

SUMMARY
In humans, mutations in the SOX10 gene are a cause of the auditory-pigmentary disorder Waardenburg syndrome type IV (WS4) and related variants. SOX10 encodes an Sry-related HMG box protein essential for the development of the neural crest; deafness in WS4 and other Waardenburg syndromes is usually attributed to loss of neural-crest-derived melanocytes in the stria vascularis of the cochlea. However, SOX10 is strongly expressed in the developing otic vesicle and so direct roles for SOX10 in the otic epithelium might also be important. Here, we examine the otic phenotype of zebrafish sox10 mutants, a model for WS4. As a cochlea is not present in the fish ear, the severe otic phenotype in these mutants cannot be attributed to effects on this tissue. In zebrafish sox10 mutants, we see abnormalities in all otic placodal derivatives. Gene expression studies indicate deregulated expression of several otic genes, including fgf8, in sox10 mutants. Using a combination of mutant and morphant data, we show that the three sox genes belonging to group E (sox9a, sox9b and sox10) provide a link between otic induction pathways and subsequent otic patterning: they act redundantly to maintain sox10 expression throughout otic tissue and to restrict fgf8 expression to anterior macula regions. Single-cell labelling experiments indicate a small and transient neural crest contribution to the zebrafish ear during normal development, but this is unlikely to account for the strong defects seen in the sox10 mutant. We discuss the implication that the deafness in WS4 patients with SOX10 mutations might reflect a haploinsufficiency for SOX10 in the otic epithelium, resulting in patterning and functional abnormalities in the inner ear.
INTRODUCTION
The Waardenburg syndromes (WS) form a family of disorders, characterised by pigmentation abnormalities and sensorineural hearing loss, with diverse genetic causes (Read and Newton, 1997; Spritz, 2006). The severity of deafness in WS individuals can range from mild to severe. Mutations in SOX10, EDN3 and EDNRB lead to WS type IV [WS4, also known as Waardenburg-Shah syndrome, or Yemenite deaf-blind syndrome (YDBS)], in which patients also present with Hirschsprung’s disease (also known as congenital megacolon) (Puffenberger et al., 1994a; Puffenberger et al., 1994b; Edery et al., 1996; Hofstra et al., 1996; Pingault et al., 1998; Southard-Smith et al., 1999; Pingault et al., 2001; Inoue et al., 2004). In humans, different heterozygous effects have been associated with different SOX10 alleles, with some resulting in WS4 (Pingault et al., 1998; Pingault et al., 2002) and others causing a more complex and severe neurocristopathy known as PCWH syndrome. This syndrome causes peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, WS and Hirschsprung’s disease (Inoue et al., 1999; Pingault et al., 2000; Inoue et al., 2002; Verheij et al., 2006). The distinction between the different alleles depends upon whether nonsense-mediated decay of the mRNA prevents the translation of truncated, dominant negative SOX10 proteins: PCWH results from dominant negative mutations, whereas WS4 results from haploinsufficiency (Inoue et al., 2004). Deletions at the SOX10 gene locus have also been found to cause both WS4 and WS2, a variant characterised by deafness and pigmentation defects, but no additional symptoms (Bondurand et al., 2007).
The aetiology of the sensorineural deafness in WS4 and YDBS individuals is likely to involve a loss of, or a reduction in the number of, melanocytes contributing to the ear (Bondurand et al., 2000). In mammals, neural-crest-derived melanocytes populate the stria vascularis of the cochlea as intermediate cells. These are thought to play a protective role and are essential for both the maintenance of endolymph composition and generation of the endocochlear potential (Steel and Barkway, 1989; Cable et al., 1992; Cable et al., 1993; Cable et al., 1994) (reviewed by Steel, 1995; Tachibana, 1999; Price and Fisher, 2001; Wangemann, 2002; Wangemann, 2006; Lang et al., 2007). In the mouse, maintenance of the endocochlear potential depends on expression of the potassium channel Kcnj10 in intermediate cells (Marcus et al., 2002; Wangemann et al., 2004). Melanocytes also populate vestibular regions and the endolymphatic apparatus in the mammalian ear (Masuda et al., 1994; Escobar et al., 1995; Peters et al., 1995; Stanchina et al., 2006), but their role here is less clear; for example, mutations in Kcnj10 have no effect on vestibular endolymph (Marcus et al., 2002). Analysis of the inner ear phenotype in murine models of WS and other auditory-pigmentary disorders has focused on the presence of intermediate cells in the stria vascularis; deafness is usually attributed to the loss of intermediate cells in this tissue, leading to a reduction or collapse of endolymph volume, a loss of the endocochlear potential and subsequent hair cell degeneration (Tachibana et al., 1992; Cable et al., 1994; Matsushima et al., 2002; Stanchina et al., 2006) (reviewed by Tachibana, 1999; Tachibana et al., 2003).
Analysis of the inner ear in Sox10Dom heterozygote mice, however, hints that deafness might result from causes other than a loss of intermediate cells because, in the few samples analysed, these are still present in the ear (Stanchina et al., 2006), and endolymphatic collapse is not observed (Tachibana et al., 2003). In addition to contributing to melanocytes, neural crest cells also migrate around the otic vesicle; in the avian embryo, they contribute to part of the cartilaginous otic capsule and form all glial cells of the spiral and vestibular ganglia (gVIII) (Couly et al., 1993; Le Douarin and Kalcheim, 1999; Evans and Noden, 2006). Because neural-crest-derived glia are also Sox10 dependent, defects in this population could contribute to the deafness found in WS4 individuals (Kelsh and Eisen, 2000; Britsch et al., 2001; Paratore et al., 2001). In addition, a conserved site of Sox10 expression is in the otic epithelium itself, suggesting that there might be a more direct role for Sox10 in the development of the inner ear (Bondurand et al., 1998; Pusch et al., 1998; Cheng et al., 2000; Watanabe et al., 2000; Dutton et al., 2001b; Aoki et al., 2003; Honoré et al., 2003; Taylor and Labonne, 2005). If this is the case, haploinsufficiency or dominant negative versions of the Sox10 protein might affect the otic epithelium directly, in addition to having effects on neural crest derivatives.
Sox proteins are classified into groups according to the degree of identity between their HMG domains; Sox10 belongs to the Sox group E (Wegner, 1999; Kamachi et al., 2000). In zebrafish, four group E sox (soxE) genes have been identified, sox8, sox9a, sox9b and sox10, although sox8 is not expressed until at least the hatching stage, and so only the latter three have been characterised in detail (Chiang et al., 2001; Dutton et al., 2001b; Li et al., 2002; Yan et al., 2002; Liu et al., 2003; Yan et al., 2005). Expression of sox9a, sox9b and sox10 has been noted in the otic placode and early otic vesicle (Chiang et al., 2001; Dutton et al., 2001b; Li et al., 2002; Liu et al., 2003; Yan et al., 2005). However, detailed characterisation and comparison of the expression patterns of these soxE genes in the developing ear have not been documented, despite the importance for interpretation of their partially overlapping functions in ear development, which have been shown by analysis of mutants (Dutton et al., 2001b; Yan et al., 2002; Liu et al., 2003; Yan et al., 2005).
Mutations have been described in all three of these soxE genes and their phenotypes have been confirmed by morpholino phenocopy (Dutton et al., 2001a; Dutton et al., 2001b; Yan et al., 2002; Liu et al., 2003; Yan et al., 2005). In jef/sox9a mutants and morphants, the early stages of inner ear development are relatively normal but at later stages the ear is slightly reduced in size, and formation of the cartilaginous capsule that normally surrounds the ear is defective (Yan et al., 2002; Yan et al., 2005). Similarly, a sox9b deletion mutant and sox9b morphants show a slightly smaller ear and reduced cartilage differentiation (Liu et al., 2003; Yan et al., 2005). Double mutants for both sox9a and sox9b either lack or have only vestigial otic vesicles, apparently owing to a failure to induce the otic placode (Liu et al., 2003; Yan et al., 2005). The tpd mutant, which has a very similar phenotype to embryos in which both sox9 genes are disrupted, appears to encode a coactivator of Sox9 function (Rau et al., 2006). Multiple alleles of colourless (cls) disrupt the sox10 gene and result in severe abnormalities of both ear and non-ectomesenchymal neural crest derivatives in homozygous mutants; all alleles are fully recessive and appear to act as hypomorphs or nulls (Kelsh et al., 1996; Whitfield et al., 1996; Kelsh and Eisen, 2000; Dutton et al., 2001b; Elworthy et al., 2003; Elworthy et al., 2005; Carney et al., 2006). These mutants also show overproliferation of placode-derived neuromast precursors as a result of the absence of posterior lateral line glial cells (Grant et al., 2005; López-Schier and Hudspeth, 2005). The ear defects in cls/sox10 mutants are more severe than the effect of removing either of the sox9 genes singly, affecting non-sensory, sensory and neural derivatives (Whitfield et al., 1996), as shown here. Detailed characterisation of the otic defects in sox10 mutants is required to evaluate the possibility that epithelial defects may contribute to deafness in cases of WS caused by mutations in SOX10.
Here, we analyse the expression of the soxE genes in development of the wild-type (WT) and sox10 mutant zebrafish otic vesicle. We reveal that, initially, sox10 expression shows an extensive overlap with that of sox9a and sox9b throughout the otic placode/vesicle. Subsequently, sox10 expression is strongly maintained in all otic epithelial cells, whereas sox9 gene expression becomes refined to specific domains. Using specific markers, we show that differentiation of most otic derivative cell-types can be observed in sox10 mutants, but that their organisation is abnormal. Abnormalities of the endolymphatic duct might contribute to the late expansion of otic vesicles in sox10 mutant embryos. To assess the interrelationship between the soxE group genes in otic development, we use mutant and morphant analysis to show that maintenance of sox10 expression in the ear is dependent upon soxE gene function, but that the loss of any one soxE gene is not sufficient to disrupt sox10 expression. We also show derepression of sox9 expression in sox10 mutant ears. Thus, soxE genes show complex regulatory relationships; for example, the sox9a; sox9b double mutant phenotype is likely to result, at least in part, from consequent loss of sox10 expression. We integrate our data into a working model of otic vesicle development. We also show, by single-cell labelling, that the contribution of neural crest cells to the otic vesicle is small and transient, which strongly indicates that the severe and varied defects in the developing sox10 mutant ear are largely dependent upon Sox10 function within the otic epithelium.
RESULTS
Group E sox genes are expressed throughout the otic epithelium from early stages
We have made a detailed analysis of the expression of the soxE genes in the developing zebrafish otic placode and vesicle. All three of the genes examined, sox9a, sox9b and sox10, are expressed from early stages in preplacodal ectoderm (Fig. 1). Initially, sox10 expression is the weakest of the three, probably reflecting a slightly later onset of expression. Expression of sox9a is restricted to an oval domain that is likely to correspond to only pre-otic tissue at the one-somite stage (Fig. 1B), whereas sox10 and, to a much greater degree, sox9b expression extends anteriorly and posteriorly in the head within the midbrain and hindbrain regions (Fig. 1A,C). Thus, this expression includes early cranial neural crest cells. By the 15-somite stage, when the otic placode has formed, sox10 expression persists throughout the placode, whereas expression of the two sox9 genes is downregulated in the anteroventral part of the placode (supplementary material Fig. S1). Subsequently, after cavitation of the vesicle and from at least 24 to 48 hours postfertilisation (hpf), sox10 continues to be ubiquitously expressed throughout the otic epithelium, whereas expression of the sox9 genes becomes restricted spatially (Fig. 2 and supplementary material Fig. S2). Thus, at 24 hpf, sox9a expression is restricted to three domains, two dorsal and one ventral, and is downregulated in regions where hair cells are differentiating in the two maculae (Fig. 2). By 48 hpf, both sox9 genes have become largely downregulated in the otic epithelium, but sox9b expression is retained in the epithelial projections that form the semicircular canal system (supplementary material Fig. S2).
Fig. 1.

Early expression of the soxE genes overlaps strongly during development of the WT otic placode. Dorsal views of flattened whole-mount one-somite stage embryos (A–D) and transverse sections through the presumptive otic region [(E–G, the plane of section is indicated by the white bar in (A–C)] showing sox10 (A,E), sox9a (B,F) and sox9b (C,G) expression. Superimposition of false-coloured whole-mount patterns (D) indicates the extensive overlap of soxE gene expression in otic and neural crest regions, but also more extensive expression of sox9b in the premigratory neural crest at this stage. o=presumptive otic region. Bars, 150 μm (A–D); 50 μm (E–G).
