

Abstract
Duocarmycin A (Duo) normally alkylates adenine N3 at the 3′ end of A+T-rich sequences in DNA. The efficient adenine alkylation by Duo is achieved by its monomeric binding to the DNA minor groove. The addition of another minor groove binder, distamycin A (Dist), dramatically modulates the site of DNA alkylation by Duo, and the alkylation switches preferentially to G residues in G+C-rich sequences. HPLC product analysis using oligonucleotides revealed a highly efficient G–N3 alkylation via the cooperative binding of a heterodimer between Duo and Dist to the minor groove. The three-dimensional structure of the ternary alkylated complex of Duo/Dist/d(CAGGTGGT)·d(ACCACCTG) has been determined by nuclear Overhauser effect (NOE)-restrained refinement using 750 MHz two-dimensional NOE spectroscopy data. The refined NMR structure fully explains the sequence requirement of such modulated alkylations. This is the first demonstration of Duo DNA alkylation through cooperative binding with another structurally different natural product, and it suggests a promising new way to alter or modify the DNA alkylation selectivity in a predictable manner.
Sequence-specific DNA recognition by naturally occurring bioactive molecules (including proteins) through a cooperative binding mode is a topic of intense current interest (1–3). For example, the initiation of transcription requires a complex set of interactions between many proteins (TATA-box binding protein, transcription factors, and transcription associated factors) and the constitutive DNA sequence (4–7).
Recently, such cooperative DNA-binding interactions have been observed for small ligands as well. An excellent example is the finding that distamycin A (Dist) can bind to the minor groove of DNA duplexes at a stretch of A·T base pairs, either in a monomeric or dimeric binding mode. Such studies have been extended to the binding of side-by-side antiparallel homo- and heterodimers of N-methylpyrrole-containing oligopeptides to the minor groove of DNA using, in particular, NMR spectroscopy (8–12), affinity cleavage experiments (13, 14), and x-ray diffraction (15). New synthetic ligands that can bind to the 5′-(A/T)GCGC(A/T)-3′ sequences through side-by-side homodimer formation have also been designed and prepared (16, 17). More recently, a synthetic polyamide peptide has been shown to recognize a longer hybrid sequence (18).
Another interesting class of DNA minor groove-interacting ligands, exemplified by the highly potent antitumor antibiotics CC-1065 and duocarmycins, alkylates DNA (19–25). Duocarmycin A (Duo) (Fig. 1A), like CC-1065, normally alkylates adenine N3 at the 3′ end of three or more consecutive A·T base pairs in DNA (19–25). We have demonstrated that the cyclopropane subunit of Duo alkylates N3 of adenine (A6) of d(CGTATACG)2 to afford a covalent adduct, which is readily decomposed to an abasic site-containing oligomer with a concomitant release of Duo-adenine adduct upon heating (23). The AT sequence preference for these alkylating agents may be understood by their matching shape to the narrow minor groove size associated with the AT sequences. However, unlike Dist, which can bind to the minor groove of the consecutive A·T base pairs in a homodimeric mode, the homodimers of duocarmycins and CC-1065 have not been observed. Although Duo prefers 3′ terminus adenine as the primary target in a consecutive sequence of A·T base pairs, it has been observed that Duo can alkylate N3 of guanine (G7) in d(GCAATTGC)2 and d(GCATATGC)2 (25). This is probably due to Duo’s greater reactivity among the seven duocarmycins in the family (21). Guanine alkylations were not observed with other alkylating ligands (e.g., CC-1065 and duocarmycin SA).
Figure 1.
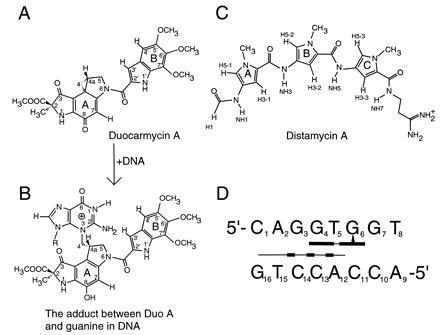
(A and B) Chemical structures of Duo and its guanine adduct at N3 site. (C) Chemical structure of Dist which normally binds to >5 base pairs of AT sequences (26). (D) Schematic representation of guanine alkylation of d(CAGGTGGT)·d(ACCACCTG) by Duo in the presence of Dist.
