the different possibilities that could apply given the latest refinements in structure (Messinger 2004; McEvoy et al. 2005; Siegbahn 2006, 2007). The defining property of the OEC is perhaps not so much that it produces O2 (catalases also do this) but that it makes an O–O bond. It is the only example in biology in which an O–O bond is formed from two H2O molecules, yet no long-lived intermediates (such as H2O2) are released. The single O–O bond, as occurs in a compound like hydrogen peroxide (H2O2, O–O bond energy 142 kJ mol−1) is very much weaker than the double bond of O2 (O=O, bond energy 494 kJ mol−1) so that once an O–O bond is formed, completion through to O2 is comparatively easy if further oxidizing capacity is available (Emsley 1998; Wiberg 2001). Indeed there is recent evidence that the latter stages of O2 production may be reversible (Clausen & Junge 2004).
To picture the general energy profile for water oxidation, an oxidation state diagram for O is shown in figure 1. The gradients between selected intermediate states show the corresponding reduction potentials for that stage (Atkins et al. 2006). I have used pH=7 because this condition is familiar to biochemists and the features are qualitatively similar regardless of pH and even using Krishtalik's corrected potentials. The graph shows why Krishtalik argued that any mechanism involving four one-electron reactions would encounter a particularly steep climb at H2O→=OH, and be energetically very unfavourable compared with two sequential two-electron transfers or a reaction in which all four electrons are transferred in a single step. Looked at in another way, formation of =OH (or any form of oxygen in the 1-state) should occur in pairs, so that most of the energy cost is recouped by simultaneous O–O bond formation.
Figure 1.

Oxidation state diagram showing the energy landscape for intermediates in the oxidation of two water molecules to a molecule of O2. These data are for pH 7 but a qualitatively similar picture is obtained for pH 0 and 14, and using Krishtalik's corrected data. Widths of horizontal block arrows indicate the thermodynamic difficulty in accomplishing that stage of the reaction.
A variety of options arise for achieving two- and four-electron transfers from metals to O-species that avoid one-electron intermediates, and some of these are sketched in figure 2. The two-electron intermediate is a peroxide, retained in the OEC, which can be formed either (a) by simultaneous oxidations of two adjacent coordinated H2O (OH−), via combination of the corresponding oxyl radicals, or (b) by attack by H2O (or OH−) on a ‘hot’ electrophilic oxygen atom on a highly oxidized [M·O] group (Limburg et al. 1999; Messinger 2004). As stated above, formation of the O–O bond alone provides approximately 142 kJ stabilization relative to two separate OH− radicals. Figure 2 shows two further scenarios: (c) that two adjacent ‘hot-O’ species, terminal [M·O] or bridging O (μ-O) could rearrange to form O2 (this would also form the basis of a concerted four-electron reaction (−4 to 0) in figure 1; Vincent & Christou 1987) and (d) attack by a cluster (bridging) O2− (or a radical-like O) on an oxyl radical attached to a high-valent metal (Siegbahn 2006, 2007).
Figure 2.
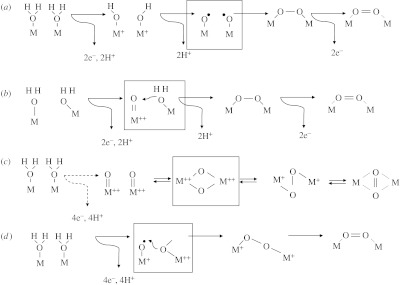
Sketches of skeletal mechanisms for formation of an O2 molecule from two molecules of H2O. Elevations in oxidation state of M are indicated by ‘+’, etc. Boxes indicate the crucial stages in each mechanism. (a,b) Ways of making an O–O bond (peroxo intermediate) that is relatively easy to oxidize further to give O2. (a) Formation of peroxo intermediate by homonuclear combination from adjacent M+−OH units deprotonated and rearranged as two oxyl radicals. (b) Formation of peroxo intermediate by attack of H2O on an electrophilic ‘O’ of [M·O]++ group. (c) Interconversion between two O2− that are either terminal ([M·O]++) or bridging ((μ-O) between two M++), an bridged between two M+, and O2 bound (weakly) between two M. (d) Attack of a bridging O2− on an oxyl radical.
3. The redox chemistry of Mn
Biology required a catalyst capable of removing four electrons and four protons from two H2O, in stages that mostly involve highly oxidizing species, with retention of all intermediates during the process. In addition to successive electron transfers, protons must be removed and transferred away rapidly and efficiently, states must exist that permit rapid binding of substrate H2O/OH− and the whole structure must be sufficiently robust to survive tens of thousands of turnovers before subunit D1 is replaced (Aro et al. 1993; Ananyev & Dismukes 2005; Cady et al. 2008). The ‘hot O’ species underlying the mechanistic ideas in figure 2 are optimized by metal ions M able to balance the ability to form an oxy species [M·O] from H2O in the first place, yet, with little further provocation, transfer O as an atom or radical and return to reduced M.