Fig. 2.

soxE gene expression in cls/sox10 mutant embryos at 24 hpf. The ears of cls/sox10 mutants (B,D,F) and WT siblings (A,C,E) are shown in a lateral view of whole-mount embryos (A–F) and in transverse sections through the anterior (A–F′), medial (A″-F″) and posterior (A‴-F‴) regions. At this stage, sox10 is expressed throughout the otic epithelium and expression is largely unchanged in cls/sox10 mutants. In WT siblings, the expression of sox9a and sox9b is lost from anteroventral and posteromedioventral regions, where hair cells are beginning to differentiate in the two maculae. Expression is retained in a central ventral region and this domain of sox9b expression appears to be slightly upregulated in cls/sox10 mutants (arrows). Arrowheads in (C) and (E‴) indicate possible sox9a and sox9b expression in the SAG. In this figure, and all subsequent figures, the developing otic structures are shown in a lateral view with anterior to the left and dorsal to the top unless stated otherwise. Bars, 75 μm (A–F); 50 μm (A′-F‴).
sox9a and sox9b are known targets of Fgf-dependent pathways of zebrafish otic induction (Phillips et al., 2001; Léger and Brand, 2002; Maroon et al., 2002). However, sox10 expression is present and appears to be relatively normal in ace/fgf8 mutants (data not shown). We have also examined the expression of sox10 in various mutant backgrounds known to affect otic epithelial patterning and development, but expression is normal in smu/smo mutants (which lack Hedgehog signalling) (Barresi et al., 2000; Hammond et al., 2003) and in vgo/tbx1, noi/pax2a and dog/eya1 mutants (data not shown), each of which lacks the function of a transcription factor expressed in early otic epithelium (Piotrowski et al., 2003; Hans et al., 2004; Mackereth et al., 2005).
Sox10 does not appear to be autoregulatory in the zebrafish ear but might regulate the expression of the sox9 genes
We extended our studies of WT ear development to examine the expression patterns of the soxE genes in the absence of sox10 function (Fig. 2 and supplementary material Fig. S2). At both 24 and 48 hpf we saw no difference in the proportion of otic vesicle cells expressing sox10 between WT and sox10 mutant embryos (Fig. 2A–B‴). Expression of sox9 genes at 24 hpf is largely comparable in sox10 mutants and their WT siblings, although sox9a expression in the dorsal otic vesicle is somewhat more extensive (Fig. 2C–C‴, D–D‴). By 48 hpf, ectopic expression of sox9a is prominent in the otic epithelium of sox10 mutants, whereas in WT siblings expression is restricted to periotic mesenchyme (supplementary material Fig. 2C–D‴). Likewise, sox9b expression appears to be slightly upregulated in the ventral otic epithelium of sox10 mutants at 24 hpf (Fig. 2E–F‴), and expression here persists at 48 hpf (supplementary material Fig. 2F‴). Note that the epithelial projections forming the semicircular canal system, which normally express sox9b strongly at 48 hpf (supplementary material Fig. 2E‴), are rudimentary in sox10 mutants (supplementary material Fig. 2F‴). Thus, although sox10 transcription appears to be independent of sox10 function, Sox10 represses sox9 gene transcription (directly or indirectly) in the otic epithelium at later stages. Note, however, that a role for sox10 in regulating its own transcription is revealed when sox9 activity is also reduced (see below).
Otic vesicle morphology is abnormal in sox10 mutants
Abnormalities in otic morphology were noted in the initial descriptions of zebrafish sox10 mutants (Whitfield et al., 1996). We have examined defects in sox10 mutant otic vesicle morphology from 27 hpf, when mutants can be identified on the basis of their pigmentation phenotype. At 27 hpf, there are subtle defects in otic vesicle shape: the dorsal epithelium of the mutant is not as thin as in the sibling, and the ventral epithelium is more uniform in thickness (Fig. 3A,B; Fig. 7A–D). By 48 hpf, the vesicle is clearly misshapen and smaller than normal, and the otoliths are small and more closely spaced than in the WT embryo (Fig. 3C–E). Development of the epithelial projections that form the semicircular canal system is initially delayed (Fig. 3C,D); they eventually develop, but are rudimentary (Fig. 3F,G). Expression of the semicircular canal tissue markers ugdh1 (Busch-Nentwich et al., 2004) and dacha (Hammond et al., 2002) is, however, normal in the epithelial projections that are present (data not shown). The otoliths remain very small and sit close to one another in the mutant (Fig. 3H–K). At 5–8 days post fertilisation (dpf), the otic phenotype is very variable; some ears remain very small but others become grossly distended, sometimes on one side of the fish only (Fig. 3J–O). Further, where semicircular canal pillars have formed, they appear thinner than normal (Fig. 3N).
Fig. 3.

Otic vesicle size and morphology is aberrant in sox10 mutants. (A–D,F–O) DIC images of the ears of live sibling (WT) and cls/sox10 mutant embryos. (A,B) At 27 hpf, the images show slight differences in vesicle morphology. (C,D) By 48 hpf, the anterior and posterior semicircular canal projections (arrows) are not visible in the mutant. (E) Quantitation of the anteroposterior (AP) length of the otic vesicle (mean±s.e.) at 48 hpf for WT and cls/sox10 mutants. n=10 for each genotype; p=0.0003 (t-test). (F–I) At 72 hpf, the epithelial projections are now fused to form pillars (arrows) in the WT embryo, but are disorganised in the mutant. (H,I) 72 hpf images focused on the otoliths (arrowheads). (J) A WT ear at 6 days postfertilisation (dpf). (K–M) Three different examples of sox10 mutant ears at 5 dpf showing the variability in size. All these ears contained two small otoliths, but they are out of the focal plane in (L) and (M). (N,O) Dorsal views of two cls/sox10 mutants at 8 dpf showing hugely distended and misshapen ears [bilateral in (N) and unilateral in (O), where the ear on the left remains small and uninflated]. A thin semicircular canal pillar (arrow) spans the enlarged lumen. All panels are lateral views, with anterior to the left, except for (N) and (O) which represent dorsal views, with anterior to the top. Mutants in (E) are the clsm618 allele; mutants in all other panels are the clst3 allele. Bars, 50 μm.
Fig. 7.

Sensory epithelia are disrupted in cls/sox10 mutants. Confocal images (projected Z sections) of FITC-phalloidin-stained ears to show sensory patch defects in clst3 mutant embryos. The age in hpf is shown for each panel. (A–H) Initial hair cell development at 28–30 hpf in both the anterior (A,C,E,G) and posterior (B,D,F,H) maculae is relatively normal in cls/sox10 mutants. (I) By 51 hpf, there are 10–20 hair cells in the WT anterior macula (arrowhead) and slightly fewer in the posterior macula (arrow). (J–L) Three examples of a cls/sox10 mutant ear at 51 hpf. The anterior macula (arrowheads) has relatively normal numbers of hair cells, but is misplaced medially; the posterior macula (arrows) has reduced numbers of hair cells. (M) In WT ears at 60 hpf, the two maculae are clearly distinct in shape and position in the vesicle (arrowhead, anterior macula; arrow, posterior macula). (N–P) Three examples of a cls/sox10 mutant ear at 60 hpf, focused on the medial wall of the vesicle. Separate maculae are no longer discernible. (Q) A lateral plane of focus at 60 hpf in a WT ear reveals hair cells in the three cristae (asterisks). (R–T) Three examples of cristae in cls/sox10 mutant ears at 60 hpf. Anterior and posterior thickenings are evident (asterisks), with variable numbers of hair cells. Right hand asterisk in (T) shows a possible lateral crista. (U,V) Sensory patches in WT and cls/sox10 mutant ears at 72 hpf. The asterisk in (V) shows a possible anterior crista. (W,X) Sensory patches in a 5 dpf WT (W) and a 6 dpf cls/sox10 mutant (X). The asterisk in (X) indicates unidentifiable crista. Panels (A–T) are lateral views with anterior to the left; (U) and (V) are dorsal views of the left-sided ears (anterior to the top); (W) and (X) are dorsal views of right-sided ears (anterior to the top). Bars, 25 μm.
Regional markers indicate patterning defects in sensory and non-sensory regions of the cls/sox10 mutant ear
The complex morphological defects coupled with the early onset of sox10 expression in otic development led us to examine molecular markers of otic patterning in sox10 mutants. Whereas the expression of some markers, such as pax2a, is unaffected in the sox10 mutant ear (Fig. 4A–D), several markers are expressed abnormally. Interestingly, these often show both a loss of expression in their normal domain and ectopic expression elsewhere. At 24 hpf, fgf8 is expressed close to its normal domain in anteroventral epithelium (with a slight extension dorsally and medially), but is also misexpressed at the posterior of the ear in sox10 mutants (Fig. 4E–H). Ectopic fgf8 expression at the posterior of the otic vesicle has also been reported in ace/fgf8 mutants (Léger and Brand, 2002), but we find that in sox10 mutants this phenotype is fully penetrant [n=14/49 (29%) embryos from an unsorted clutch], whereas in ace/fgf8 embryos a posterior patch of fgf8 expression was only seen in two out of 12 identified mutants (data not shown). By 30 hpf, expression is lost in the anteroventral epithelium and, instead, is displaced to a broad region over the medial wall of the vesicle, which persists through to at least 49 hpf (Fig. 4I–J and data not shown). If the staining reaction is allowed to develop further, an occasional faint trace of fgf8 expression is seen at the posterior of sibling otic vesicles at 24 hpf, but in all cases the ectopic expression in mutant otic vesicles is at comparable or higher levels to that found in the normal anterior domain. pax5, which is also normally expressed in an anteroventral domain corresponding to the anterior (utricular) macula, is also misexpressed in a similar pattern (Fig. 4K–L). fsta, a marker of non-sensory posterior otic epithelium, is overexpressed at the posterior of the ear at 24 hpf, and is later widely misexpressed in medial otic epithelium at 49 hpf when the normal posterior expression domain is very weak (Fig. 4M–P). Therefore, the medial wall of the ear shows both abnormal and strong expression of markers of both anterior and posterior identity. The expression of central, ventral otic epithelium markers is also abnormal; otx1 expression is slightly expanded and the sharp anterior boundary of its expression domain is lost, whereas wnt4a expression is more diffuse than normal (Fig. 4U–V and data not shown).
Fig. 4.

Otic vesicle patterning is disrupted in cls/sox10 mutants. Whole-mount in situ hybridisations showing the changes in gene expression domains in the clst3 (cls) mutant otic vesicle. (A,C,E,G,O,P,S–BB) are lateral views, with anterior to the left; (B,D,F,I,J,H,M,N,Q,R) are dorsal views, with anterior to the top. The age in hpf is shown for each panel. (A–D) pax2a expression at 24 hpf appears normal in sox10 mutants, marking the medial side of the vesicle. (E–H) fgf8 expression at 24 hpf. In mutants, expression in the normal anteroventral domain is expanded (arrows) and ectopic expression is observed at the posterior of the vesicle (arrowheads). (I,J) fgf8 expression at 30 hpf. In the mutant, the normal anteroventral expression [(I) arrows] extends all around the medial side of the mutant vesicle (J). (K,L) Partially dissected whole-mounts showing pax5 expression at 50 hpf in the otic vesicle. WT expression of pax5 at this stage [(K), dorsal view, anterior to the left] marks the extreme medial edge of the utricular macula (um), with no expression in the cristae (ac, anterior crista) or saccular macula (sm). Ectopic expression is present in the posterior of the mutant otic vesicle [(L), dorsolateral view, anterior to the left, arrowhead]. (M,N) The posterior expression domain of fsta is stronger in sox10 mutants at 24 hpf. (O,P) By 49 hpf, WT otic fsta expression is very weak; in the mutant, ectopic fsta expression stretches over the medial wall of the otic vesicle (arrowhead). (Q,R) bmp4 expression at 24 hpf is much stronger in the mutant otic vesicle. (S,T) At 50 hpf, bmp4 expression normally marks the three cristae (asterisks) and the endolymphatic duct (ED). In the mutant, lateral crista and ED expression domains are lost; expression in the remaining two cristae is expanded (asterisks). (U,V) Expression of wnt4a (bracket) is slightly expanded in the mutant. (W,X) aldh1a3 expression normally marks the three cristae (asterisks), anterior macula (arrow) and ED at 72 hpf; in the mutant, the lateral crista domain is very weak, and the anterior macula and ED domains are absent [see (S) and (T), above]. (Y,Z) Expression of connexin27.5 is lost in the lateral crista (asterisk) of the mutant at 3 dpf. (AA–BB) Expression of connexin33.8 is lost in the lateral crista (asterisk) and anterior macula (arrow) of the mutant at 3 dpf. Bars, 25 μm.
bmp4 is normally expressed at the anterior and posterior ends of the otic vesicle at 24–27 hpf, but by 48 hpf, bmp4 expression marks all cristae and a dorsal domain corresponding to the endolymphatic duct (Mowbray et al., 2001) (L.A. and T.T.W., unpublished) (Fig. 4Q,S). In sox10 mutants, the level of bmp4 expression is upregulated at 24–27 hpf but the pattern is largely normal, appearing concentrated in the anterior and posterior regions on the lateral wall of the vesicle (Fig. 4R). However, at 48 hpf, the lateral crista domain and dorsal endolymphatic duct domain are lost in the mutant ear, whereas the anterior and posterior crista domains of expression are hugely expanded (Fig. 4T). bmp2b and msxc, which also mark cristae at 48 hpf, also indicate loss of the lateral crista domain (Whitfield et al., 1996) (data not shown). Expression of a second marker of cristae and the endolymphatic duct, aldh1a3, also reveals two abnormal crista expression domains at 48 hpf; expression in the lateral crista is reduced to a trace and expression in the endolymphatic duct is missing (Fig. 4W,X).