Unexpectedly, it was discovered that the addition of another minor groove binder, Dist (Fig. 1C), markedly modulates the site of alkylation by Duo in longer DNA fragments where the major alkylation occurs at G residues in G+C-rich sequences, although the molecular mechanism for the G-alkylation has not been established (27). To understand the molecular basis of how this highly efficient G–N3 alkylation is achieved through the cooperative binding of the heterodimer between Duo and Dist to the minor groove of DNA, we analyzed the detailed results from the alkylation reactions between Duo and a series of DNA oligonucleotides in the absence and presence of Dist. In addition, the three-dimensional structure of the ternary alkylated complex of Duo/Dist/d(CAGGTGGT)·d(ACCACCTG) has been determined by nuclear Overhauser effect (NOE)-restrained refinement using 750 MHz two-dimensional NOE spectroscopy (2D-NOESY) data.
MATERIALS AND METHODS
A series of DNA oligonucleotide hairpins whose stems contain the preferred target sequence as deduced (27) have been synthesized on an automated DNA synthesizer. The alkylation of DNA oligonucleotides was carried out according to the following procedure. A reaction mixture (50 μl) containing Duo (0.1 mM), Dist (0.1 mM), and the hairpin oligonucleotides (1 mM base concentration) in 50 mM sodium cacodylate buffer (pH 7.0) was incubated at 0°C for 3 hr. The reaction was monitored by HPLC using a Cosmosil 5C18 MS column (4.6 × 150 mm) (Nacalai Tesque, Kyoto, Japan). Elution was performed with 0.05 M ammonium formate and 0–50% acetonitrile linear gradient (0–40 min) at a flow rate of 1.0 ml/min. Detection was at 254 nm. Different peaks were assigned on the basis of UV absorption spectrum. The production of Duo-guanine adduct together with the abasic site-containing nucleotides upon brief heating (90°C for 5 min) was established using previous procedures (25).
For structural analysis, a large-scale preparation of the ternary complex was carried out using a reaction mixture (511 μl) containing Duo (0.68 mM), Dist (0.68 mM), and d(CAGGTGGT)·d(ACCACCTG) (0.68 mM of duplex) in 6.8 mM sodium phosphate buffer (pH 7.0) incubated at 0°C for 1 hr and then lyophilized. The NMR solution was prepared by dissolving 3.18 mg of lyophilized powder in 0.55 ml of H2O containing 20 mM phosphate buffer (pH 7.0), resulting in 1.4 mM of duplex solution. NMR spectra were collected either on a Varian model VXR500 500 MHz spectrometer or a Varian model 750 MHz spectrometer, and the data were processed with felix version 1.1 (Hare Research, Woodinville, WA). The nonexchangeable 2D-NOE spectra were collected at 15°C at a mixing time of 200 ms and a total recycle delay of 2.273 s. The data were collected by the States/time proportional phase incrementation technique (28) with 400 t1 increments and 2048 t2 complex points, each the average of 32 transients. 2D-NOESY spectra at 2°C in 90% H2O were collected with the 1–1 pulse sequence as the read pulse of the NOESY. Sixty-four transients were averaged with a recycle delay of 3.0 s and a mixing time of 200 ms. The excitation offset was set to one quarter of the spectral bandwidth, which was set to 12,000 Hz so that the imino resonances around 13 ppm were nearly maximally excited. The 2D-NOESY and total correlated spectroscopy in D2O and 2D-NOESY spectra in H2O were used to assign the resonances of all nonexchangeable and exchangeable protons.
The initial model of the 1:1:1 complex was constructed by docking Duo and Dist in a side-by-side orientation to the minor groove of the octamer B-DNA duplex using quanta version 4.0 (Molecular Simulation, Burlington, MA), based on the NOE data and the sites of DNA alkylation observed experimentally. The model subsequently was energy minimized. The three-dimensional structure was obtained by a combined spectral-driven refinement (29–31) and NOE-constrained molecular dynamics refinement (32) with an NMR R-factor (Σ|No − Nc|/ΣNo, where No and Nc are the experimental and calculated NOE integrals, respectively) of 24%.