The underlying redox chemistry of Mn is appreciated from the stabilities of simple compounds with oxygen and water. These stabilities are portrayed in the oxidation state diagram given in figure 3, where the inset compares ionization energies (IEs) of Mn and Fe. The diagram reveals a rich redox chemistry in which all states above Mn(II) are strongly oxidizing under some conditions. Most obviously Mn(II) sits in a local recession and is thus inherently stable under all conditions, Mn(III) is inherently unstable under acid conditions and Mn(V) is unstable under all conditions. I will return to this diagram after reviewing possibilities that are offered by other d-block metals.
Figure 3.

Oxidation state diagram for Mn aqua species at pH 0 and 14, and the stabilization afforded as Mn species are transferred from pH 0 to 14. Inset shows ionization energies (IE) for Mn (black bars) and Fe (white bars). Reduction potentials for Mn(V) and Mn (VI) aqua species in acidic solutions are subject to large uncertainties; hence the large spread in volt-equivalent values for these species at pH 0.
Biology uses many first-row d-block elements, V, Mn, Fe, Co, Ni, Cu and Zn, but not Ti nor to any certain degree Cr. As we move across the 3d row, the highest accessible oxidation states as well as the number of accessible oxidation states increase up to Mn then decrease to Zn where only the 2+ state is observed (Atkins et al. 2006). This is chiefly a function of IEs which increase across the row and for each electron removed. Not only does Mn display a greater range of oxidation states than other 3d metals, but all the simple higher states, from Mn(III) upwards, are hydroxo- or oxo-species with reduction potentials in the ranges of the O-transformation reactions we have just outlined. Both V and Cr have several oxidation states available; however, V(V) ([V(O)2]+) is only moderately oxidizing and although Cr(V) and Cr(VI) are strongly oxidizing, Cr(II) is powerfully reducing and the most stable-state Cr(III) is immobilized because it undergoes ligand exchange too slowly to be incorporated into a protein as a metal cofactor. Hence these metals are inherently unsuitable.
Beyond Mn, the chemistry of Fe most resembles that of Mn. The ferryl entity [FeO]2+ is well known as an intermediate in the reductive activation of O2 and it can even be generated as an aqua ion species by reacting Fe2+ with ozone (Pestovsky et al. 2005). Of the higher oxidation states, Fe(V) has proved elusive but a complex has recently been reported (Tiago de Oliveira et al. 2007), and Fe(VI) exists as the ferrate ion that is produced by oxidation in concentrated alkali (Wiberg 2001). For the later 3d elements, complexes in the higher oxidation states tend to be binuclear, consisting of a diamond-shaped core with two bridging oxo ligands [M2(μ-O)2]n+ that resemble sections of the OEC (Que & Tolman 2002). A particularly interesting example of these diamond-core complexes is found for Cu, for which isomerism exists between species formulated as [μ-η2-n2-peroxo Cu(II)] and bis[(μ-oxo) Cu(III)]. This chemistry is relevant as Cu(III) is expected to be powerfully oxidizing; yet, in the common reversible O2-binding protein haemocyanin, the incoming O2 molecule is coordinated centrally between two Cu(I) giving an adduct that is formulated as [μ-η2-n2-peroxo Cu(II)] (Atkins et al. 2006). A plausible transformation from bis[(μ-oxo) Cu(III)] to 2Cu(I)+O2 would resemble the mechanism in figure 2c.
Of the second and third row d-block elements, only Mo and W are used in biology and neither of these is sufficiently oxidizing even in the VI state ([M(O)2]2+). In studies to identify artificial catalysts for water splitting the best metal so far has appeared to be Ru, which is functional in compounds featuring the oxidation states III, IV and V (Meyer & Huynh 2003; Alstrum-Acevedo et al. 2005; Liu et al. 2007). But there is no evidence that Ru is used by biology. Clearly, efforts to make catalysts based on Mn (or Fe) are highly desirable because Ru is much rarer and more expensive than Mn.
The inherent stability of Mn(II) arises from the electron configuration 3d5 in which the d-shell is half filled—a favoured configuration due to optimal exchange energy. (The fact that a free Mn atom has the electron configuration 4s23d5, again with a stable half-filled d-shell, is one reason why metallic Mn has an unusually low heat of atomization and electropositive character among the d-block elements; Atkins et al. 2006.) The exchange energy makes it difficult to remove the fifth d-electron, so Mn(III) compounds are oxidizing—Mn3+ particularly so:
| (3.1) |
The oxidation state diagram in figure 3 is shown for pH 0 and pH 14, and the difference between the two datasets indicates the stabilization afforded by OH−/O2− coordination. There is clearly a shortage of data for Mn species that are unstable in crude aqueous media but could be stabilized in special environments.
