

Abstract
The (1-naphthyl)propargyl group is introduced as a sterically unintrusive alcohol protecting group that is cleaved in a single step by exposure to dichlorodicyanoquinone in wet dichloromethane. In conjunction with a 4,6-O-benzylidene protecting group and the use of the sulfoxide glycosylation method, 3-O-naphthylpropargyl protected mannosyl donors are extremely β-selective.
The apposite use of protecting groups continues to be an essential element in preparative carbohydrate and oligosaccharide synthesis, with considerable effort devoted to their development in recent years.1 This is due to the central role of protecting groups in modulating reactivity of both glycosyl donors and acceptors, and critically, in the control of regioselectivity, 2 and stereoselectivity. 3 In response to a problem arising from the influence of protecting groups size on the stereoselectivity of a glycosylation reaction, 4 we recently described the successful application of propargyl ethers as sterically unintrusive donor protecting groups for β-mannosylation.5 However, while the propargyl ethers were readily introduced, and had the anticipated effect on stereoselectivity, they required a two step deprotection protocol: an initial treatment with base followed by catalytic osmoylation of the resulting allenyl ether (Scheme 1).
Scheme 1.
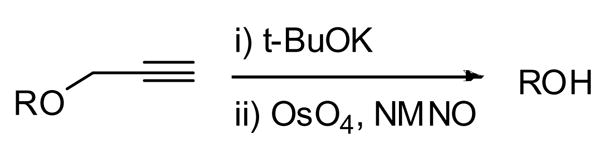
Deprotection of Propargyl Ethers
We considered that the advantages of the propargyl ether protecting system would be significantly enhanced if it could be modified in such a way as to be cleavable in a single step, orthogonal to the ubiquitous benzyl ethers. We report here on the successful accomplishment of this goal through the use of the naphthylpropargyl system.
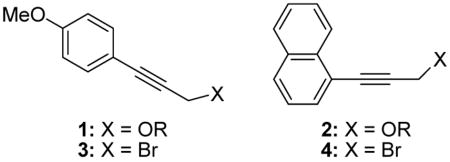
The p-methoxybenzyl6 and naphthylmethyl7 ethers are widely employed as benzyl ether surrogates, cleavable under oxidative conditions. We reasoned that the insertion of an acetylenic group into the aryl-methylene bond of either the PMB or naphthylmethyl system would afford a system combining the steric advantages of the propargyl ether with the facile oxidative cleavage of the PMB and naphthylmethyl ethers. This line of thought led us to the ethers 1 and 2, which we assumed could be assembled from the known bromides 3 and 4.8,9
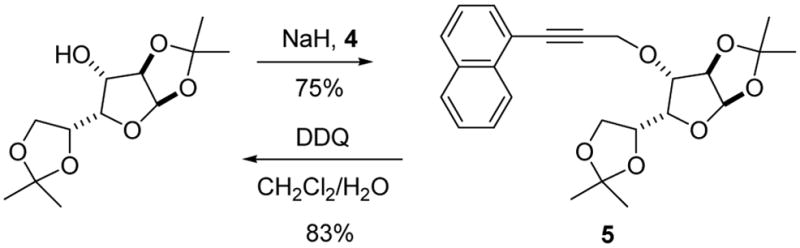
Alkylation of 1,2;5,6-diacetone-D-glucofuranose with sodium hydride and bromide 4 gave the model ether 5 (Scheme 2). Treatment of this compound with DDQ in wet dichloromethane, typical conditions for the removal of PMB and naphthylmethyl ethers, returned the alcohol in 83% yield, thereby establishing proof of principle. Directly analogous transformations with the p-methoxyphenylproparyl protected system were also successful. However, it subsequently became clear that the more electron rich p-methoxyphenylpropargyl group 1 was incompatible with various glycosylation conditions leading to our subsequent preference for system 2.
Scheme 2.
Deprotection of a Naphthylpropargyl Ether
To examine the effect of the new protecting group 2 on stereoselectivity of glycoslation reactions, when located at both O2 and O3, we prepared donors 9 and 10 from known diol 610 by standard means as set out in Scheme 3.