RESULTS
Duo Alkylation.
Based on our previous observations (27), we selected the octamer duplex d(CAGGTGGT)·d(ACCACCTG) to investigate the DNA alkylation by Duo in the presence and absence of Dist. HPLC analysis of the reaction mixture after 1 hr of incubation in the absence of Dist showed that the three peaks of d(CAGGTGGT), d(ACCACCTG), and Duo remained unchanged with no appreciable alkylation product. However, addition of Dist to the reaction system instantaneously initiated alkylation, and after 1 min of incubation ≈80% of d(CAGGTGGT) was converted to the alkylated adduct. The reaction was complete in ≈30 min. The chemical structures of the alkylated adducts were confirmed by heat-dependent decomposition of the adducts and their corresponding abasic sites (25). The alkylation site is G6 on the d(CAGGTGGT) strand. These results showed that, in the presence of Dist, N3 of G6 nucleophilically attacks the cyclopropane subunit of Duo to produce the alkylated G efficiently (Fig. 1 A and B). In contrast, prolonged incubation without Dist gave only a trace amount of the alkylated G. The yields of Duo adducts of d(CAGGTGGT)·d(ACCACCTG) with Dist and without Dist are 80% (1 min) and 2.1% (5 hr), respectively. Thus, Dist markedly accelerated the G-alkylation by Duo by a factor of ≈104. For comparison, the yield of Duo adduct of d(CGTATACG)2 (at A6 site) is 60% in 4.5 hr.
The extent of G-alkylation increased with increasing Dist concentration and reached a maximum at a 1:1 molar ratio of Dist and Duo. Interestingly, excess Dist actually retarded the rate of G-alkylation by Duo. This is probably due to the homodimeric binding of Dist to the DNA minor groove, which may compete with the heterodimeric binding responsible for G-alkylation.
To determine the consensus sequence for the Dist-dependent guanine alkylation, a series of DNA hairpin oligodeoxynucleotides (ODN) with a T5 loop—i.e., d(CAGGTGGT)·d(ACCACCTG) and mutated sequences linked with a T5 loop (Table 1)—were prepared. The systematic replacement or deletion of various base pairs enabled us to determine the binding efficiency of Duo to DNA double helices of different sequences. By HPLC analysis of the reaction products, the chemical structures of the alkylated adducts were confirmed by heat-dependent formation shown in Fig. 1B. ODN 1 was efficiently alkylated by Duo in the presence of Dist, consistent with the results for d(CAGGTGGT)·d(ACCACCTG). The mutated hairpin ODNs had varying alkylation efficiencies. Change of A2·T15 (in Table 1, the numbering of the hairpin nucleotides in the stem follows that of the octamer duplex) to a T·A base pair reduced the extent of alkylation by 35%, whereas replacement of this A·T base pair by a G·C base pair reduced alkylation by a factor of 8. The exchanges of G3·C14, G4·C13, and T5·A12 to C·G, C·G, and A·T base pairs, respectively, markedly reduced the extent of alkylation by a factor of 5–25 (ODN 4–6). Deletion of one of the base pairs at the -G3G4- site completely inhibited the alkylation (ODN 7). However, the alkylation was not changed by deletion of the G7·C10 base pair (ODN 8). These results from Table 1 clearly establish that d[(A/T)GGTG*] is the consensus sequence, with G* as the alkylation site. In all the experiments performed above, no homodimeric binding of Duo to the hairpin oligonucleotides was found.
Table 1.