In contrast to the inherent instability of simple Mn(III) compounds, simple Mn(IV) chemistry is represented under all aqueous conditions as the insoluble compound MnO2, which has a high lattice energy and is effectively a thermodynamic sink. Mn(III) aqua species tend to disproportionate to Mn(II) and Mn(IV). This happens naturally in solution and would be predicted to occur in multinuclear O-bridged structures such as Mn(III)–Mn(III) complexes or in Mn(III) minerals, although it need not occur if single Mn(III) species are isolated. This tendency contrasts with Fe where the ‘rust-like’ [Fe–O–Fe] entity with Fe(III) ions bridged by O2− is very stable (Fraústo da Silva & Williams 2001). An example of where this prediction holds is the mixed-valence oxide Mn5O8, which contains Mn(II) and Mn(IV), rather than Mn(III) (Wiberg 2001).
A major gap in our knowledge of how the OEC works is the complete lack of electrochemical data for any stage of the S-cycle. Despite our ignorance in this area, it is now widely accepted that state S2, the source of the most characteristic electron paramagnetic resonance (EPR) signature during catalysis, should be formulated as [Mn(III) : 3Mn(IV)] rather than [3Mn(III) : 1Mn(IV)] as favoured earlier (McEvoy & Brudvig 2006). The elevated-oxidation-level assignment is supported by reductive titration of the OEC with hydroxylamine, during which states S−1 and S−2 were formed and characterized (Kuntzleman & Yocum 2005). State S0 is formulated as [3Mn(III), Mn(IV)] and there is spectroscopic evidence that it does not contain Mn(II) (so disproportionation of two Mn(III) has not occurred; Kulik et al. 2005). It follows that the ‘power stroke’ driving O–O bond formation does not stem from reduction of Mn(III), which is instead stabilized in the OEC. We look instead to higher oxidation states and note that state S4, from which O2 is released, must be at a level one equivalent above 4Mn(IV); consequently, the crucial species for O–O bond formation is likely to include Mn(V) (or Mn(IV)-oxyl radical) (Shimazaki et al. 2004).
Of the highest oxidation states, only Mn(VII) is important in everyday chemistry. The familiar purple ion, which exemplifies the stabilizing effect of O2− as an anionic, strong π-donor ligand, liberates O2 from H2O in a slow process. The anhydrous oxide Mn2O7 (O3MnOMnO3) is extremely reactive and decomposes at 0°C (explosively at 95°C) yielding O2 and MnO2 (Wiberg 2001). However, it is very difficult to consider how a single metal ion could be harnessed catalytically to carry out a demanding four-electron reaction, and biology's use of a metal-oxo cluster to provide the necessary capacity to do this ‘made good sense’. The data given in figure 3 for Mn(V) and Mn(VI) are reliable only for pH 14, because the aqua species are unstable in acid (Carrington & Symons 1956; Zordan & Hepler 1968). The simple aqua species of Mn(V), inherently the least stable oxidation state of Mn, can be studied only in concentrated alkaline solutions which contain a mixture of blue species , MnO3(OH)− and MnO2(OH)2 (Rush & Bielski 1995). The data entries at pH 0 for Mn(V) and Mn(VI) have estimated ranges: Mn(V) should exist as H3MnO4 (more correctly assigned as (HO)3Mn·O, ‘hypomanganous’ acid, analogous to phosphoric acid); likewise, for Mn(VI), the green ‘manganate’ ion should exist as (more likely assigned as [(HO)Mn(·O)3]−, by analogy with hydrogen sulphate). Despite the instability of simple Mn(V) species, many Mn(V)-oxo complexes have been prepared or implicated in catalytic reactions (Lansky & Goldberg 2006). Interestingly, it is possible to isolate Mn(V) complexes that do not contain oxo ligands. These include imido complexes, where the RN2− group is a good π-donor ligand like O2− (Danopoulos et al. 1994). These complexes are quite stable and do not react with H2O.
4. The aqueous coordination chemistry of Mn species
In aqueous solution, the chemistry of Mn is dominated by the aqua ion which remains unhydrolysed up to pH 9. Owing to its lack of ligand field stabilization, Mn(II) forms weaker complexes than other divalent d-block metals and it behaves more like Group 2 metal ions for which the only truly stable complexes in water are those with chelating ligands (Fraústo da Silva & Williams 2001). In size, Mn lies between Mg and Ca with radii (Shannon and Prewitt values) typically: Mn2+=97 pm, Mg2+=86 pm, Ca2+=114 pm (Wiberg 2001). With regard to ligand preference, Mn(II) is usually found with O-donors, although compared with either Mg or Ca, it does have a greater tendency to bind to N-donors (Fraústo da Silva & Williams 2001). The favourable exchange energy for d5 means that only very strong-field ligands such as CN− are able to cause spin pairing. Mn2+ has high rates of ligand exchange (H2O exchange occurs at a rate of 107 s−1) and is thus easily mobilized.