In addition, we documented changes in the expression patterns of two connexin genes, cx27.5 (homologous to mammalian CX32/GJB1) and cx33.8 (homologous to CX26/GJB2 and CX30/GJB6) (Eastman et al., 2006). At 72–74 hpf, expression of both genes is lacking in the cls/sox10 mutant ear (Fig. 4Y–BB). Both genes are normally expressed in the lateral crista, which is missing in the mutant, probably accounting for the loss of this expression domain; however, cx33.8 is also expressed in the anterior macula at this stage (Fig. 4AA, arrow), and this domain of expression is also missing in the sox10 mutant ear (Fig. 4BB).
cls/sox10 mutant otic phenotypes are enhanced by knockdown of sox9 genes
Although sox10 may not be absolutely dependent upon itself for maintenance of its expression, it is plausible that sox9 gene expression compensates for the loss of sox10. To test this, embryos from crosses of sox10 carriers were injected with sox9a or sox9b morpholinos, or mixtures of the two, and patterning of otic vesicles was then observed at 30 hpf. Note that, in each case, equimolar mixtures of pairs of gene-specific morpholinos were used at doses phenocopying the described jaw and body morphology phenotypes of sox9 mutants (Liu et al., 2003; Yan et al., 2005). As expected from previous studies (Liu et al., 2003; Yan et al., 2005), knockdown of sox9 function resulted in smaller otic vesicles, with knockdown of both sox9 genes often resulting in tiny patches of otic epithelia, especially when performed in a sox10 mutant background (Fig. 5G,H). In some cases, such morphants showed no indication of an otic epithelium. Occasionally, sox9-morpholino-treated embryos developed small, but duplicated, otic vesicles, as detected using markers including sox10, fgf8 or pax2a (data not shown).
Fig. 5.
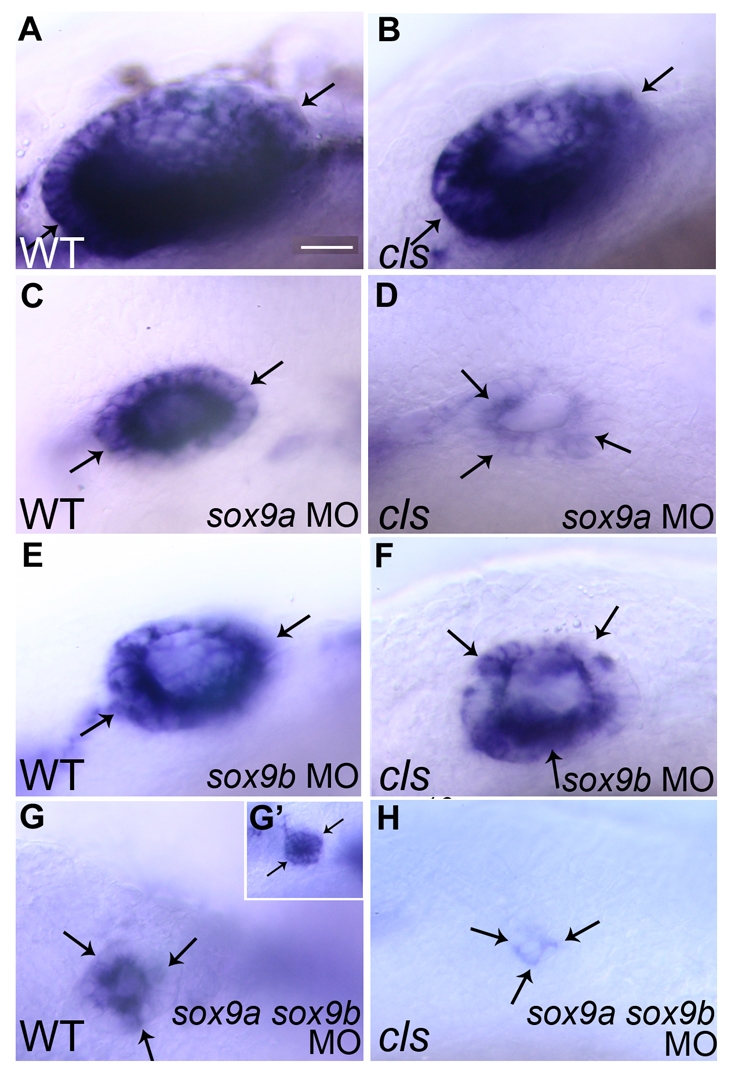
sox9 knockdown results in loss of sox10 expression in otic epithelia. In situ hybridisations showing sox10 expression (arrows) in the otic vesicle of 30 hpf WT (A,C,E,G,G′) and sox10 mutant (cls) (B,D,F,H) zebrafish injected with sox9a morpholinos (sox9a MO) (C,D), sox9b morpholinos (sox9b MO) (E,F) or sox9a and sox9b morpholinos (sox9a sox9b MO) (G,G′,H). Bars, 20 μm (A–G); 40 μm (G’).
We asked specifically whether soxE gene function is required for maintenance of sox10 expression in the otic epithelium. Although injection of any one sox9 morpholino into sibling embryos did not affect the ubiquitous strong expression of sox10 in otic epithelial cells, knockdown in sox10 mutants resulted in reduced levels of sox10 expression (Fig. 5C–F). Furthermore, the remaining expression was often patchy in its distribution through the epithelium (Fig. 5C–F). These phenotypes were noted when the function of any two soxE genes was disrupted, and enhanced when the function of all three genes was disrupted. These observations suggest that the soxE group genes function redundantly in the maintenance of sox10 gene expression in the otic epithelium.
We extended these studies by looking at other early otic vesicle markers and comparing the changes in expression patterns at 30 hpf when one or more soxE group genes were inhibited. At this stage, pax2a expression is essentially unchanged in sox10 mutants (Fig. 4A–D) and is retained in the otic cells of sox10 mutants and in their WT siblings injected with either sox9a or sox9b morpholinos, or both sox9 gene morpholinos (data not shown; see also Liu et al., 2003). Thus, otic pax2a expression does not seem to be regulated by soxE gene function. In contrast, otic expression of fgf8 becomes increasingly deregulated as soxE gene function is decreased (Fig. 6). Knockdown of a single sox9 gene results in limited ectopic fgf8 expression at the posterior of the ear; a double sox9 knockdown results in similar ectopic posterior expression to that observed in sox10 mutants, whereas the injection of sox9 morpholinos into sox10 mutants results in a further expansion of fgf8 expression. When the function of all three soxE genes is perturbed, almost all cells in the residual otic tissue express fgf8 (Fig. 6H). We conclude that fgf8 expression in the developing otic vesicle is normally restricted to the developing anterior macula by the activity of soxE gene products. Expression of foxi1 is more variable, even in WT embryos at this stage, but nevertheless a similar pattern of increasing deregulation of gene expression is seen (data not shown).
Fig. 6.

fgf8 expression becomes increasingly deregulated as soxE gene function is impaired. Expression of fgf8 (dark blue, arrows) in the otic vesicle of 30 hpf WT (A,C,E,G) and sox10 mutant (cls; B,D,F,H) zebrafish injected with sox9a (sox9a MO; C,D) or sox9b (sox9b MO; E,F), or with sox9a and sox9b morpholinos (sox9a sox9b MO; G,H) was determined using whole-mount in situ hybridisation. Bar, 20 μm.
Sensory epithelia differentiate in cls/sox10 mutant ears, but are not patterned correctly
We examined hair cell development in the cls/sox10 mutant ear by using FITC-phalloidin staining, which marks the actin-rich stereociliary bundle on the apical surface of each hair cell (Haddon and Lewis, 1996). At 28–30 hpf, hair cell production in the cls/sox10 mutant ear appears relatively normal; two to four hair cells are present in each macula, as in the WT embryo (Fig. 7A–H). The otic vesicle is more rounded than normal, and the epithelium of the otic vesicle appears disrupted in places. However, by 48–51 hpf, there is no longer a clear distinction between anterior and posterior maculae; the patches are misplaced around the medial wall, and are too close together, sometimes forming a continuous patch.
By 60 hpf, five separate sensory patches are evident in the WT zebrafish ear: two maculae, each containing 30±2 hair cells (n=3 ears), and three cristae. In the cls/sox10 mutant, there is some variability in the hair cell phenotype. Macular hair cells appear to occupy one contiguous strip covering the medial wall of the vesicle (Fig. 7M–P), possibly correlating with the misexpression of anterior macula markers in this region (pax5 and fgf8; Fig. 4I–L). Anterior and posterior foci of hair cells are also present; these occupy the positions of the anterior and posterior cristae, but often have higher numbers of hair cells than would be expected for cristae at this stage (Fig. 7Q–T). In some ears they are contiguous with the medial patch of hair cells, forming a single patch of hair cells that extends round from the anterior to the posterior of the vesicle. This was confirmed by assessing the expression of the hair cell marker pou4f3 (data not shown). The ear is so misshapen at this stage that is difficult to resolve whether the anterior and posterior hair cell groups reflect enlarged cristae (which might correlate with the enlarged domains of bmp4 expression at the anterior and posterior of the vesicle; Fig. 4T) or displaced macular hair cells (which might correlate with the dorsomedially displaced fgf8 and pax5 expression), or both. In some ears the lateral crista appears to be missing, whereas in others it is present but appears apically constricted, possibly in the process of being extruded from the epithelium (e.g. Fig. 7T), which may correlate with the loss of bmp4 expression in this domain. At 72 hpf, hair cells occupy the entire medial wall of the vesicle; in the example shown, only one crista is present. A similar pattern is found at 5–6 dpf (Fig. 7W,X), although in many cases the ear is grossly distended (see Fig. 3); if this is the case, organisation of hair cells into separate sensory patches is not evident (data not shown).
The neurogenic domain is expanded in cls/sox10 mutant ears
The statoacoustic ganglion (SAG) arises by delamination of neuroblasts from the otic epithelium. In the neural crest, sox10 has been shown to have a role in sensory neuron specification and survival (Britsch et al., 2001; Sonnenberg-Riethmacher et al., 2001; Carney et al., 2006). Hence, we asked whether sox10 mutants might show disrupted sensory neuron formation in the SAG. We used two markers of early delaminating neuroblasts, neurogenin1 (ngn1) and neuroD, to examine 30 hpf embryos. In WT embryos, ngn1 is expressed in otic sensory neuron precursors before delamination, whereas neuroD is rarely expressed in the otic epithelium itself at this stage; both are robustly expressed in the nascent ganglion (Fig. 8A,C; data not shown). In cls/sox10 mutants, both ngn1 and neuroD appear to be expressed at higher levels, or in supernumerary sensory neuron precursors, in both the otic epithelium and the nascent ganglion (Fig. 8A–D); this phenotype continues until at least 36 hpf (Fig. 8G,H). However, expression of snail2, which also appears to mark delaminating otic neuroblasts – albeit possibly a different population to the ngn1- and neuroD-positive cells, is unaffected (Fig. 8E,F). To test whether the abnormal expression of ngn1 and neuroD in the mutants correlates with an abnormal number of neurons in the SAG, we stained embryos with antibodies to Isl1 and the Hu antigen, both of which mark differentiating and mature neurons (Haddon et al., 1998; Marusich et al., 1994). We found that the overall levels of Isl1 protein expression are decreased in the SAG ofcls/sox10 mutants (Fig. 8I,J), although Hu levels are indistinguishable (Fig. 8K,L). However, counts of Isl1-positive nuclei and Hu-positive cells in the SAG revealed no significant differences in the number of neurons in cls/sox10 mutants (Isl1: WT, 46.1±9.9; cls/sox10 mutant: 46.8±9.1; p=0.85, t-test; n=12 ears for each genotype. Hu: WT, 21.80±6.268; cls/sox10 mutant: 26.57±3.359; p=0.0877, t-test; n=10 and n=7 ears, respectively). We conclude that the abnormal expression of ngn1 and neuroD in the cls/sox10 mutant otic vesicle and developing SAG does not result in a significant overproduction of SAG neurons, possibly owing to increased cell death (see below).