Alkylation of DNA hairpins by Duo A in the presence of Dist A
| ODN | Sequence | Relative extent of alkylation | Comments |
|---|---|---|---|
| 1 | CAGGTG*GT\ T5 | 1.0 | Wild-type control |
| GTCCAC CA/ | |||
| 2 | CTGGTG*GT\ T5 | 0.65 | Relatively good H-bond between N3 of A15 and NH7 of Dist A; not as good as in wild type |
| GACCAC CA/ | |||
| 3 | CGGGTG*GT\ T5 | 0.13 | Amidinium tail of Dist A clashes with G2 amino |
| GCCCAC CA/ | |||
| 4 | CACGTG*GT\ T5 | 0.18 | NH5 of Dist A not in good H-bond position |
| GTGCAC CA/ | |||
| 5 | CAGCTG*GT\ T5 | 0.04 | NH3 of Dist A not in good H-bond position |
| GTCGAC CA/ | |||
| 6 | CAGGAG*GT\ T5 | 0.09 | A5 is too big resulting in clashes with H51/H52 of DuoA |
| GTCCTC CA/ | |||
| 7 | CAG_TG GT\ T5 | 0 | Amidinium tail of Dist A extrudes beyond the end of stem |
| GTC_AC CA/ | |||
| 8 | CAGGTG*_T\ T5 | 0.94 | Base pairs beyond G* site on 3′ end are not critical |
| GTCCAC _A/ |
The numbering of the nucleotides in the stem follows that of the octamer duplex. The mutated base pairs are shown in boldfaced type and the deletions are shown with underlines. The alkylated guanine is marked as G*.
Structure of Duo/Dist/DNA Ternary Complex.
To gain further insight into the molecular basis of the specific G-alkylation, the three-dimensional structure of the 1:1:1 alkylated complex of Duo, Dist, and d(CAGGTGGT)·d(ACCACCTG) (Fig. 1D) was determined by using 2D-NMR and NOE-restrained refinement (31). The one dimensional NMR spectrum displays a remarkable chemical shift dispersion, which minimizes the overlap of crosspeaks in the 2D spectra and simplifies the assignment process.
Except for the terminal T8 imino proton, all other seven imino protons from DNA are well resolved in the H2O spectrum (data not shown). The sequential imino proton connectivity can be traced from G16 on one end to G7 on the other end without interruption. T8 imino proton is not seen due to the end fraying. T5 and G6 imino protons shift significantly upfield to 13.2 ppm and downfield to 14.2 ppm, respectively, which presumably is caused by the Duo alkylation to the N3 of G6. The NOE connectivities between the imino protons and amino protons and the base nonexchangeable proton (AH2)—e.g., G-imino to C-amino across the G·C base pair; T-imino to AH2 and A-amino protons across the A·T base pair—establish the Watson–Crick pairing at all base pairs.
The intermolecular NOEs were used to position Dist and Duo in the DNA minor groove. The crosspeaks between drug protons and DNA sugar protons indicate their spatial proximity. In Fig. 2, there are several strong NOEs (DistH3-1 × C13H1′, DistH3-2 × C14H1′, DistH3-3 × T15H1′, DistH3-1 × A12H2), plus a number of weaker NOEs (DistH3-1 × A12H1′, DistH3-2 × C13H1′, DistH3-3 × C14H1′, DistH3-3 × A2H2, DistH5-1 × C13H1′, DistH5-2 × C14H1′). Other NOEs (not shown) between Dist and DNA include very strong crosspeaks [e.g., DistNH1 × A12H1′, DistNH3 × C13H1′, DistNH5 × C14H1′, DistNH7 × T15H1′ (from 2D-NOESY in H2O)] and weak NOE crosspeaks (e.g., DistH5-3 × T15H1′). Also, DistH1 has crosspeaks with C11H1′ and A12H1′ with equal intensities. The amidinium tail methylene protons show NOEs to A2H2. These intermolecular NOEs firmly establish the position of Dist relative to the DNA.
Figure 2.

Expanded regions of the observed and simulated 2D-NOESY spectrum of the ternary complex in D2O showing many important drugs-to-DNA crosspeaks. (A and B) Drug to H1′/H5 crosspeaks. (C and D) Drug-to-DNA aromatic protons. Selected crosspeaks: (a) T15H1′ × DistH3-3, (b) C13H1′ × DistH5-1, (c) C13H1′ × DistH3-1, (d) C13H1′ × DistH3-2, (e) T5H1′ × DuoH3′, (f) T5H1′ × T5H6 (g) T5H1′ × DuoH4′, (h) C14H1′ × DistH5-2, (i) A12H1′ × DistH3-1, (j) C14H1′ × DistH3-3, (k) C14H1′ × DistH3-2, (l) G4H1′ × T5H6, (m) G4H1′ × DuoH4′, (n) DistH5-3 × DuoH4′, (p) DistH5-3 × DistH3-3, (q) G6H8 × T5H6, (r) A2H2 × DistH3-3, (s) G4H8 × T5H6, (t) DistH1 × DistH5-1, (u) A12H2 × DuoH3′, (v) DuoH7 × DistH5-2, (w) A12H2 × DistH3-1.