The difficulty in stabilizing Mn(III) starts to recede once we improve the complexing power of the aqueous solvent, either by increasing the pH or introducing ligands with good donor properties. An important example (see below) is the stabilization of Mn(III) by bicarbonate , a ubiquitous ligand in natural waters, which lowers the IV/III reduction potential from 1.5 to 0.6 V (Kozlov et al. 2004; Dasgupta et al. 2006). Owing to its d4 configuration the coordination sphere of Mn(III) is subject to Jahn–Teller distortion, which allows Mn(III) to have fast rates of ligand exchange, like Cu(II) or Cr(II), for monodentate ligands like H2O.
Mn(IV) complexes are mainly found with bridging or terminal oxo-ligands, and polynuclear Mn–O clusters of mixed valency are common. Of these, compounds of the type [Mn4O4L6], based on a Mn4O4 cubane cluster and containing both Mn(III) and Mn(IV), have attracted interest as structural and functional models for the OEC (Rüttinger & Dismukes 1997). These clusters assemble spontaneously when L is a suitable ligand like phosphinate , which resembles bicarbonate in the arrangement of its donor orbitals. A relevant note here is that the oxidation levels of polynuclear Mn–O clusters, including the OEC, are usually considered in terms of localized Mn oxidation states, although this may be a mistaken generality (Glatzel et al. 2004). Owing to its d3 configuration, Mn(IV) should favour octahedral geometry and this is observed for each subsite of (Wu et al. 2006). Normally we would expect simple Mn(IV) complexes to display much slower rates of ligand exchange than Mn(III), a property that is well established for another d3 ion Cr(III) (Atkins et al. 2006). Studies on model μ-oxo dimanganese complexes have suggested that this is the case when considering exchange of bridging oxo-ligands (Tagore et al. 2006). However, for terminal ligands, the strong π-donor ability of terminal or bridging O is expected to labilize a trans position, thus terminal water exchange at Mn(IV) could be fast.
5. Availability of Mn in biology
Manganese is the twelfth most abundant element in the Earth's crust (approx. 950 ppm), the third most abundant transition element, and it occurs in seawater as the Mn2+ aqua ion at a concentration of approximately 10−4 ppm (Wiberg 2001). It is likely that in the archaean Earth there would have been extensive complexation by bicarbonate ions due to the very high level of CO2 existing at that time, and Mn(II)-bicarbonate clusters of the type [Mn(HCO3)2]n would have existed (Dismukes et al. 2001). In minerals, Mn is almost entirely found as oxides, or with other O-containing anions such as silicates. The most common sources are MnO2 (pyrolusite), Mn3O4 (hausmannite) and MnCO3 (rhodochrosite). Other naturally occurring oxides include manganite (MnO(OH)) and MnO (manganosite; Wiberg 2001). Rich deposits of manganese are found as ‘manganese nodules’ on the ocean floor: compacted colloidal particles of Mn/Fe oxides containing 15–20% Mn. Thus, manganese is a plentiful and freely available resource.
Owing to the difficulty in oxidizing Mn2+ in simple aqueous environments, it is almost certain that this form is the one available to organisms, although it has recently been proposed that dissolved Mn(III) species may persist in certain ‘suboxic’ marine environments where insoluble Mn(IV) oxides at the seabed are subject to reduction by inorganic sulphides like H2S (Trouwborst et al. 2006). I remarked above that the Mn2+ cation is very stable, exchanges ligands rapidly and forms soluble salts; these factors, coupled with its natural abundance and the fact that (unlike Mg) a cell's requirement for Mn is very low, means that supply will usually exceed demand. For plants, soil acidity increases the availability of Mn and leads to toxicity, whereas alkaline environments can actually result in Mn deficiency. Once taken up by an organism, Mn will tend to remain as Mn2+ unless it is captured by ligands that favour its oxidation to Mn(III) and Mn(IV).
The mechanisms of Mn2+ transport and homeostasis are best understood in micro-organisms, and only recently have Mn2+ transport pathways begun to be identified in plants (Chandler et al. 2003; Pittman 2005). These pathways include specific plasma membrane ‘Nramp’ Mn2+ transporters, as well as broad specificity systems that transport Mn2+ along with Ca2+, Mg2+ and Fe2+. In plant cells, excess Mn2+, which is highly toxic, accumulates in vacuoles as a result of ‘housekeeping’ mechanisms that are linked with Fe homeostasis. It is not clear how Mn is transported into chloroplasts.