Fig. 8.

cls/sox10 mutants show abnormal neurogenic development in the ear. Lateral views of WT (A,C,E) and sox10t3 mutant (B,D,F) embryos at 30 hpf showing the otic epithelium and the statoacoustic ganglion (SAG). Note that, although snail2 expression (E,F) is comparable between genotypes, both neuroD-(A,B) and neurogenin1- (C,D) expressing cells are more prominent in the mutant otic epithelia (the arrows indicate selected positive cells in the epithelium). At this stage, neuroD is strongly expressed in the developing SAG but only rarely in cells within the otic epithelium; (A) shows a rare individual with detectable neuroD expression in one or two neuroblasts before they have delaminated from the epithelium. The mutant in (B) is typical. (G,H) Transverse section of WT (G) and sox10t3 mutant (H) embryos showing the location of neuroD-expressing cells in the mutant otic epithelium. (I–L) Lateral views showing expression of the neuronal markers Islet-1 (I,J) and Hu (K,L) at 30 hpf in WT (I,K) and sox10 mutant (J,L) embryos. (M–P) Cell death in the developing ear. (M,N) 35 hpf embryos labelled for anti-Hu (brown) and TUNEL (dark blue) show prominent dying cells (arrows) in both the otic epithelium and the SAG (outlined with a black line) of clst3 mutants (N), whereas such cells are usually absent from WT siblings (M). Note that WT embryos have TUNEL+ cells outside of the ear. (O,P) Quantitation (mean±s.e.) of TUNEL+ dying cells in the otic epithelium (O) and of TUNEL+/Hu+ SAG neurons (P) in WT (n=12) and sox10 mutant (n=10) embryos at 30 hpf (see also Tables 1 and 2). Bar, 20 μm [the bar in (A) applies to (A–H,K–N) and the bar in (I) applies to (I, J)].
Levels of cell death are increased in the cls/sox10 mutant ear
Sox10 has been shown to have a key role in promoting neural crest cell survival: in zebrafish, death of neural crest cells in sox10 mutants occurs principally between 35 and 45 hpf (Dutton et al., 2001b). Therefore, we used TUNEL staining to assess whether apoptosis contributed to the otic phenotypes in sox10 mutants. Embryos were examined between 25 and 40 hpf; at all time points, cell death in the otic epithelium and/or SAG was significantly elevated over WT siblings (Tables 1 and 2; Fig. 8G–J). Although WT and sox10 mutant embryos cannot be easily distinguished morphologically at 25 hpf, 25% of the embryos in a batch have apoptotic cells in the otic epithelium and/or SAG. We infer that sox10 mutants already have elevated cell death in the ear at this stage. At all stages up to at least 40 hpf, elevated cell death was prominent in both the otic epithelium and the SAG, although individual embryos differed widely in the number of apoptotic cells they displayed.
Table 1.
Apoptotic cell death is elevated in the otic vesicle of sox10 mutants
| Number of TUNEL-labelled cells in the otic vesicle, mean±s.e.
|
|||
|---|---|---|---|
| Embryo stage (hpf) | WT | sox10 mutant | P |
| 25 | 0±0 (n=29) | 2.11±0.63 (n=9) | 0.0103 |
| 30 | 0.33±0.25 (n=12) | 1.80±0.47 (n=10) | 0.0165 |
| 35 | 0±0 (n=11) | 2.91±0.80 (n=11) | 0.0047 |
| 40 | 0.33±0.26 (n=12) | 4.46±0.96 (n=11) | 0.0013 |
Table 2.
Apoptotic cell death is elevated in the statoacoustic ganglion of sox10 mutants
| Number of TUNEL-labelled cells in the SAG, mean±s.e.
|
|||
|---|---|---|---|
| Embryo stage (hpf) | WT | sox10 mutant | P |
| 25 | 0±0 (n=29) | 0.89±0.31 (n=9) | 0.0207 |
| 30 | 0.25±0.13 (n=12) | 1.5±0.40 (n=10) | 0.0368 |
| 35 | 0±0 (n=11) | 0.82±0.23 (n=11) | 0.0047 |
| 40 | 0.25±0.18 (n=12) | 0.64±0.25 (n=11) | 0.0519 |
Neural crest cells occasionally contribute to the ear and can give rise to several otic derivatives
Unlike mammals, the teleost ear has no cochlea and, therefore, no stria vascularis. Furthermore, neither melanophores nor melanoblasts are present in the zebrafish ear during embryonic stages (data not shown). Nevertheless, we tested whether there was any neural crest contribution to the zebrafish ear. We used iontophoretic labelling with rhodamine-dextran to label single neural crest cells in the region of rhombomere 4 (Dutton et al., 2001b); cells were labelled in the embryo at around the seven-somite stage (12 hpf) and, thus, were premigratory (Raible et al., 1992; Schilling and Kimmel, 1994), but they were already clearly distinguishable morphologically from otic placode cells. After labelling, embryos were recovered and then re-examined approximately one hour after labelling to confirm that single cells were indeed labelled. These cells all showed a mesenchymal morphology characteristic of neural crest cells (Fig. 9A–C). Embryos were then re-examined at around 36 hpf and the number and fate of labelled cell clones was recorded (Table 3). These cells gave rise to all expected fates, including craniofacial cartilage, multiple pigment cell-types, glia and some neurons of the cranial ganglia. In addition, a small proportion (5%; Table 3) of clones contained progeny that were clearly incorporated within the otic epithelium, and that in at least one case formed part of a sensory macula. This was confirmed by confocal microscopy using BODIPY ceramide to reveal tissue morphology (Fig. 9D–G). This contribution to the otic vesicle was, however, transient because we were unable to find labelled cells in the epithelium at 72 hpf. In one case where a clone had contributed to the otic epithelium, we did later see a single labelled sensory neuron in the SAG. We conclude that, although the majority of otic cells derive from the otic placode, a small subset of otic cells are derived from the neural crest, but that most or all of these might be lost. In part, this might result from delamination during SAG formation, although this inference will need to be tested rigorously using further approaches.
Fig. 9.
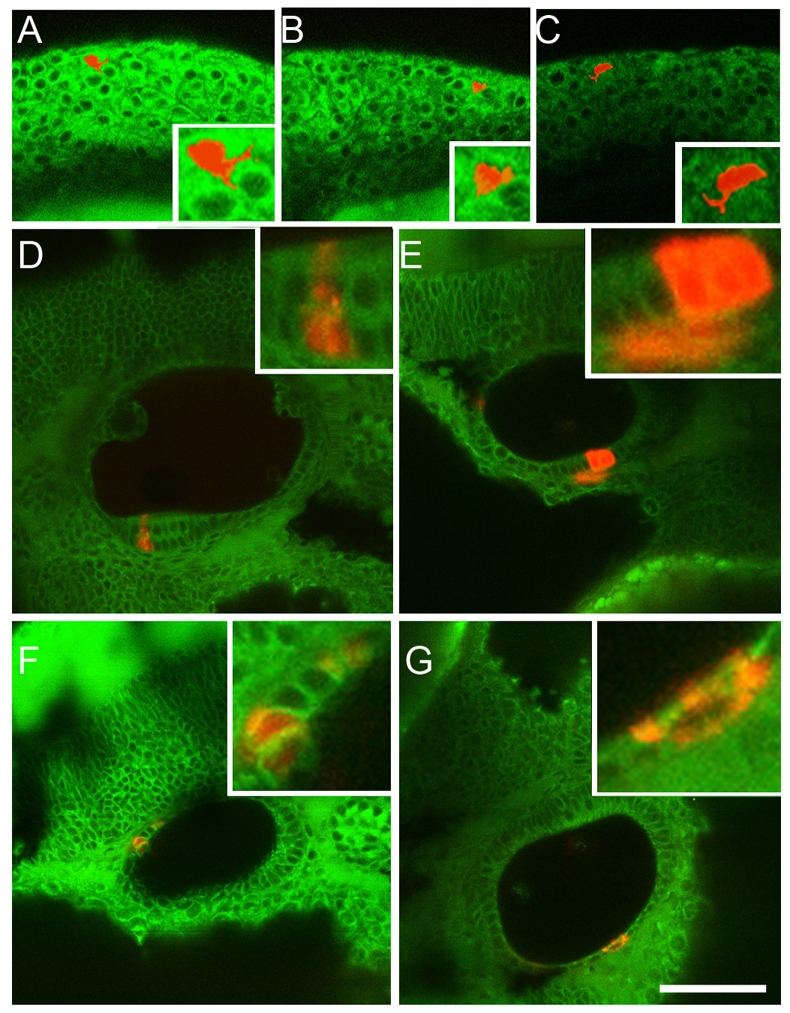
Neural crest cells make a minor, and transient, contribution to the otic vesicle. Single premigratory neural crest cells in the rhombomere 4 region of WT embryos that have been labelled iontophoretically with rhodamine dextran. The cells are shown at the 10-somite stage, approximately 1 hour after labelling (A–C, three different cells). The progeny of single labelled cells that contribute to the otic epithelium are shown at 36 hpf, approximately 24 hours after labelling (D–G, four different examples). The insets show closeups of each cell or clone. Note that occasional progeny incorporated into the otic epithelium include cells in the sensory epithelium of the anterior macula (D). The clone in (E) includes three cells in the otic epithelium, plus one lying outside the epithelium. The fate of this latter cell is not known. Bar, 25 μm.
Table 3.
Fates of single neural crest cells in the rhombomere 4 region
| Fate | n | % | Notes |
|---|---|---|---|
| Cartilage/connective tissue | 41 | 38 | Includes six cells (6%) that contributed to otic capsule |
| Xanthophores | 17 | 16 | |
| Melanophores | 12 | 11 | |
| Glia | 9 | 8 | |
| Neurons | 5 | 5 | |
| Otic placode | 5 | 5 | By 72 hpf, one cell became a neuron in the SAG, the others were lost |
| Died | 6 | 6 | |
| Unidentified | 12 | 11 | Cells either lost or difficult to image |
| TOTAL | 107 | 100 |
This table shows the fate of labelled, single cell clones at 35-72 hpf, as deduced from cell morphology and location.
DISCUSSION
Sox10 participates in a feedback loop with sox9 genes to establish otic-specific patterns of gene expression
Current models of otic induction in zebrafish place the maintenance of otic sox9a and sox9b expression downstream of two convergent otic induction pathways, each dependent on Fgf activity (Phillips et al., 2001; Léger and Brand, 2002; Maroon et al., 2002; Nissen et al., 2003; Hans et al., 2004; Solomon et al., 2004; Hans et al., 2007). The exact requirements for initiation of soxE gene expression in the otic region are less clear. Otic expression of sox9a, and to a lesser extent sox9b, is dependent on Fgf signals from the central nervous system (CNS) (Liu et al., 2003), and expression of the sox9 genes is reduced, but not completely lost, in the absence of either foxi1 or dlx3b activity (Liu et al., 2003; Hans et al., 2004). We found that zebrafish sox10 is not dependent on Fgf8 function, as expression is normal in ace (fgf8) mutants (data not shown), although treatment of Xenopus embryos with the Fgf inhibitor SU5402 eliminates both otic and neural crest expression of sox10 (Honoré et al., 2003). All three soxE genes are coexpressed with dlx3b from early stages (one somite) in domains where neural crest and otic precursors overlap. The timing of the onset of soxE gene expression is similar to that of pax8, which marks the otic domain by the 10 hpf (tailbud) stage (Phillips et al., 2001) and precedes the onset of pax2a expression in otic cells at the three-somite stage (Fig. 1). pax2a expression appears to be independent of soxE function in zebrafish since, even in triple mutant/morphants, residual otic cells show pax2a expression (data not shown). Disruption of pax gene function at stages before the onset of pax2a expression suggests that Pax8 plays an early role in the initiation or maintenance of sox9 gene expression (Hans et al., 2004). However, in Xenopus, pax8 expression in the otic placode is dependent on sox9 function (Saint-Germain et al., 2004). It is therefore difficult to place soxE factors either upstream or downstream of pax factors in a simple linear pathway in the ear; dissection of the gene regulatory network underlying otic induction will require an exhaustive analysis of transcription factor binding to the regulatory regions of all these genes.