Positioning of Duo in the minor groove is straightforward, since C4 of the cyclopropane ring of Duo is covalently linked to the N3 of G6 after the three-membered ring is opened under nucleophilic attack by the N3 of G6. Intermolecular NOEs between Duo and DNA (e.g., DuoH3′ × T5H1′, DuoH4′ × T5H1′, DuoH3′ × A12H2, DuoH4′ × G4H1′ in Fig. 2) reinforce the positioning of Duo in the minor groove. Other NOEs observed include Duo5′CH3 × G4H1′ (strong), Duo5′CH3 × G4H4′, Duo5′CH3 × T5H1′, Duo7′CH3 × G6H5′/H5", and DuoH7 × G7H5′/H5".
Many intermolecular NOEs between Duo and Dist further confirm the relative positioning of the two drugs in the minor groove. These NOEs include DuoH3′ × DistH3-1, DuoH3′ × DistH3-2, DuoH3′ × DistH5-2, DuoH4′ × DistH3-2, DuoH7 × DistH5-2, DuoH7 × DistCH3-1, Duo5′CH3 × DistH3-3, Duo5′CH3 × DistH5-3, Duo6′CH3 × DistH5-3, and Duo7′CH3 × DistCH3-2. These data allowed a good starting model to be constructed, which was subjected to the NOE-restrained refinement. The model was subsequently refined to an NMR R-factor of 24%. The final refined structure is shown in Fig. 3 and the key hydrogen bonds are illustrated in Fig. 4.
Figure 3.
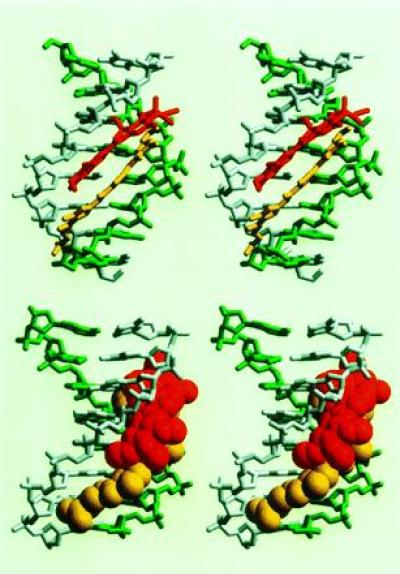
Refined structure of the heterodimer of Duo (red) and Dist (yellow) binding in the minor groove of d(CAGGTGGT) (gray)·d(ACCACCTG) (green). (Upper) View into the minor groove, showing the side-by-side binding of the two drugs. (Lower) Side view with the drugs in van der Waals representations.
Figure 4.
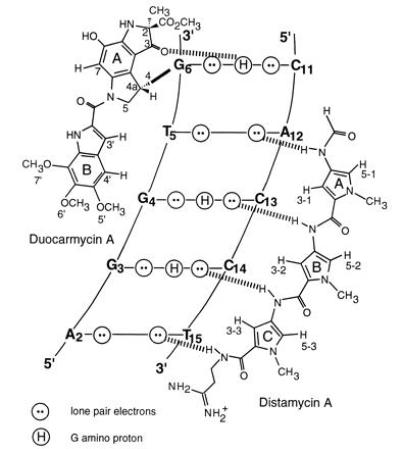
Schematic representation of the heterodimeric binding model of Duo and Dist to the minor groove of d(CAGGTGGT)·d(ACCACCTG). Hydrogen bonds between drugs and DNA are illustrated by dashed lines. The hydrogen bond between the O3 of Duo and the N2 amino group of G6 helps anchor the drug in its position.