6. Assembly and maintenance of the OEC
How did the OEC come into existence? Dismukes and co-workers have considered this question and drawn attention to the important role that bicarbonate might have played as a Mn2+ chaperone in the archaean period (some 3–4 Gyr ago) when atmospheric CO2 levels were extremely high (Ananyev et al. 2001). The concentration of in the oceans would have been high enough for [Mn(HOCO2)]+ and higher complexes to exist in solution (Baranov et al. 2000). It has also been argued that bicarbonate, at these high concentrations, would have been a more efficient substrate than H2O for O2 production (Dismukes et al. 2001). As mentioned earlier, complexes formed by reaction of Mn2+ with are quite easily oxidized to Mn(III) and an intermediate of this nature is proposed to be important in OEC assembly (Baranov et al. 2000). There is indeed evidence that a carbonate is retained as a ligand in the intact OEC (Ferreira et al. 2004). The indigenous protein ligands lining the Mn4CaOx binding site, mainly glutamates and aspartates, are also highly suited to coordinating a cluster of ‘hard’ metal ions, and it has been proposed that the OEC might originally have been a Ca site (Fraústo da Silva & Williams 2001). Even without much ‘chemical’ intervention, Mn2+ ions may have become trapped in such sites by photooxidation, due to the higher solar UV intensity of the pre-oxygenic/ozonic Earth (Allen & Martin 2007).
Since the mainstay of the S-state cycle involves interconversions between Mn(III) and Mn(IV) the OEC avoids formation of the labile Mn2+ and remains intact. The OEC is stabilized structurally by Mn(IV)–O bonding and accumulates charge over a range of Mn(III)/Mn(IV) states leading finally to Mn(IV)/Mn(V) then back to S0, which does not include the labile Mn(II). An Fe–O cluster would behave differently in this respect. The binding constants for high-spin Fe(II) with O-donor ligands are only marginally higher than for Mn(II), and much less than Mn(III) (Fraústo da Silva & Williams 2001). Also, Fe(IV) is too oxidizing to provide a stable intermediate (compare the lack of Fe(IV) oxides with the abundance of Mn(IV) oxides). An Fe–O cluster equivalent of the OEC able to accumulate oxidizing power by sequential oxidations would therefore be one in which the subsites cycle between Fe(III) and Fe(II). Although similar redox cycling occurs in Fe–S clusters as part of their physiological function, it is clear that clusters consisting mainly (or entirely) of Fe(II) are more labile. An Fe–O cluster might be prone to loss of labile Fe2+ in the same way as the Fe-storage protein ferritin releases Fe upon reduction (Atkins et al. 2006). Loss of Fe2+ would occur particularly during darkness where a reduced, resting state would prevail. It follows that Mn(II) may not be relevant, apart from providing a source of Mn, and the oxidation of Mn(II) to Mn(III) is important only for the purpose of assembling the OEC. Figure 4 summarizes and integrates the useful properties of the various oxidation states of Mn, as they are relevant to the function of the OEC. There is no single property of manganese that suits it for its role—rather there are several attributes, ranging from its elemental abundance and availability as a stable aqua-cation to its unrivalled repertoire of redox chemistry under highly oxidizing conditions.
Figure 4.

Summary of the roles played by each oxidation state of Mn. The Mn(II) is an abundant mobile building unit which is taken up into the OEC and trapped by oxidation; Mn(III) and Mn(IV) provide a stable framework within which oxidizing equivalents are accumulated by increasing the Mn(IV) content; Mn(V) (or an oxyl radical) hosts the ‘hot’ O-atom generated and spent during the ‘power stroke’ of the S-cycle (S3→S4). No labile Mn2+ is generated at any part of the cycle.
Discussion
J. Barber (Imperial College London). Nature chose Mn not only due to its chemical properties but because it is an abundant transition metal in the environment. If you could choose any metal from the periodic table to replace Mn, independent of its abundance, which would it be?
F. Armstrong. From a simple angle of catalytic competence, it would be Ru, because compounds of this element are well known to have high activities in O2 evolution (see papers by T. J. Meyer). However, there is a lack of simple Ru species (labile aqua ions) that could be easily processed by a living organism. I would therefore suggest that Fe is the best alternative.
W. Junge (University of Osnabrück). You are reporting on Mn-energetics ‘in aquo’. How much are these figures affected not only by the number of ligands but also their chemical nature, in short by ‘the protein’?
F. Armstrong. The basis of energetics in coordination chemistry is the competition between a ligand and the one or more water molecules that it replaces in an interchange reaction. This competition is reflected in equilibrium constants and the change in the reduction potential of the half-cell reaction. Higher oxidation states are favoured (i.e. the reduction potential is lowered) when the ligand is a strong σ-donor, strong π-donor and negatively charged, whereas lower oxidation states are favoured by neutral ligands, particularly if they are π-acceptors. Ligands of the latter type include not only the exchangeable molecules CO and O2, but also either of the two imidazole-N atoms of a histidine side chain. In a protein, there is also a tendency for the most stable oxidation state to be the one that confers ‘lowest internal electrical charge’, although this is usually achieved by deprotonating an aqua/hydroxo ligand if one is present. Oxide (O2−) is a particularly important ligand for higher oxidation states of transition metals like manganese as it is a good π-donor and its −2 charge enables it to provide very economical coordination: for example, stabilization of Mn(VII) by F− would require at least seven F− ligands (which would be sterically unfavourable) rather than just four O2−, as in MnO4−).