Once expression of pax and soxE gene expression is established in the otic region, their expression patterns are maintained or refined by complex cross-regulatory actions. Our results with morpholino disruption of the sox9 genes corroborate previous observations in which morpholino knockdown of sox9b in jefhi1134 (sox9a) mutants results in the loss of the otic vesicle, with just a few scattered patches of pax2a-positive cells at 36 hpf (Liu et al., 2003). We show that within the soxE group, the activity of any two soxE genes is required for the maintenance of sox10 expression throughout the placode, and that an important early role of all three sox genes is to restrict fgf8 expression to anterior macula regions. Note that cavitation to form a vesicle can occur with the loss of one or even two soxE factors, despite the reduction in overall otic tissue (data not shown). This is in contrast to the mouse Sox9 mutant, in which an otic placode forms but fails to invaginate to form a vesicle (Barrionuevo et al., 2008). Since vesicle formation in the zebrafish involves cavitation, rather than invagination, it is perhaps not surprising that this role for Sox9 is not conserved.
As development proceeds, sox10 appears to be required for the downregulation of sox9 gene expression in the otic vesicle. Thus, sox10 provides a link between the otic induction pathways, otic specification factors (pax and sox9 genes) and subsequent otic patterning. This key function is demonstrated by multiple instances of abnormal marker gene expression in the sox10 mutant otic vesicle. Thus, we show shifts in anteroventral (utricular macula) expression domains to more dorsomedial positions (fgf8, pax5); deregulation or ectopic expression of several markers (fgf8, pax5, fsta, bmp4); loss of markers of the lateral crista and endolymphatic duct (bmp4, aldh1a3); and precocious neurogenesis in the otic epithelium (ngn1, neuroD). Although the abnormal patterning of sensory patches reflects these changes in gene expression, specification of sensory hair cells within the maculae appears to be unaffected since differentiated hair cells appear on schedule. Failure of endolymphatic duct development or function might account for the late, and variable, otic swelling seen in mutants.
The otic phenotype of sox10 and hnf1b mutant embryos is very similar
We note the striking similarity of the sox10 mutant otic phenotype to that described for the hnf1b mutant (formerly vhnf1 or tcf2) (Lecaudey et al., 2007). Many aspects appear similar at the 24–26 hpf stage, in particular the misexpression of fgf8 and pax5. Both mutants also show a transient increase in otic neurogenesis and tend to form only two cristae. The hnf1b mutant phenotype has been interpreted as an anteriorisation of the otic vesicle (Lecaudey et al., 2007). We do not suggest that the sox10 mutant otic phenotype is a straightforward anteriorisation, because although anterior markers (fgf8 and pax5) are expressed ectopically, the posterior group of hair cells in sox10 mutants does not resemble an anterior macula; in addition, and in contrast to the hnf1b mutant, expression of the posterior marker fst is upregulated. hnf1b codes for a transcription factor that is expressed in the hindbrain and not in the ear; in the hnf1b mutant, rhombomere 5 (r5) fails to develop correctly and the r5-specific expression of val/mafb, wnt1, wnt3a and wnt8b is lost (Hans et al., 2007; Lecaudey et al., 2007). Given the similarity of the otic phenotypes in the sox10 and hnf1b mutants, it is likely that they function in the same pathway. In Xenopus, injection of a dominant negative Wnt8 mRNA eliminates Sox10 expression in the neural crest and otic placode (Honoré et al., 2003). However, we suggest it is unlikely that, in zebrafish, sox10 is a direct target of a localised signal (such as a Wnt) from r5, since our preliminary data (not shown) suggest that sox10 expression is unaffected in hnf1b mutants. Instead, we propose that sox10 might be required for competence of otic tissue to respond to a hindbrain signal. Alternatively, Hnf1b activity could be required for the expression of a partner of Sox10 in the otic epithelium.
Neural crest cells make a small contribution to the zebrafish ear
The expression patterns of soxE genes in zebrafish suggest that neural crest cells and otic placode cells are initially intermingled. In the chick, fate-mapping studies have shown that otic, neural crest, epidermal, epibranchial and CNS precursors are initially intermixed (Streit, 2002). Placodal precursors for the otic vesicle, lens, olfactory epithelium, cranial ganglia and lateral line are also initially intermixed at the 50–60% epiboly (5–6 hpf) stage in zebrafish, but there is already clear anteroposterior organisation in the fate map (Kozlowski et al., 1997). After cell sorting through extensive cell movements, neural crest cells in the chick embryo contribute to the glia of the VIIIth ganglion and nerve, and to the cartilaginous otic capsule (Le Lièvre, 1978; Noden, 1978; D’Amico-Martel and Noden, 1983; Couly et al., 1993; Le Douarin and Kalcheim, 1999). In quail/chick chimeras, neural crest cells were observed to contribute to a minority of cells in the vestibular, but not the acoustic, ganglion (D’Amico-Martel and Noden, 1983); however, this study was not able to follow whether these cells entered the otic epithelium first before differentiating. Neural crest cells have not been shown previously to contribute to the otic epithelium itself, apart from the melanocytes that populate the stria vascularis, vestibular epithelium and endolymphatic duct of the mammalian ear (Steel and Barkway, 1989). Nevertheless, in fish and amphibian embryos, neural crest cells have been shown to contribute to sensory and supporting cells of the lateral line system (Collazo et al., 1994), and previous observations have also shown that the neural crest occasionally contributes to the zebrafish ear (T. Schilling, personal communication). In the chick, it has been reported that cells migrating from ventral regions of the neural tube can contribute to the otic vesicle (Ali et al., 2003).
Although melanophores migrate around, and settle close to, the otic vesicle in the zebrafish embryo, they never appear to penetrate the otic epithelium itself – at least during the first five days of development. Furthermore, it is unlikely that melanophores play a role in the early stages of zebrafish otic development, as homozygous nacre/mitfa embryos, which lack melanophores, have no obvious otic defects or behavioural abnormalities that are indicative of hearing or vestibular dysfunction (Lister et al., 1999; O’Malley et al., 2004). However, using single-cell labelling, we find that a small population of neural crest cells does contribute to the otic epithelium. Our sample is small, but indicates that these cells usually only transiently incorporate into the otic epithelium; in one case, such cells contributed a neuron to the SAG, but in all other cases (n=4) these cells subsequently disappeared. It is perhaps not surprising that neural crest cells can contribute to the otic epithelium, since otic and neural crest precursors initially share the expression of several genes (including the soxE genes). We estimate that the neural crest contribution to the ear is small, with the vast majority of cells still being placodal in origin. Therefore, we think it unlikely that loss of these cells contributes significantly to the otic phenotype in sox10 mutant embryos.
The cause of deafness in WS4 individuals
The aetiology of the deafness in WS4 individuals who carry SOX10 mutations is not straightforward. In the Waardenburg syndromes, together with other pigmentation and deafness variant syndromes, hearing loss is generally attributed to the loss of intermediate cells (melanocytes) in the stria vascularis of the cochlea, where they play a vital role in the maintenance of endolymph volume and the endocochlear potential and, ultimately, in hair cell survival (Steel and Barkway, 1989; Tachibana, 1999). Phenotypic analysis of mouse models of the WS syndromes has, thus, concentrated almost exclusively on the melanocyte phenotype. For example, in mice carrying homozygous mutations in the Ednrb or Mitf genes, the stria vascularis lacks melanocytes, leading to an endolymphatic collapse and degeneration of hair cells in the organ of Corti (Tachibana et al., 1992; Matsushima et al., 2002; Tachibana et al., 2003).
Sox10 differs from other pigmentation pathway genes (Edn3, Ednrb, Pax3 and Mitf) in that it is strongly expressed both in the neural crest and the otic epithelium, a pattern that is highly conserved in both fish and mammals. As a result, it is possible that deafness in WS4 individuals who carry SOX10 mutations not only results from a deficit of melanocytes in the ear, but also reflects additional roles for SOX10 in the otic epithelium. Interestingly, in the mouse models, hearing loss does not always correlate with melanocyte defects: melanocytes are still present in the heterozygous Sox10Dom mouse ear and the endolymph volume appears normal. In occasional individuals with hearing loss, vacuoles were found in the stria and spiral ligament, and hair cells were absent in the organ of Corti (Tachibana et al., 2003; Stanchina et al., 2006). However, a role for Sox10 in melanocytes of the stria is revealed in Sox10Dom/+;Ednrbs/s double mutants, in which a severe loss, or absence, of melanocytes in the ear is seen, representing an enhanced phenotype from that observed in the Ednrbs/s single mutant. In single and double mutants at birth, analysis of hair cells in the organ of Corti revealed normal hair cell formation, even in melanocyte-free ears, suggesting that any subsequent hair cell loss is the result of degeneration (Stanchina et al., 2006).
Although we do not detect haploinsufficiency in the zebrafish sox10 mutant, in humans, SOX10 mutations generally show dominant inheritance, owing to either haploinsufficiency or dominant negative mutations. Our work indicates that it is possible, even likely, that dominant forms of deafness in WS resulting from SOX10 mutations involve inner ear malformations as well as a deficit of melanocytes. Computed tomography (CT) and other scan studies involving WS individuals have given variable results, with some studies indicating that inner ear malformations (including semicircular canal malformations) might be common in WS (Nemansky and Hageman, 1975; Higashi et al., 1992; Madden et al., 2003) and others indicating that cochlear defects in WS are rare, making these individuals good candidates for cochlear implant therapy (Cullen et al., 2006; Daneshi et al., 2005) (reviewed by Oysu et al., 2001). Temporal bone CT scans have been described for four patients known to be carrying mutations or heterozygous deletions in SOX10. Interestingly, three of these patients had severe morphological abnormalities of the inner ear: a WS4 patient carrying a SOX10 mutation had bilateral semicircular canal agenesis (Sznajer et al., 2008); a PCWH patient, carrying a large heterozygous deletion encompassing the entire SOX10 gene, had bilateral semicircular canal hypoplasia and vestibular malformation; and a WS2 patient, carrying a smaller heterozygous deletion within the SOX10 gene, had semicircular canal hypoplasia and dilatation, together with vestibular malformation and dilatation (Bondurand et al., 2007). These structural abnormalities correspond well to the defects that might be predicted from our analysis of the zebrafish model.
Even if structural abnormalities are not present, the loss or misexpression of Sox10 target genes in the ear, especially those implicated in deafness such as the connexin genes, could contribute to hearing loss. In the zebrafish sox10 mutant ear, many of the genes that we examined show complex patterns of misexpression, with loss of normal domains of expression and derepression, or ectopic expression, elsewhere. In mammalian glia, the connexin genes CX32/GJB1 and CX47 are known targets of Sox10 function (Bondurand et al., 2001; Schlierf et al., 2006). Connexins play crucial roles in the inner ear: mutations in CX26/GJB2 and CX30/GJB6 account for the most common form of inherited deafness (DFNB1) in humans, whereas mutations in CX32/GJB1 have been implicated in causing the sensorineural deafness in Charcot-Marie-Tooth disease (Stojkovic et al., 1999; Lee et al., 2002). Our data indicate that expression of the orthologous connexin genes cx27.5 and cx33.8 is lost in zebrafish sox10 mutant ears at 72–74 hpf. Although loss of expression may partly be explained by the loss of the lateral crista, it is possible that the direct or indirect downregulation of connexin gene expression could contribute to the hearing loss in individuals carrying SOX10 mutations.
Studies of Sox10 targets in mammalian neural crest derivatives suggest that Sox10 function is dose sensitive in the mammal, raising the possibility that similar patterning defects might occur in the ears of mammals heterozygous for Sox10 mutations. Any structural abnormalities may be attributed to a requirement for Sox10 in early patterning of the otic vesicle. Disruption to endolymph homeostasis might result from the loss of melanocytes in the stria vascularis, and/or through abnormal functioning of gap junctions owing to downregulation of connexin gene expression, or, alternatively, from abnormalities in the endolymphatic duct. We conclude that deafness in mammals carrying Sox10 mutations is likely to be complex in origin, reflecting a direct role for Sox10 in otic placodal derivatives.