DISCUSSION
The intra- and intermolecular NOE crosspeaks for Duo/Dist/d(CAGGTGGT)·d(ACCACCTG) ternary complex clearly support the formation of a Dist/Duo heterodimer that binds to the minor groove in a side-by-side arrangement. Some important NOEs (experimental and calculated) are summarized in Fig. 2. The refined structure (Fig. 3) shows the relative positioning of the two drugs in the minor groove. Ring A and ring B of Duo are stacked on ring A and ring B of Dist, respectively. The heterodimer fits snugly in the minor groove, making the groove width slightly wider than the G+C-rich B-DNA duplex, and much wider than the A+T-rich B-DNA duplex bound with Dist. The averaged P–P distance across the groove is 15.3 Å, in contrast to 12.3 Å in the d(CATGGCCATG) duplex (33), 13.3 Å in d(CCAGGCCTGG) (34), and 9.9 Å in the 1:1 complex of Dist-d(CGCAAATTTGCG) (26). The refined structure also explains the dramatic upfield chemical shift changes (some >2 ppm) associated with some protons, due to the extreme ring current effect from drugs. The H4′ (1.24 ppm)/H2′ (1.09 ppm)/H2" (1.28 ppm) of the T5 residue in the d(CAGGTGGT) strand, all having large upfield shifts, are situated directly above ring B of Duo (Fig. 3). G6H1′ (4.64 ppm) shifts upfield because of the ring A of Duo. Similarly, C13H4′ (2.47 ppm), C14H4′ (1.92 ppm), and T15H4′ (2.90 ppm) are greatly shifted upfield because they are positioned directly above rings A, B, and C of Dist, respectively.
There are specific hydrogen bonds in the heterodimeric binding of Duo and Dist to d(CAGGTG*GT)·d(ACCACCTG) as illustrated in Fig. 4. Note that the O3 (ring A) of Duo receives a hydrogen bond from N2 amino group of G6. Such a hydrogen bond may help Duo anchor its ring A into a favorable position for the nucleophilic attack from the G–N3 onto its C4 site. It has been concluded that the G–N2 amino proton hydrogen bonding to the imidazole rings in several synthetic polyamide peptides (8–14, 16–18) provides the sequence-specific recognition for G+C-rich sequences. If this hydrogen bond (O3 of Duo to N2 amino of G6) favors the recognition of G over A for the alkylation by Duo, elimination of this hydrogen bond may retard the Duo alkylation. This may be tested by replacing the G with an inosine (I); this work is in progress.
The four amide protons of Dist form four hydrogen bonds with DNA, specifically, NH1 with A12N3, NH3 with C13O2, NH5 with C14O2, and NH7 with T15O2 (Fig. 4). These hydrogen bonds contribute to Dist binding to the right side of the groove and position Dist in such a way that ring A and ring B of Duo stack on the ring A and ring B of Dist. The stacking energy may help stabilize the heterodimer in the minor groove.
The refined structure provides satisfactory explanations for the observed G-alkylation efficiencies of various DNA hairpins in Table 1, where the molecular bases for such observations are listed as “comments.” We have performed simple computer modeling studies using all the DNA sequences (stems only) except for ODN 1 in Table 1. The standard B-DNA sequences were used initially, and the heterodimer of the two drugs was taken from the refined Duo/Dist/d(CAGGTGGT)·d(ACCACCTG) ternary complex and positioned in the respective DNA minor groove so that all the hydrogen bonds were in good position. The steepest decent energy minimizations were subsequently performed. The resulting models were compared with the refined ternary complex. The comments were made, based on the initial models and final energy-minimized models, to explain why these sequences have varying alkylation efficiencies.