Scheme 3.

Preparation of Donors 9 and 10
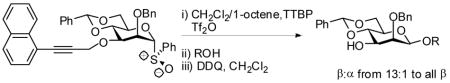
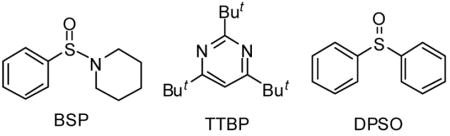
Attempted activation of donors 9 and 10 by our standard treatment with 1-benzenesulfinyl piperidine (BSP) and trifluoromethanesulfonic anhydride11 in the presence of the hindered base tri-tert-butylpyrimidine (TTBP) 12 was unproductive affording little produts or no reaction. We turned, therefore, to the more potent combination of diphenyl sulfoxide (DPSO) and triflic anhydride13 when consumption of the donors was observed, but complex reaction mixtures were obtained. Study of the several products indicated that electrophilic attack on the arylpropargyl system was the root of the problem.
Precedent suggested, however, the activation of glycosyl sulfoxides with Tf2O to be compatible with electron rich aromatic systems, especially when used in conjunction with an electrophile scavenger. 16 Accordingly donor 9 was oxidized to the sulfoxide 11 (Scheme 3), which was formed as a single diastereomer whose configuration rests on analogy.17
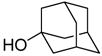
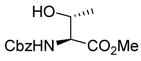
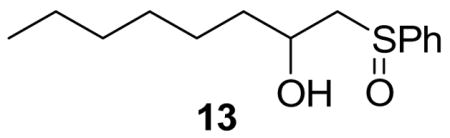
Treatment of 11 with triflic anhydride in the presence of TTBP at −78 °C in a 3:1 mixture of CH2Cl2 and 1-octene, to give an intermediate glycosyl triflate, 18 followed by addition of 1-adamantanol finally resulted in the formation of the β-mannoside 12a with impeccable selectivity (Table 1, entry 1). That 1-octene fulfilled its role of trapping of extraneous thiophilic species was established by isolation of 13.
Table 1.
Coupling Reactions of Donor 11
| entry | acceptor | coupled product 12 | yielda | ratiob β:α |
|---|---|---|---|---|
| 1 |
|
12a |
77% | β only |
| 2 |
|
12b |
46% | β only |
| 3 |
|
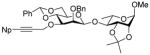
12c |
87% | 13:1 |
| 4 |
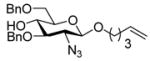
|
12d |
43% | β only |
| 5 |
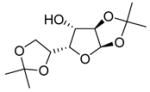
|
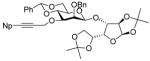
12e |
74% | β only |
| 6 |
|
12f |
85% | β only |
Isolated yields after column chromatography.
Ratio was determined by 1H-NMR of crude reaction mixtures.
A number of couplings were then conducted with more standard glycosyl acceptors, leading to the yields and selectivities collected in Table 1. The influence of the 3-O-naphthylpropargyl group on selectivty is best illustrated in entry 4 (Table 1): previously coupling of the. identical acceptor to the 3-O-tert-butyldimethylsilyl analog of 11 resulted in the formation of a 1.8:1 mixture of glycosides favoring the α-anomer.4
Oxidation of thioglycoside 10 afforded the sulfoxide 14, as a single diastereomer, in 94% yield. Activation of 14 under the conditions employed for 11 afforded β-mannosides with excellent selectivity (Table 2). Unfortunately, the reaction mixtures were relatively complex and included a significant byproduct, ketone 15, resulting from cyclization of the protecting group onto the activated glycosyl donor. In the face of this problem couplings to donor 14 were not pursued further.
Table 2.
Coupling Reactions of Donor 14
| entry | acceptor | coupled product 16 yielda, β:αb |
|---|---|---|
| 1 |
|
16a
67%, β only |
| 2 |
|
16b
38%, β only |
| 3 |
|
16c
13%, β only |
Isolated yields after column chromatography.