METHODS
Fish husbandry
Zebrafish (Danio rerio) embryos were obtained through natural crosses and were staged by morphology (Kimmel et al., 1995). The mutant alleles used in this study were: aceti282a, clst3, clsm618, dogtc257e, hnf1bhi2169Tg, noitu29a, smub641 and vgotm90b. Chorions were removed with watchmaker’s forceps and the embryos were maintained in embryo medium (Westerfield, 2000). Living embryos were mounted in 1.2% low melting point agarose (Eisen et al., 1989), for single cell iontophoretic injection of fluorescent dye, or in 3% methyl cellulose dissolved in embryo medium for later observations, and observed using Nomarski (DIC) optics. Where necessary, embryos were anaesthetised using a dilute solution of tricaine methylsulfonate (MS-222; Sigma). All experiments complied with institutional and national animal welfare laws, guidelines and policies. Procedures involving fish older than 5 dpf were undertaken under licence from the UK Home Office.
Morpholino injections
Morpholinos (Gene Tools; sequences in Table 4) were used to knock down sox9a and sox9b gene function, both individually and in combination, in clsm618 embryos and their siblings, as described previously (Dutton et al., 2001a). Two morpholinos were used in combination to knock down the expression of each of sox9a and sox9b, and all four morpholinos were used in combination to knock down the expression of sox9a and sox9b simultaneously (Table 4). Morpholinos were injected into the yolk of embryos prior to the 16-cell stage using a Drummond Nanoject II apparatus (Drummond Scientific Co., Broomall, PA). Initial experiments showed that individual morpholinos generated similar, consistent phenotypes affecting jaw development (sox9a and sox9b) and a curly tail down phenotype (sox9b) over a range of doses; these were fully consistent with the published mutant and morpholino phenotypes (Liu et al., 2003; Yan et al., 2005). Subsequently, each gene-specific morpholino was used at a relatively low dose, but in combination to avoid off-target effects. Each embryo received 4.6 ng of each morpholino, or 2.3 ng where both sox9a and sox9b were knocked down simultaneously. The injection solution was labelled with Phenol Red to allow the distribution of the injected morpholinos to be visualised. Injected and uninjected control embryos were incubated at 28.5°C, before processing for marker expression.
Table 4.
Sox9 morpholinos used in this study
| Gene | Morpholino name | Morpholino sequence (5′-3′) |
|---|---|---|
| sox9a | Sox9a | GGGTCGAGGAGATTCATGCGAACAC |
| Sox9a.b | CGTAGATGGACGATCTCAGAGAATT | |
| sox9b | Sox9b | TGCAGAGAGAGAGTGTTTGAGTGTG |
| Sox9b.b-3A | AGCTGCTGAAACACACACAGATCCT |
Whole-mount in situ hybridisation
RNA in situ hybridisation was performed as described previously (Kelsh et al., 2000) with clst3 or clsm618 mutants and their phenotypically WT siblings. The probes used were: aldh1a3 (Pittlik et al., 2008), bmp4 (Hammerschmidt et al., 1996), fgf8 (Guo et al., 1999), fst (Bauer et al., 1998), foxd3 (Odenthal and Nuesslein-Volhard, 1998), foxi1 (Hans et al., 2004), neuroD (Korzh et al., 1998), ngn1 (Blader et al., 1997), pax2a (Hans et al., 2004), pax8 (Hans et al., 2004), sna2 (Thisse et al., 1995), sox9a (Chiang et al., 2001), sox9b (Chiang et al., 2001) and sox10 (Dutton et al., 2001b).
Sectioning
Embryos were post-fixed, embedded in paraffin and sectioned at a thickness of 7 μm. Sections were counterstained with Fast Red. Sections in Fig. 8G,H were hand cut (approximately 50 μm).
Immunocytochemistry
Antibody staining with anti-Hu mAb 16A11 (1:500) (Marusich et al., 1994) and anti-Isl1 (DSHB 40.2D6, 1:2000) was performed using an anti-mouse Alexa 546 fluorescent secondary or an anti-mouse-HRP antibody and developed with DAB. Both antibodies stain two groups of cells in the vicinity of the otic vesicle, one in an anteroventral position and the other in a posteromedial position. Isl1-positive nuclei (chromogenic stain) were counted individually in each group and the numbers for each individual ear were then combined. Hu-positive cells (fluorescent stain) were counted from single confocal sections of the anterior and posterior SAG cell groups containing the largest number of cells. Counts from each group were then combined for each ear.
Whole-mount double staining for TUNEL and Hu antibody
Embryos were developed for TUNEL staining using a fluorescein label (Dutton et al., 2001b) incubated with alkaline-phosphatase-conjugated anti-fluorescein (Boehringer Mannheim) and developed using 4-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl phosphate (Boehringer Mannheim). The embryos were subsequently processed for anti-Hu as detailed in Immunocytochemistry, and developed with DAB.
Microscopy and photography
Embryos were examined using standard DIC optics or fluorescence, as appropriate on the Nikon Eclipse E800, and photographed using either a Canon digital SLR or SPOT camera, or examined on an Olympus BX51 and photographed using either an Olympus C-3030ZOOM camera and AnalySIS software or a CELLB camera and software. Confocal images were taken using a Zeiss LSM 510 or a Leica SP confocal microscope. Images were processed using Adobe Photoshop. Panels in Fig. 1A–D and Fig. 8D–F are composites of different focal planes.
Data analysis
Data were analysed using Prism3.0.
Single-cell labelling
Individual neural crest cells were labelled by iontophoresis, as described previously (Dutton et al., 2001b). Briefly, cells were injected intracellularly with a 5% solution of lysinated rhodamine dextran (10,000 MW; Molecular Probes, Eugene, OR) dissolved in 0.2 M KCl, using glass micropipettes that were pulled on a Flaming-Brown micropipette puller model P-97 (Sutter Instrument Co., Novato, CA). Micropipette tips were filled with 3–5% lysinated rhodamine dextran in 0.2 M KCl and the needles backfilled with 0.2 M KCl. Premigratory crest cells that were segregated from, and lateral to, the neural keel between the posterior eye and the anterior boundary of somite one were labelled in embryos at the eight-somite stage. Only embryos with single labelled crest cells were analysed. Labelled cells were monitored visually using both DIC and fluorescence on a model BX50WI microscope (Olympus), but documented using confocal imaging with an LSM 510 (Zeiss), then processed using Adobe Photoshop. Cells were then monitored twice daily for 3 days until the progeny could be identified based on their morphology and location. Progeny fates were identified by the morphological characteristics described previously (Raible et al., 1992; Raible and Eisen, 1994; Schilling and Kimmel, 1994; Dutton et al., 2001b). Thus, melanophores contained melanin granules, xanthophores were visibly yellow and fluoresced at a wavelength near that of fluorescein, iridophores contained iridescent granules, cartilage formed characteristic stacks of vacuolated cells, neurons had axons and were appropriately positioned, and glia were associated with axonal processes. Otic cells were defined as those incorporated into the otic epithelium. Although connective tissue has been described as a fate for neural crest cells in the head (Schilling and Kimmel, 1994), these cells, along with cells in positions with limited optical resolution or that were not morphologically identifiable as one of the above cell types, were categorised as unidentified.
FITC-phalloidin staining
Embryos were fixed, permeabilised and stained with 2.5 μg/ml FITC-phalloidin as described previously (Haddon and Lewis, 1996). For dorsal views in older embryos, ears were dissected before mounting. Embryos or dissected ears were mounted in Vectashield (Vector labs) and imaged with a Leica SP confocal microscope.
Supplementary Material
Acknowledgments
This work was funded by the Wellcome Trust (063998). L.A. and C.M. were funded by the MRC (G0300196, G78/5898). M.N. was funded by a Wellcome Trust VIP award (083770). We thank Iryna Whittington for expert sectioning, Richard Squire and Marc Shedden for expert fish care in Bath, Dora Sapède for sending hnf1b mutants, and many members of the zebrafish community for sharing probes. We thank Melanie Cotterill, Laina Murphy and Jill Shepherd for technical support and Lisa van Hateren and the Sheffield aquarium staff for care of the Sheffield fish stocks. Confocal imaging for Fig. 7 was carried out in the Sheffield Light Microscopy Facility, funded by grants from Yorkshire Cancer Research and the Wellcome Trust (GR077544AIA), and for Figs 8 and 9 in the Bath Bioimaging Centre, funded by the Wellcome Trust (GR077454/Z/05/Z). The CDBG zebrafish facility was supported by the MRC (G0400100, G0700091) and the EU FP6 (ZF-MODELS). The University of Bath zebrafish aquarium was supported by a Wellcome Trust Equipment Grant (066326).
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/cgi/content/full/2/1-2/68/suppl/DC1
REFERENCES
- Ali M. M., Jayabalan S., Machnicki M., Sohal G. S. (2003). Ventrally emigrating neural tube cells migrate into the developing vestibulocochlear nerve and otic vesicle. Int. J. Dev. Neurosci. 21, 199–208 [DOI] [PubMed] [Google Scholar]
- Aoki Y., Saint-Germain N., Gyda M., Magner-Fink E., Lee Y. H., Credidio C., Saint-Jeannet J. P. (2003). Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev. Biol. 259, 19–33 [DOI] [PubMed] [Google Scholar]
- Barresi M. J., Stickney H. L., Devoto S. H. (2000). The zebrafish slow-muscle-omitted gene product is required for Hedgehog signal transduction and the development of slow muscle identity. Development 127, 2189–2199 [DOI] [PubMed] [Google Scholar]
- Barrionuevo F., Naumann A., Bagheri-Fam S., Speth V., Taketo M. M., Scherer G., Neubuser A. (2008). Sox9 is required for invagination of the otic placode in mice. Dev. Biol. 317, 213–224 [DOI] [PubMed] [Google Scholar]
- Bauer H., Meier A., Hild M., Stachel S., Economides A., Hazelett D., Harland R. M., Hammerschmidt M. (1998). Follistatin and noggin are excluded from the zebrafish organizer. Dev. Biol. 204, 488–507 [DOI] [PubMed] [Google Scholar]
- Blader P., Fischer N., Gradwohl G., Guillemot F., Strahle U. (1997). The activity of Neurogenin1 is controlled by local cues in the zebrafish embryo. Development 124, 4557–4569 [DOI] [PubMed] [Google Scholar]
- Bondurand N., Kobetz A., Pingault V., Lemort N., EnchaRazavi F., Couly G., Goerich D. E., Wegner M., Abitbol M., Goossens M. (1998). Expression of the SOX10 gene during human development. FEBS Lett. 432, 168–172 [DOI] [PubMed] [Google Scholar]
- Bondurand N., Pingault V., Goerich D. E., Lemort N., Sock E., Caignec C. L., Wegner M., Goossens M. (2000). Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum. Mol. Genet. 9, 1907–1917 [DOI] [PubMed] [Google Scholar]
- Bondurand N., Girard M., Pingault V., Lemort N., Dubourg O., Goossens M. (2001). Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 10, 2783–2795 [DOI] [PubMed] [Google Scholar]
- Bondurand N., Dastot-Le Moal F., Stanchina L., Collot N., Baral V., Marlin S., Attie-Bitach T., Giurgea I., Skopinski L., Reardon W., et al. (2007). Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 81, 1169–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britsch S., Goerich D. E., Riethmacher D., Peirano R. I., Rossner M., Nave K. A., Birchmeier C., Wegner M. (2001). The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch-Nentwich E., Söllner C., Roehl H., Nicolson T. (2004). The deafness gene dfna5 is crucial for ugdh expression and HA production in the developing ear in zebrafish. Development 131, 943–951 [DOI] [PubMed] [Google Scholar]