It is intriguing to note that all polyamide-containing drugs bind to GC sequence in a 2:1 ratio by forming a dimer (preferentially antiparallel) (8–18) in the DNA minor groove. However, these drugs bind consecutive TA sequence in either 1:1 or 2:1 ratios. One could argue that in a 1:1 drug to DNA-binding mode, GC-containing sequence has protruding amino protons from G in the minor groove that present steric hindrance to the pyrrole-containing drug and prevent its binding to the minor groove floor. Moreover, the GC sequence generally is associated with a wider minor groove width than that of AT sequence. This makes the single drug fit to GC sequences less snug, resulting in less favorable hydrophobic interaction between the drug and DNA minor groove walls. On the other hand, GC sequences can accommodate two drug molecules without significant additional widening of the minor groove. The two drug molecules would move to the respective DNA strands away from the center, minimizing the steric contact with the protruding G amino proton and forming favorable hydrogen bonds with bases. Also, a direct benefit of two drug molecules moving away from the center is that the drug molecules can effectively interact with the DNA minor groove wall (H1′, H4′, H5′/5") hydrophobically, stabilizing the complex. With these arguments taken together, it is clear that GC sequences prefer to bind dimers in the minor groove, whether as a homodimer or as a heterodimer, consistent with the refined structure observed in this work.
It is of interest to note that in Duo/Dist/d(CAGGTGGT)·d(ACCACCTG) ternary complex, rings A and B of Duo directly stack on top of rings A and B of Dist, respectively. This suggests that the stacking may play an important part in the stabilization of the dimer in the minor groove.
In conclusion, the Duo/Dist heterodimer specifically recognizes the sequence 5′-AGGTG*-3′ in the minor groove of DNA, resulting in a highly efficient guanine alkylation. To the best of our knowledge, this is the first demonstration of efficient DNA alkylation by bioactive natural products through a cooperative heterodimer formation. The novel DNA alkylation through heterodimer formation may hold promises for designing new alkylating agents with significantly higher DNA sequence selectivity and efficiency. For example, a covalently linked hybrid dimer may be prepared for such a purpose.
Acknowledgments
We thank Prof. C. W. Hilbers and Dr. H. Heus of Nijmegen University for assisting with the 2D-NOESY data collection on the Varian 750 MHz NMR spectrometer in Utrecht, and Dr. H. Robinson for the SPEDREF structure refinement package. The Kyoto part of this work was supported by a grant-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture, Japan; the Urbana part was supported by American Cancer Society Grant DHP-114 to A.H.-J.W. C.L. was supported by a National Institutes of Health institutional National Research Service Award in molecular biophysics (GM-08276). The Varian VXR500 NMR spectrometer at the University of Illinois at Urbana–Champaign was supported in part by National Institutes of Health shared instrumentation Grant 1S10RR06243.
Footnotes
Abbreviations: Duo, duocarmycin A; Dist, distamycin A; 2D-NOESY, two-dimensional nuclear Overhauser effect spectroscopy; ODN, oligodeoxynucleotide.
References
- 1.Hill T L. Cooperativity Theory in Biochemistry: Steady State and Equilibrium Systems. New York: Springer; 1985. [Google Scholar]
- 2.Ptashne M. A Genetic Switch. 2nd Ed. Palo Alto, CA: Blackwell Scientific; 1992. [Google Scholar]
- 3.Adhya S. Annu Rev Genet. 1989;23:227–250. doi: 10.1146/annurev.ge.23.120189.001303. [DOI] [PubMed] [Google Scholar]
- 4.Roeder R G. Trends Biochem Sci. 1991;16:402–408. doi: 10.1016/0968-0004(91)90164-q. [DOI] [PubMed] [Google Scholar]