Ratio was determined based on 1H-NMR of crude reaction mixtures.
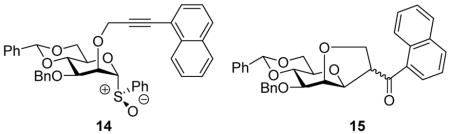
The excellent stereoselectivity obtained with the 3-O-naphthylpropargyl protected donor 11 contrasts with the poor selectivity delivered by the corresponding 3-O-propargyl donor.5b On the other hand, 4,6-O-benzylidene mannosyl donors carrying a 2-O-propargyl group were previously found to be highly efficient, in contrast to the 2-O-naphthylpropargyl system 14, and highly β-selective.5 Thus, in addition to their different requirements for deprotection, the propargyl and naphthylpropargyl systems are highly complementary.
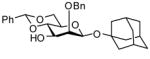
In accordance with the model experiments (Scheme 2), selective deprotection of the glycosides 12 was accomplished with DDQ in CH2Cl2/H2O (20:1) over a period of 2–3 h at room temperature in excellent yield as reported in Table 3. The employment of other solvent systems recommended for the cleavage of 2-naphthylmethyl ethers, such as CH2Cl2/CH3OH, 19 and CHCl3/H2O, and CH2Cl2 alone20 was less satisfactory.
Table 3.
Cleavage of Naphthylpropargyl Ethers
| entry | deprotected product 17 | yielda |
|---|---|---|
| 1 |
17a |
86% |
| 2 |
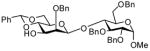
17b |
80% |
| 3 |
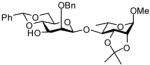
17c |
89% |
| 4 |
17d |
90% |
| 5 |
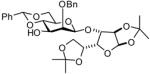
17e |
84% |
| 6 |
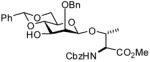
17f |
86% |
Isolated yields after column chromatography.
To conclude, we report the development of the naphthylpropargyl ether system. In conjunction with the sulfoxide glycosylation method, when introduced on the 3-position of 4,6-O-benzylidene protected mannosyl donors this system affords extremely β-selective coupling reactions, and the possibility of orthogonal cleavage in a single step with DDQ. We anticipate that this group will find application in oligosaccharide synthesis and, because of its minimal steric character and ease of deprotection, beyond the confines of carbohydrate chemistry.
Supplementary Material
Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.
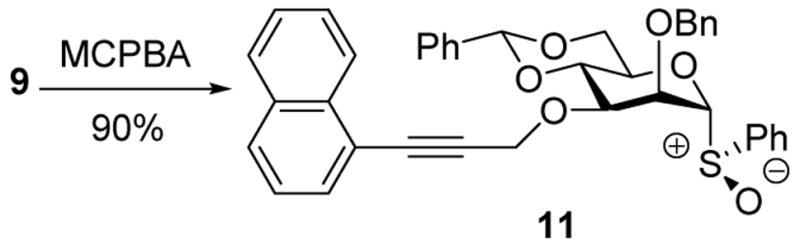
Preparation of Sulfoxide 11
Acknowledgments
We thank the NIH(GM 57335) for support of our work in this area.