- Cable J., Barkway C., Steel K. P. (1992). Characteristics of stria vascularis melanocytes of viable dominant spotting (Wv/Wv) mouse mutants. Hear. Res. 64, 6–20 [DOI] [PubMed] [Google Scholar]
- Cable J., Jackson I. J., Steel K. P. (1993). Light (Blt), a mutation that causes melanocyte death, affects stria vascularis function in the mouse inner ear. Pigment Cell Res. 6, 215–225 [DOI] [PubMed] [Google Scholar]
- Cable J., Huszar D., Jaenisch R., Steel K. P. (1994). Effects of mutations at the W locus (c-kit) on inner ear pigmentation and function in the mouse. Pigment Cell Res. 7, 17–32 [DOI] [PubMed] [Google Scholar]
- Carney T. J., Dutton K. A., Greenhill E., Delfino-Machin M., Dufourcq P., Blader P., Kelsh R. N. (2006). A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development 133, 4619–4630 [DOI] [PubMed] [Google Scholar]
- Cheng Y., Cheung M., Abu-Elmagd M. M., Orme A., Scotting P. J. (2000). Chick Sox10, a transcription factor expressed in both early neural crest cells and central nervous system. Brain Res. Dev. Brain Res. 121, 233–241 [DOI] [PubMed] [Google Scholar]
- Chiang E. F., Pai C. I., Wyatt M., Yan Y. L., Postlethwait J., Chung B. (2001). Two sox9 genes on duplicated zebrafish chromosomes: expression of similar transcription activators in distinct sites. Dev. Biol. 231, 149–163 [DOI] [PubMed] [Google Scholar]
- Collazo A., Fraser S. E., Mabee P. M. (1994). A dual embryonic origin for vertebrate mechanoreceptors. Science 264, 426–430 [DOI] [PubMed] [Google Scholar]
- Couly G. F., Coltey P. M., Le Douarin N. M. (1993). The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development 117, 409–429 [DOI] [PubMed] [Google Scholar]
- Cullen R. D., Zdanski C., Roush P., Brown C., Teagle H., Pillsbury H. C., 3rd, Buchman C. (2006). Cochlear implants in Waardenburg syndrome. Laryngoscope 116, 1273–1275 [DOI] [PubMed] [Google Scholar]
- D’Amico-Martel A., Noden D. M. (1983). Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. Am. J. Anat. 166, 445–468 [DOI] [PubMed] [Google Scholar]
- Daneshi A., Hassanzadeh S., Farhadi M. (2005). Cochlear implantation in children with Waardenburg syndrome. J. Laryngol. Otol. 119, 719–723 [DOI] [PubMed] [Google Scholar]
- Dutton K., Dutton J. R., Pauliny A., Kelsh R. N. (2001a). A morpholino phenocopy of the colourless mutant. Genesis 30, 188–189 [DOI] [PubMed] [Google Scholar]
- Dutton K. A., Pauliny A., Lopes S. S., Elworthy S., Carney T. J., Rauch J., Geisler R., Haffter P., Kelsh R. N. (2001b). Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128, 4113–4125 [DOI] [PubMed] [Google Scholar]
- Eastman S. D., Chen T. H.-P., Falk M. M., Mendelson T. C., Iovine M. K. (2006). Phylogenetic analysis of three complete gap junction gene families reveals lineage-specific duplications and highly supported gene classes. Genomics 87, 265–274 [DOI] [PubMed] [Google Scholar]
- Edery P., Atti’e T., Amiel J., Pelet A., Eng C., Hofstra R. M., Martelli H., Bidaud C., Munnich A., Lyonnet S. (1996). Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 12, 442–444 [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Pike S. H., Debu B. (1989). The growth cones of identified motoneurons in embryonic zebrafish select appropriate pathways in the absence of specific cellular interactions. Neuron 2, 1097–1104 [DOI] [PubMed] [Google Scholar]
- Elworthy S., Lister J. A., Carney T. J., Raible D. W., Kelsh R. N. (2003). Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development 130, 2809–2818 [DOI] [PubMed] [Google Scholar]
- Elworthy S., Pinto J. P., Pettifer A., Cancela M. L., Kelsh R. N. (2005). Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech. Dev. 122, 659–669 [DOI] [PubMed] [Google Scholar]
- Escobar C., Zuasti A., Ferrer C., Garcia-Ortega F. (1995). Melanocytes in the stria vascularis and vestibular labyrinth of the Mongolian gerbil (Meriones unguiculatus). Pigment Cell Res. 8, 271–278 [DOI] [PubMed] [Google Scholar]
- Evans D. J., Noden D. M. (2006). Spatial relations between avian craniofacial neural crest and paraxial mesoderm cells. Dev. Dyn. 235, 1310–1325 [DOI] [PubMed] [Google Scholar]
- Grant K. A., Raible D. W., Piotrowski T. (2005). Regulation of latent sensory hair cell precursors by glia in the zebrafish lateral line. Neuron 45, 69–80 [DOI] [PubMed] [Google Scholar]
- Guo S., Brush J., Teraoka H., Goddard A., Wilson S. W., Mullins M. C., Rosenthal A. (1999). Development of noradrenergic neurons in the zebrafish hindbrain requires BMP, FGF8, and the homeodomain protein soulless/Phox2a. Neuron 24, 555–566 [DOI] [PubMed] [Google Scholar]
- Haddon C., Lewis J. (1996). Early ear development in the embryo of the zebrafish, Danio rerio. J. Comp. Neurol. 365, 113–123 [DOI] [PubMed] [Google Scholar]
- Haddon C., Jiang Y.-J., Smithers L., Lewis J. (1998). Delta-Notch signalling and the patterning of sensory cell differentiation in the zebrafish ear: evidence from the mind bomb mutant. Development 125, 4637–4644 [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M., Serbedzija G. N., McMahon A. P. (1996). Genetic analysis of dorsoventral pattern-formation in the zebrafish: requirement of a BMP-like ventralizing activity and its dorsal repressor. Genes Dev. 10, 2452–2461 [DOI] [PubMed] [Google Scholar]
- Hammond K., Hill R. E., Whitfield T. T., Currie P. D. (2002). Isolation of three zebrafish dachshund homologues and their expression in sensory organs, the central nervous system and pectoral fin buds. Mech. Dev. 112, 183–189 [DOI] [PubMed] [Google Scholar]
- Hammond K. L., Loynes H. E., Folarin A. A., Smith J., Whitfield T. T. (2003). Hedgehog signalling is required for correct anteroposterior patterning of the zebrafish otic vesicle. Development 130, 1403–1417 [DOI] [PubMed] [Google Scholar]
- Hans S., Liu D., Westerfield M. (2004). Pax8 and Pax2a function synergistically in otic specification, downstream of the Foxi1 and Dlx3b transcription factors. Development 131, 5091–5102 [DOI] [PubMed] [Google Scholar]
- Hans S., Christison J., Liu D., Westerfield M. (2007). Fgf-dependent otic induction requires competence provided by Foxi1 and Dlx3b. BMC Dev. Biol. 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi K., Matsuki C., Sarashina N. (1992). Aplasia of posterior semicircular canal in Waardenburg syndrome type II. J. Otolaryngol. 21, 262–264 [PubMed] [Google Scholar]
- Hofstra R. M., Osinga J., Tan-Sindhunata G., Wu Y., Kamsteeg E. J., Stulp R. P., van Ravenswaaij-Arts C., Majoor-Krakauer D., Angrist M., Chakravarti A., et al. (1996). A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat. Genet. 12, 445–447 [DOI] [PubMed] [Google Scholar]
- Honoré S. M., Aybar M. J., Mayor R. (2003). Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev. Biol. 260, 79–96 [DOI] [PubMed] [Google Scholar]
- Inoue K., Tanabe Y., Lupski J. R. (1999). Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Ann. Neurol. 46, 313–318 [DOI] [PubMed] [Google Scholar]
- Inoue K., Shilo K., Boerkoel C. F., Crowe C., Sawady J., Lupski J. R., Agamanolis D. P. (2002). Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation. Ann. Neurol. 52, 836–842 [DOI] [PubMed] [Google Scholar]
- Inoue K., Khajavi M., Ohyama T., Hirabayashi S., Wilson J., Reggin J. D., Mancias P., Butler I. J., Wilkinson M. F., Wegner M., et al. (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36, 361–369 [DOI] [PubMed] [Google Scholar]
- Kamachi Y., Uchikawa M., Kondoh H. (2000). Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. 16, 182–187 [DOI] [PubMed] [Google Scholar]
- Kelsh R. N., Eisen J. S. (2000). The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development 127, 515–525 [DOI] [PubMed] [Google Scholar]
- Kelsh R. N., Brand M., Jiang Y. J., Heisenberg C. P., Lin S., Haffter P., Odenthal J., Mullins M. C., van Eeden F. J., Furutani-Seiki M., et al. et al. (1996). Zebrafish pigmentation mutations and the processes of neural crest development. Development 123, 369–389 [DOI] [PubMed] [Google Scholar]
- Kelsh R. N., Schmid B., Eisen J. S. (2000). Genetic analysis of melanophore development in zebrafish embryos. Dev. Biol. 225, 277–293 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Korzh V., Sleptsova I., Liao J., He J., Gong Z. (1998). Expression of zebrafish bHLH genes ngn1 and nrd defines distinct stages of neural differentiation. Dev. Dyn. 213, 92–104 [DOI] [PubMed] [Google Scholar]
- Kozlowski D. J., Murakami T., Ho R. K., Weinberg E. S. (1997). Regional cell movement and tissue patterning in the zebrafish embryo revealed by fate mapping with caged fluorescein. Biochem. Cell Biol. 75, 551–562 [PubMed] [Google Scholar]
- Lang F., Vallon V., Knipper M., Wangemann P. (2007). Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell Physiol. 293, C1187–C1208 [DOI] [PubMed] [Google Scholar]
- Le Douarin N. M., Kalcheim C. (1999). The Neural Crest. Cambridge: Cambridge University Press [Google Scholar]
- Le Lievre C. S. (1978). Participation of neural crest-derived cells in the genesis of the skull in birds. J. Embryol. Exp. Morphol. 47, 17–37 [PubMed] [Google Scholar]
- Lecaudey V., Ulloa E., Anselme I., Stedman A., Schneider-Maunoury S., Pujades C. (2007). Role of the hindbrain in patterning the otic vesicle: a study of the zebrafish vhnf1 mutant. Dev. Biol. 303, 134–143 [DOI] [PubMed] [Google Scholar]
- Lee M. J., Nelson I., Houlden H., Sweeney M. G., Hilton-Jones D., Blake J., Wood N. W., Reilly M. M. (2002). Six novel connexin32 (GJB1) mutations in X-linked Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 73, 304–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léger S., Brand M. (2002). Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech. Dev. 119, 91–108 [DOI] [PubMed] [Google Scholar]
- Li M., Zhao C., Wang Y., Zhao Z., Meng A. (2002). Zebrafish sox9b is an early neural crest marker. Dev. Genes Evol. 212, 203–206 [DOI] [PubMed] [Google Scholar]
- Lister J. A., Robertson C. P., Lepage T., Johnson S. L., Raible D. W. (1999). nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 126, 3757–3767 [DOI] [PubMed] [Google Scholar]
- Liu D., Chu H., Maves L., Yan Y. L., Morcos P. A., Postlethwait J. H., Westerfield M. (2003). Fgf3 and Fgf8 dependent and independent transcription factors are required for otic placode specification. Development 130, 2213–2224 [DOI] [PubMed] [Google Scholar]
- López-Schier H., Hudspeth A. J. (2005). Supernumerary neuromasts in the posterior lateral line of zebrafish lacking peripheral glia. Proc. Natl. Acad. Sci. USA 102, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackereth M. D., Kwak S. J., Fritz A., Riley B. B. (2005). Zebrafish pax8 is required for otic placode induction and plays a redundant role with Pax2 genes in the maintenance of the otic placode. Development 132, 371–382 [DOI] [PubMed] [Google Scholar]
- Madden C., Halsted M. J., Hopkin R. J., Choo D. I., Benton C., Greinwald J. H. J. (2003). Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope 113, 2035–2041 [DOI] [PubMed] [Google Scholar]
- Marcus D. C., Wu T., Wangemann P., Kofuji P. (2002). KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell Physiol. 282, C403–C407 [DOI] [PubMed] [Google Scholar]
- Maroon H., Walshe J., Mahmood R., Kiefer P., Dickson C., Mason I. (2002). Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development 129, 2099–2108 [DOI] [PubMed] [Google Scholar]
- Marusich M. F., Furneaux H. M., Henion P. D., Weston J. A. (1994). Hu neuronal proteins are expressed in proliferating neurogenic cells. J. Neurobiol. 25, 143–155 [DOI] [PubMed] [Google Scholar]