Acknowledgments
This research is supported by the BBSRC and EPSRC. The Leverhulme Trust and St Johns College, Oxford are also gratefully acknowledged.
Footnotes
One contribution of 20 to a Discussion Meeting Issue ‘Revealing how nature uses sunlight to split water’.
References
- Allen J.F, Martin W. Out of thin air. Nature. 2007;445:610–612. doi: 10.1038/445610a. doi:10.1038/445610a [DOI] [PubMed] [Google Scholar]
- Alstrum-Acevedo J.H, Brennaman M.K, Meyer T.J. Chemical approaches to artificial photosythesis: 2. Inorg. Chem. 2005;44:6802–6827. doi: 10.1021/ic050904r. doi:10.1021/ic050904r [DOI] [PubMed] [Google Scholar]
- Ananyev G, Dismukes G.C. How fast can photosystem II split water? Kinetic performance at high and low frequencies. Photosynth. Res. 2005;84:355–365. doi: 10.1007/s11120-004-7081-1. doi:10.1007/s11120-004-7081-1 [DOI] [PubMed] [Google Scholar]
- Ananyev G.M, Zaltsman L, Vasko C, Dismukes G.C. The inorganic biochemistry of photosynthetic oxygen evolution/water oxidation. Biochim. Biophys. Acta. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. doi:10.1016/S0005-2728(00)00215-2 [DOI] [PubMed] [Google Scholar]
- Anderson A.B, Albu T.V. Catalytic effect of platinum on oxygen reduction: an ab initio model including electrode potential dependence. J. Electrochem. Soc. 2000;147:4229–4238. doi:10.1149/1.1394046 [Google Scholar]
- Aro E.-M, Virgin I, Andersson B. Photoinhibition of photosystem II inactivation, protein damage and turnover. Biochim. Biophys. Acta. 1993;1143:113–134. doi: 10.1016/0005-2728(93)90134-2. doi:10.1016/0005-2728(93)90134-2 [DOI] [PubMed] [Google Scholar]
- Atkins P.W, Overton T.L, Rourke J.P, Weller M.T, Armstrong F.A. 4th edn. Oxford University Press; Oxford, UK: 2006. Shriver & Atkins: inorganic chemistry. [Google Scholar]
- Baranov S.V, Ananyev G.N, Klimov V.V, Dismukes G.C. Bicarbonate accelerates assembly of the inorganic core of the water-oxidizing complex in manganese-depleted photosystem II: a proposed biogeochemical role for atmoshpheric carbon dioxide in oxygenic photosynthesis. Biochemistry. 2000;39:6060–6065. doi: 10.1021/bi992682c. doi:10.1021/bi992682c [DOI] [PubMed] [Google Scholar]
- Barber J. Biological solar energy. Phil. Trans. R. Soc. A. 2007;365:1007–1023. doi: 10.1098/rsta.2006.1962. doi:10.1098/rsta.2006.1962 [DOI] [PubMed] [Google Scholar]
- Cady C.W, Crabtree R.H, Brudvig G.W. Functional models for the oxygen-evolving complex of photosystem II. Coord. Chem. Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. doi:10.1016/j.ccr.2007.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington A, Symons M.C.R. Structure and reactivity of the oxy-anions of transition metals. Part 1. The manganese oxy-anions. J. Chem. Soc. 1956;42:3373–3380. doi:10.1039/jr9560003373 [Google Scholar]
- Chandler L.E, Bartsevich V.V, Pakrasi H.B. Regulation of manganese uptake in Synechocystis 6803 by RfrA, a member of a novel family of proteins containing a repeated five-residues domain. Biochemistry. 2003;42:5508–5514. doi: 10.1021/bi027113a. doi:10.1021/bi027113a [DOI] [PubMed] [Google Scholar]
- Clausen J, Junge W. Detection of an intermediate of photosynthetic water oxidation. Nature. 2004;430:480–483. doi: 10.1038/nature02676. doi:10.1038/nature02676 [DOI] [PubMed] [Google Scholar]
- Danopoulos, A. A., Wilkinson, G., Sweet, T. K. N. & Hursthouse, M. B. 1994 Non-oxo chemistry of manganese in high oxidation states. Part I. Mononuclear tert-butylimido compounds of manganese-(VII) and -(VI). J. Chem. Soc. Dalton Trans. 1037–1049.