- 5.Hori R, Carey M. Curr Opin Genet Dev. 1994;4:236–244. doi: 10.1016/s0959-437x(05)80050-4. [DOI] [PubMed] [Google Scholar]
- 6.Goodrich L A, Tjian R. Curr Opin Cell Biol. 1994;6:403–409. doi: 10.1016/0955-0674(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 7.Tanese N, Tjian R. Cold Spring Harbor Symp Quant Biol. 1993;58:179–185. doi: 10.1101/sqb.1993.058.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Pelton J G, Wemmer D E. Proc Natl Acad Sci USA. 1989;86:5723–5727. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelton J G, Wemmer D E. J Am Chem Soc. 1990;112:1393–1399. [Google Scholar]
- 10.Mrksich M, Wade W S, Dwyer T J, Geierstanger B H, Wemmer D E, Dervan P B. Proc Natl Acad Sci USA. 1992;89:7586–7590. doi: 10.1073/pnas.89.16.7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geierstanger B H, Jacobsen J-P, Mrksich M, Dervan P B, Wemmer D E. Biochemistry. 1994;33:3055–3062. doi: 10.1021/bi00176a039. [DOI] [PubMed] [Google Scholar]
- 12.Geierstanger B H, Dwyer T J, Bathini Y, Lown J W, Wemmer D E. J Am Chem Soc. 1993;115:4474–4482. [Google Scholar]
- 13.Wade W S, Mrksich M, Dervan P B. Biochemistry. 1993;32:11385–11389. doi: 10.1021/bi00093a015. [DOI] [PubMed] [Google Scholar]
- 14.Mrksich M, Dervan P B. J Am Chem Soc. 1993;115:2572–2576. [Google Scholar]
- 15.Chen X, Ramakrishman B, Rao S T, Sundaralingam M. Nat Struct Biol. 1994;1:169–175. doi: 10.1038/nsb0394-169. [DOI] [PubMed] [Google Scholar]
- 16.Geierstanger B H, Mrksich M, Dervan P B, Wemmer D E. Science. 1994;766:646–650. doi: 10.1126/science.7939719. [DOI] [PubMed] [Google Scholar]
- 17.Mrksich M, Dervan P B. J Am Chem Soc. 1995;117:3325–3332. [Google Scholar]
- 18.Geierstanger B H, Mrksich M, Dervan P B, Wemmer D E. Nat Struct Biol. 1996;3:321–324. doi: 10.1038/nsb0496-321. [DOI] [PubMed] [Google Scholar]
- 19.Boger D L. Acc Chem Res. 1995;28:20–29. [Google Scholar]
- 20.Boger D L, Johnson D S, Yun W. J Am Chem Soc. 1994;116:1635–1656. [Google Scholar]
- 21.Boger D L, Johnson D S. Proc Natl Acad Sci USA. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boger D L, Ishizaki T, Zarrinmayeh H, Kitos P A, Suntornwat O. J Am Chem Soc. 1990;112:8961–8971. [Google Scholar]
- 23.Sugiyama H, Hosoda M, Saito I, Asai A, Saito H. Tetrahedron Lett. 1990;31:7197–7200. [PubMed] [Google Scholar]
- 24.Boger D L, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645–6649. [Google Scholar]
- 25.Sugiyama H, Ohmori K, Chan K L, Hosoda M, Asai A, Saito H, Saito I. Tetrahedron Lett. 1993;34:2179–2182. [PubMed] [Google Scholar]
- 26.Coll M, Frederick C A, Wang A H-J, Rich A. Proc Natl Acad Sci USA. 1987;84:8385–8389. doi: 10.1073/pnas.84.23.8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto K, Sugiyama H, Kawanishi S. Biochemistry. 1993;32:1059–1066. doi: 10.1021/bi00055a010. [DOI] [PubMed] [Google Scholar]
- 28.States D J, Haberkorn R A, Ruben D J. J Magn Reson. 1982;48:286–292. [Google Scholar]
- 29.Robinson H, Wang A H-J. Biochemistry. 1992;31:3524–3533. doi: 10.1021/bi00128a029. [DOI] [PubMed] [Google Scholar]
- 30.Robinson H, van der Marel G A, van Boom J H, Wang AH-J. Biochemistry. 1992;31:10510–10517. doi: 10.1021/bi00158a014. [DOI] [PubMed] [Google Scholar]
- 31.Robinson H, Wang A H-J. Proc Natl Acad Sci USA. 1993;90:5224–5228. doi: 10.1073/pnas.90.11.5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brünger, A. (1993) x-plor (The Howard Hughes Medical Institute and Yale University, New Haven, CT), Version 3.1.
- 33.Goodsell D S, Kopka M L, Cascio D, Dickerson R E. Proc Natl Acad Sci USA. 1993;90:2930–2934. doi: 10.1073/pnas.90.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, van der Marel A, van Boom J H, Wang A H-J. Biochemistry. 1991;30:9922–9931. doi: 10.1021/bi00105a016. [DOI] [PubMed] [Google Scholar]