- Masuda M., Yamazaki K., Kanzaki J., Hosoda Y. (1994). Ultrastructure of melanocytes in the dark cell area of human vestibular organs: functional implications of gap junctions, isolated cilia, and annulate lamellae. Anat. Rec. 240, 481–491 [DOI] [PubMed] [Google Scholar]
- Matsushima Y., Shinkai Y., Kobayashi Y., Sakamoto M., Kunieda T., Tachibana M. (2002). A mouse model of Waardenburg syndrome type 4 with a new spontaneous mutation of the endothelin-B receptor gene. Mamm. Genome 13, 30–35 [DOI] [PubMed] [Google Scholar]
- Mowbray C., Hammerschmidt M., Whitfield T. T. (2001). Expression of BMP signalling pathway members in the developing zebrafish inner ear and lateral line. Mechanisms of Development 108, 179–184 [DOI] [PubMed] [Google Scholar]
- Nemansky J., Hageman M. J. (1975). Tomographic findings of the inner ears of 24 patients with Waardenburg’s syndrome. Am. J. Roentgenol. Radium Ther. Nucl. Med. 124, 250–255 [DOI] [PubMed] [Google Scholar]
- Nissen R. M., Yan J., Amsterdam A., Hopkins N., Burgess S. M. (2003). Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development 130, 2543–2554 [DOI] [PubMed] [Google Scholar]
- Noden D. M. (1978). The control of avian cephalic neural crest cytodifferentiation. I. Skeletal and connective tissues. Dev. Biol. 67, 296–312 [DOI] [PubMed] [Google Scholar]
- O’Malley D. M., Sankrithi N. S., Borla M. A., Parker S., Banden S., Gahtan E., Detrich H. W., 3rd (2004). Optical physiology and locomotor behaviors of wild-type and nacre zebrafish. Methods Cell Biol. 76, 261–284 [DOI] [PubMed] [Google Scholar]
- Odenthal J., Nuesslein-Volhard C. (1998). fork head domain genes in zebrafish. Dev. Genes Evol. 208, 245–258 [DOI] [PubMed] [Google Scholar]
- Oysu C., Oysu A., Aslan I., Tinaz M. (2001). Temporal bone imaging findings in Waardenburg’s syndrome. Int. J. Pediatr. Otorhinolaryngol. 58, 215–221 [DOI] [PubMed] [Google Scholar]
- Paratore C., Goerich D. E., Suter U., Wegner M., Sommer L. (2001). Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 128, 3949–3961 [DOI] [PubMed] [Google Scholar]
- Peters T. A., Kuijpers W., Tonnaer E. L., van Muijen G. N., Jap P.H. (1995). Distribution and features of melanocytes during inner ear development in pigmented and albino rats. Hear. Res. 85, 169–180 [DOI] [PubMed] [Google Scholar]
- Phillips B. T., Bolding K., Riley B. B. (2001). Zebrafish fgf3 and fgf8 encode redundant functions required for otic placode induction. Dev. Biol. 235, 351–365 [DOI] [PubMed] [Google Scholar]
- Pingault V., Bondurand N., Kuhlbrodt K., Goerich D. E., Prehu M. O., Puliti A., Herbarth B., Hermans-Borgmeyer I., Legius E., Matthijs G., et al. (1998). SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173 [DOI] [PubMed] [Google Scholar]
- Pingault V., Guiochon-Mantel A., Bondurand N., Faure C., Lacroix C., Lyonnet S., Goossens M., Landrieu P. (2000). Peripheral neuropathy with hypomyelination, chronic intestinal pseudo-obstruction and deafness: a developmental “neural crest syndrome” related to a SOX10 mutation. Ann. Neurol. 48, 671–676 [PubMed] [Google Scholar]
- Pingault V., Bondurand N., Le Caignec C., Tardieu S., Lemort N., Dubourg O., Le Guern E., Goossens M., Boespflug-Tanguy O. (2001). The SOX10 transcription factor: evaluation as a candidate gene for central and peripheral hereditary myelin disorders. J. Neurol. 248, 496–499 [DOI] [PubMed] [Google Scholar]
- Pingault V., Girard M., Bondurand N., Dorkins H., Van Maldergem L., Mowat D., Shimotake T., Verma I., Baumann C., Goossens M. (2002). SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum. Genet. 111, 198–206 [DOI] [PubMed] [Google Scholar]
- Piotrowski T., Ahn D. G., Schilling T. F., Nair S., Ruvinsky I., Geisler R., Rauch G. J., Haffter P., Zon L. I., Zhou Y., et al. (2003). The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development 130, 5043–5052 [DOI] [PubMed] [Google Scholar]
- Pittlik S., Domingues S., Meyer A., Begemann G. (2008). Expression of zebrafish aldh1a3 (raldh3) and absence of aldh1a1 in teleosts. Gene Exp. Patt. 8, 141–147 [DOI] [PubMed] [Google Scholar]
- Price E. R., Fisher D. E. (2001). Sensorineural deafness and pigmentation genes: melanocytes and the Mitf transcriptional network. Neuron 30, 15–18 [DOI] [PubMed] [Google Scholar]
- Puffenberger E., Kauffman E., Bolk S., Matise T., Washington S., Angrist M., Weissenbach J., Garver K., Mascari M., Ladda R., et al. (1994a). Identity-by-descent and association mapping of a recessive gene for Hirschsprung’s disease on human chromosome 13q22. Hum. Mol. Genet. 3, 1217–1225 [DOI] [PubMed] [Google Scholar]
- Puffenberger E. G., Hosoda K., Washington S. S., Nakao K., deWit D., Yanagisawa M., Chakravarti A. (1994b). A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 79, 1257–1266 [DOI] [PubMed] [Google Scholar]
- Pusch C., Hustert E., Pfeifer D., Sudbeck P., Kist R., Roe B., Wang Z., Balling R., Blin N., Scherer G. (1998). The SOX10/Sox10 gene from human and mouse: sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum. Genet. 103, 115–123 [DOI] [PubMed] [Google Scholar]
- Raible D. W., Eisen J. S. (1994). Restriction of neural crest cell fate in the trunk of the embryonic zebrafish. Development 120, 495–503 [DOI] [PubMed] [Google Scholar]
- Raible D. W., Wood A., Hodsdon W., Henion P. D., Weston J. A., Eisen J. S. (1992). Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev. Dyn. 195, 29–42 [DOI] [PubMed] [Google Scholar]
- Rau M. J., Fischer S., Neumann C. J. (2006). Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development. Dev. Biol. 296, 83–93 [DOI] [PubMed] [Google Scholar]
- Read A. P., Newton V. E. (1997). Waardenburg syndrome. J. Med. Genet. 34, 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Germain N., Lee Y.-H., Zhang Y., Sargent T. D., Saint-Jeannet J.-P. (2004). Specification of the otic placode depends on Sox9 function in Xenopus. Development 131, 1755–1763 [DOI] [PubMed] [Google Scholar]
- Schilling T. F., Kimmel C. B. (1994). Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development 120, 483–494 [DOI] [PubMed] [Google Scholar]
- Schlierf B., Werner T., Glaser G., Wegner M. (2006). Expression of connexin47 in oligodendrocytes is regulated by the Sox10 transcription factor. J. Mol. Biol. 361, 11–21 [DOI] [PubMed] [Google Scholar]
- Solomon K. S., Kwak S.-J., Fritz A. (2004). Genetic interactions underlying otic placode induction and formation. Dev. Dyn. 230, 419–433 [DOI] [PubMed] [Google Scholar]
- Sonnenberg-Riethmacher E., Miehe M., Stolt C. C., Goerich D. E., Wegner M., Riethmacher D. (2001). Development and degeneration of dorsal root ganglia in the absence of the HMG-domain transcription factor Sox10. Mech. Dev. 109, 253–265 [DOI] [PubMed] [Google Scholar]
- Southard-Smith E. M., Collins J. E., Ellison J. S., Smith K. J., Baxevanis A. D., Touchman J. W., Green E. D., Dunham I., Pavan W. J. (1999). Comparative analyses of the Dominant megacolon-SOX10 genomic interval in mouse and human. Mamm. Genome 10, 744–749 [DOI] [PubMed] [Google Scholar]
- Spritz R. A. (2006). Melanoblast Development and Associated Disorders. In The Pigmentary System: Physiology and Pathophysiology (eds Nordlund J. J., Boissy R. E., Hearing V. J., King R. A., Oetting W. S., Ortonne J.-P.), pp. 140–154 New York: Oxford University Press [Google Scholar]
- Stanchina L., Baral V., Robert F., Pingault V., Lemort N., Pachnis V., Goossens M., Bondurand N. (2006). Interactions between Sox10, Edn3 and Ednrb during enteric nervous system and melanocyte development. Dev. Biol. 295, 232–249 [DOI] [PubMed] [Google Scholar]
- Steel K. P. (1995). Inherited hearing defects in mice. Ann. Rev. Genet. 29, 675–701 [DOI] [PubMed] [Google Scholar]
- Steel K. P., Barkway C. (1989). Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107, 453–463 [DOI] [PubMed] [Google Scholar]
- Stojkovic T., Latour P., Vandenberghe A., Hurtevent J. F., Vermersch P. (1999). Sensorineural deafness in X-linked Charcot-Marie-Tooth disease with connexin 32 mutation (R142Q). Neurology 52, 1010–1014 [DOI] [PubMed] [Google Scholar]
- Streit A. (2002). Extensive cell movements accompany formation of the otic placode. Dev. Biol. 249, 237–254 [DOI] [PubMed] [Google Scholar]
- Sznajer Y., Coldéa C., Meire F., Delpierre I., Sekhara T., Touraine R. L. (2008). A de novo SOX10 mutation causing severe type 4 Waardenburg syndrome without Hirschsprung disease. Am. J. Med. Genet. A 146, 1038–1041 [DOI] [PubMed] [Google Scholar]
- Tachibana M. (1999). Sound needs sound melanocytes to be heard. Pigment Cell Res. 12, 344–354 [DOI] [PubMed] [Google Scholar]
- Tachibana M., Hara Y., Vyas D., Hodgkinson C., Fex J., Grundfast K., Arnheiter H. (1992). Cochlear disorder associated with melanocyte anomaly in mice with a transgenic insertional mutation. Mol. Cell. Neurosci. 3, 433–445 [DOI] [PubMed] [Google Scholar]
- Tachibana M., Kobayashi Y., Matsushima Y. (2003). Mouse models for four types of Waardenburg syndrome. Pigment Cell Res. 16, 448–454 [DOI] [PubMed] [Google Scholar]
- Taylor K. M., Labonne C. (2005). SoxE factors function equivalently during neural crest and inner ear development and their activity is regulated by SUMOylation. Dev. Cell 9, 593–603 [DOI] [PubMed] [Google Scholar]
- Thisse C., Thisse B., Postlethwait J. H. (1995). Expression of snail2, a second member of the zebrafish snail family, in cephalic mesendoderm and presumptive neural crest of wild-type and spadetail mutant embryos. Dev. Biol. 172, 86–99 [DOI] [PubMed] [Google Scholar]
- Verheij J. B., Sival D. A., van der Hoeven J. H., Vos Y. J., Meiners L. C., Brouwer O. F., van Essen A. J. (2006). Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur. J. Paediatr. Neurol. 10, 11–17 [DOI] [PubMed] [Google Scholar]
- Wangemann P. (2002). K+ cycling and the endocochlear potential. Hear. Res. 165, 1–9 [DOI] [PubMed] [Google Scholar]
- Wangemann P. (2006). Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J. Physiol. 576, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P., Itza E. M., Albrecht B., Wu T., Jabba S. V., Maganti R. J., Lee J. H., Everett L. A., Wall S. M., Royaux I. E., et al. (2004). Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Takeda K., Katori Y., Ikeda K., Oshima T., Yasumoto K., Saito H., Takasaka T., Shibahara S. (2000). Expression of the Sox10 gene during mouse inner ear development. Brain Res. Mol. Brain Res. 84, 141–145 [DOI] [PubMed] [Google Scholar]
- Wegner M. (1999). From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 27, 1409–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. (2000). The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). Eugene: Univ. of Oregon Press [Google Scholar]
- Whitfield T., Granato M., van Eeden F., Schach U., Brand M., Furutani-Seiki M., Haffter P., Hammerschmidt M., Heisenberg C., Jiang Y., et al. (1996). Mutations affecting development of the zebrafish inner ear and lateral line. Development 123, 241–254 [DOI] [PubMed] [Google Scholar]
- Yan Y. L., Miller C. T., Nissen R. M., Singer A., Liu D., Kirn A., Draper B., Willoughby J., Morcos P. A., Amsterdam A., et al. (2002). A zebrafish sox9 gene required for cartilage morphogenesis. Development 129, 5065–5079 [DOI] [PubMed] [Google Scholar]
- Yan Y. L., Willoughby J., Liu D., Crump J. G., Wilson C., Miller C. T., Singer A., Kimmel C., Westerfield M., Postlethwait J. H. (2005). A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development 132, 1069–1083 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}