- Dasgupta J, Tyryshkin A.M, Kozlov Y.N, Klimov V.V, Dismukes G.C. Carbonate complexation of Mn2+ in the aqueous phase: redox behaviour and ligand binding modes by electrochemistry and EPR spectroscopy. J. Phys. Chem. B. 2006;110:5099–5111. doi: 10.1021/jp055213v. doi:10.1021/jp055213v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dismukes G.C, Klimov V.V, Baranov S.V, Kozlov Y.N, Dasgupta J, Tyryshkin A. The origin of atmospheric oxygen on earth: the innovation of oxygenic photosynthesis. Proc. Natl Acad. Sci. USA. 2001;98:2170–2175. doi: 10.1073/pnas.061514798. doi:10.1073/pnas.061514798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley J. 3rd edn. Oxford University Press; Oxford, UK: 1998. The elements. [Google Scholar]
- Ferreira K.N, Iverson T.M, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen evolving centre. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. doi:10.1126/science.1093087 [DOI] [PubMed] [Google Scholar]
- Fraústo da Silva J.J.R, Williams R.J.P. 2nd edn. Oxford University Press; Oxford, UK: 2001. The biological chemistry of the elements. [Google Scholar]
- Glatzel P, et al. The electronic structure of Mn in oxides, coordination complexes and the oxygen-evolving complex of photosystem II studied by resonant inelastic X-ray scattering. J. Am. Chem. Soc. 2004;126:9946–9959. doi: 10.1021/ja038579z. doi:10.1021/ja038579z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabolle M, Dau H. Energetics of primary and secondary electron transfer photosystem II membrane particles of spinach revisited on basis of recombination-fluorescence measurements. Biochim. Biophys. Acta. 2005;1708:209–218. doi: 10.1016/j.bbabio.2005.03.007. doi:10.1016/j.bbabio.2005.03.007 [DOI] [PubMed] [Google Scholar]
- Kozlov Y.N, Zharmukhamedov S.K, Tikhonov K.G, Dasgupta J, Kazakova A.A, Dismukes G.C, Klimov V. Oxidation potentials and electron donation to photosystem II of manganese complexes containing bicarbonate and carboxylate ligands. Phys. Chem. Chem. Phys. 2004;6:4905–4911. doi:10.1039/b406569g [Google Scholar]
- Krishtalik L.I. Energetics of multielectron reactions—photosynthetic oxygen evolution. Biochim. Biophys. Acta. 1986;849:162–171. doi:10.1016/0005-2728(86)90107-6 [Google Scholar]
- Kulik L.V, Epel B, Lubitz W, Messinger J. 55Mn pulse ENDOR at 34 GHz of the S0 and S2 states of the oxygen-evolving complex in photosystem II. J. Am. Chem. Soc. 2005;127:2392–2393. doi: 10.1021/ja043012j. doi:10.1021/ja043012j [DOI] [PubMed] [Google Scholar]
- Kuntzleman T, Yocum C.F. Reduction-induced inhibition and Mn(II) release from the photosystem II oxygen evolving complex by hydroquinone or NH2OH are consistent with a Mn(III)/Mn(III)Mn(IV)/Mn(IV) oxidation state for the dark-adapted enzyme. Biochemistry. 2005;44:2129–2142. doi: 10.1021/bi048460i. doi:10.1021/bi048460i [DOI] [PubMed] [Google Scholar]
- Lansky D.E, Goldberg D.P. Hydrogen atom abstraction by a high-valent Mn(V)-oxo corrolazine. Inorg. Chem. 2006;45:5119–5125. doi: 10.1021/ic060491+. doi:10.1021/ic060491+ [DOI] [PubMed] [Google Scholar]
- Lewis N.S, Nocera D.G. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. USA. 2006;103:15 729–15 735. doi: 10.1073/pnas.0603395103. doi:10.1073/pnas.0603395103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limburg J, Vrettos J.S, Liable-Sands L.M, Rheingold A.L, Crabtree R.H, Brudvig G.W. A functional model for O–O bond formation by the O2-evolving complex in photosystem II. Science. 1999;283:1524–1527. doi: 10.1126/science.283.5407.1524. doi:10.1126/science.283.5407.1524 [DOI] [PubMed] [Google Scholar]
- Liu F, Cardolaccia T, Hornstein B.J, Schoonhover J.R, Meyer T.J. Electrochemical oxidation of water by an adsorbed μ-oxo-bridged Ru complex. J. Am. Chem. Soc. 2007;129:2446–2447. doi: 10.1021/ja068630+. doi:10.1021/ja068630+ [DOI] [PubMed] [Google Scholar]
- McEvoy J.P, Brudvig G.W. Water splitting chemistry of photosystem II. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. doi:10.1021/cr0204294 [DOI] [PubMed] [Google Scholar]
- McEvoy J.P, Gascon J.A, Batista V.S, Brudvig G.W. The mechanism of photosynthetic water splitting. Photochem. Photobiol. 2005;4:940–949. doi: 10.1039/b506755c. doi:10.1039/b506755c [DOI] [PubMed] [Google Scholar]
- Messinger J. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys. Chem. Chem. Phys. 2004;6:4764–4771. doi:10.1039/b406437b [Google Scholar]
- Meyer T.J, Huynh M.H.V. The remarkable reactivity of high oxidation state ruthenium and osmium polypyridyl complexes. Inorg. Chem. 2003;42:8140–8160. doi: 10.1021/ic020731v. doi:10.1021/ic020731v [DOI] [PubMed] [Google Scholar]
- Pestovsky O, Stoian S, Bominaar E.L, Shan X, Münck E, Que L, Jr, Bakac A. Aqueous FeIV =O: spectroscopic identification and oxo-group exchange. Angew. Chem. Int. Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. doi:10.1002/anie.200502686 [DOI] [PubMed] [Google Scholar]
- Pittman J.K. Managing the manganese: molecular mechanisms of manganese transport and homeostasis. New Phytol. 2005;167:733–742. doi: 10.1111/j.1469-8137.2005.01453.x. doi:10.1111/j.1469-8137.2005.01453.x [DOI] [PubMed] [Google Scholar]
- Pushkar Y, Yano J, Glatzel P, Messinger J, Lewis A, Sauer K, Bergmann U, Yachandra V. Structure and orientation of the Mn4Ca cluster in plant photosystem II membranes studied by polarized range-extended X-ray absorption spectroscopy. J. Biol. Chem. 2007;282:7198–7208. doi: 10.1074/jbc.M610505200. doi:10.1074/jbc.M610505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L, Jr, Tolman W.B. Bis(μ-oxo)dimetal ‘diamond’ cores in copper and iron complexes relevant to biocatalysis. Angew. Chem. Int. Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. doi:10.1002/1521-3773(20020402)41:7<1114::AID-ANIE1114>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- Rush J.D, Bielski B.H.J. Studies of manganate (V), -(VI), and -(VII) tetraoxyanions by pulse radiolysis. Optical spectra of protonated forms. Inorg. Chem. 1995;34:5832–5838. doi:10.1021/ic00127a022 [Google Scholar]
- Rüttinger W, Dismukes G.C. Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation. Chem. Rev. 1997;97:1–24. doi: 10.1021/cr950201z. doi:10.1021/cr950201z [DOI] [PubMed] [Google Scholar]
- Shimazaki Y, Nagano T, Takesue H, Ye B.-H, Tani F, Naruta Y. Characterization of a dinuclear MnV=O complex and its efficient evolution of O2 in the presence of water. Angew. Chem. Int. Ed. 2004;43:98–100. doi: 10.1002/anie.200352564. doi:10.1002/anie.200352564 [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M. O–O bond formation in the S4 state of the oxygen-evolving complex in photosystem II. Chem. Eur. J. 2006;12:9217–9227. doi: 10.1002/chem.200600774. doi:10.1002/chem.200600774 [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M. Mechanism and energy diagram for O–O bond formation in the oxygen-evolving complex in photosystem II. Phil. Trans. R. Soc. B. 2007;363:1221–1228. doi: 10.1098/rstb.2007.2218. doi:10.1098/rstb.2007.2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagore R, Chen H, Crabtree R.H, Brudvig G.W. Determination of μ-oxo exchange rates in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J. Am. Chem. Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. doi:10.1021/ja061348i [DOI] [PubMed] [Google Scholar]
- Tiago de Oliveira F, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Jr, Bominaar E.L, Münck E, Collins T.J. Chemical and spectroscopic evidence for an FeV-oxo complex. Science. 2007;315:835–838. doi: 10.1126/science.1133417. doi:10.1126/science.1133417 [DOI] [PubMed] [Google Scholar]
- Trouwborst R.E, Clement B.G, Tebo B.M, Glazer B.T, Luther G.W., III Soluble Mn(III) in suboxic zones. Science. 2006;313:1955–1957. doi: 10.1126/science.1132876. doi:10.1126/science.1132876 [DOI] [PubMed] [Google Scholar]
- Vincent J.B, Christou G. A molecular ‘double-pivot’ mechanism for water oxidation. Inorg. Chim. Acta. 1987;136:L41–L43. doi:10.1016/S0020-1693(00)81142-1 [PubMed] [Google Scholar]
- Wiberg N. English version. 34th edn. Academic Press; San Diego, CA: 2001. Holleman–Wiberg: lehrbuch der anorganischen chemie. [Google Scholar]
- Wu J.-Z, De Angelis F, Carrell T.G, Yap G.P.A, Sheats J, Car R, Dismukes G.C. Tuning the photoinduced O2-evolving reactivity of Mn4O47+ Mn4O46+, and Mn4O3(OH)6+ manganese-oxo cubane complexes. Inorg. Chem. 2006;45:189–195. doi: 10.1021/ic051587r. doi:10.1021/ic051587r [DOI] [PubMed] [Google Scholar]
- Yano J, et al. Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. doi:10.1126/science.1128186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan T.A, Hepler L.G. Thermochemistry and oxidation potentials of manganese and its compounds. Chem. Rev. 1968;68:737–745. doi:10.1021/cr60256a003 [Google Scholar]