References
- 1.(a) Grindley TB. In: Modern Methods in Carbohydrate Chemistry. Khan SH, O’Neill RA, editors. Harwood Academic; Amsterdam: 1996. p. 225. [Google Scholar]; (b) Green LG, Ley SV. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. p. 427. [Google Scholar]; (c) Fraser-Reid B, Lopez JC, Gomez AM, Uriel C. Eur J Org Chem. 2004:1387. doi: 10.1021/jo202335n. [DOI] [PubMed] [Google Scholar]
- 2.Ley SV, Baeschlin DK, Dixon DJ, Foster AC, Ince SJ, Priepke HWM, Reynolds DJ. Chem Rev. 2001;101:53. doi: 10.1021/cr990101j. [DOI] [PubMed] [Google Scholar]
- 3.(a) Crich D, Vinod AU. J Org Chem. 2005;70:1291. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Dudkin V. J Am Chem Soc. 2001;121:6819. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]; (c) Crich D, Jayalath P. J Org Chem. 2005;70:7252. doi: 10.1021/jo0508999. [DOI] [PubMed] [Google Scholar]; (d) Crich D, Yao Q. J Am Chem Soc. 2004;126:8232. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]; (e) Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asymmetry. 2005;16:105. [Google Scholar]; (f) Crich D, Vinod AU, Picione J. J Org Chem. 2003;68:8453. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]; (g) Crich D, Vinod AU, Picione J, Wink DJ. ARKIVOC. 2005;vi:339. [PMC free article] [PubMed] [Google Scholar]; (h) Kim JH, Yang H, Boons GJ. Angew Chem, Int Ed. 2005;44:947. doi: 10.1002/anie.200461745. [DOI] [PubMed] [Google Scholar]; (i) Kim JH, Yang H, Park J, Boons GJ. J Am Chem Soc. 2005;127:12090. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]; (j) Smoot JT, Pornsuriyasak P, Demchenko AV. Angew Chem, Int Ed. 2005;44:7123. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]; (k) Bowers SG, Coe DM, Boons GJ. J Org Chem. 1998;63:4570. [Google Scholar]; (l) Jiao H, Hindsgaul O. Angew Chem, Int Ed. 1999;38:346. doi: 10.1002/(SICI)1521-3773(19990201)38:3<346::AID-ANIE346>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (m) Crich D, Banerjee A. J Am Chem Soc. 2006;128:8078. doi: 10.1021/ja061594u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Crich D, Bowers AA. J Org Chem. 2006;71:3452. doi: 10.1021/jo0526688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crich D, Dudkin V. Tetrahedron Lett. 2000;41:5643. [Google Scholar]
- 5.(a) Crich D, Jayalath P. Org Lett. 2005;7:2277. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Jayalath P, Hutton TK. J Org Chem. 2006;71:3064. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Bhattacharyya S, Magnusson B, Wellmar U. J Chem Soc, Perkin Trans 1. 2000;8:886. [Google Scholar]; (b) Maruyama M, Takeda T, Shimizu N. Carbohydr Res. 2000;325:83. doi: 10.1016/s0008-6215(99)00315-8. [DOI] [PubMed] [Google Scholar]; (c) Yan L, Kahne D. Synlett. 1995:523. [Google Scholar]; (d) Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. John Wiley & Sons, Inc; Hoboken: 1991. p. 86. [Google Scholar]; (d) Kocienski PJ. Protecting Groups. 3. Thieme; Stuttgart, Germany: 2005. Chapter 4; p. 257. [Google Scholar]; (e) Wuts PGM. In: Handbook of Reagents for Organic Synthesis. Crich D, editor. Wiley; Hoboken: 2005. p. 425. [Google Scholar]
- 7.(a) Wright JA, Yu J, Spencer JB. Tetrahedron Lett. 2001;42:4033. [Google Scholar]; (b) Csávás M, Szabó ZB, Borbás A, Lipták A. In: Handbook of Reagents for Organic Synthesis. Crich D, editor. Wiley; Hoboken: 2005. p. 459. [Google Scholar]; (c) Szabo ZB, Borbas A, Bajza I, Liptak A. Tetrahedron: Asymmetry. 2005;16:83. [Google Scholar]; (d) Csavas M, Borbas A, Szilagyi L, Liptak A. Synlett. 2002;6:887. [Google Scholar]
- 8.Bromide 3 was prepared according to: Batey RA, Shen M, Lough AJ. Org Lett. 2002;7:1411. doi: 10.1021/ol017245g.
- 9.Bromide 4 was prepared according to: Banerjee M, Roy S. Org Lett. 2004;6:2137. doi: 10.1021/ol0493352.
- 10.Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 11.Crich D, Smith M. J Am Chem Soc. 2001;123:9015. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001;2:323. [Google Scholar]
- 13.Codee JDC, Litjens REJN, Den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- 16.(a) Yan L, Kahne D. J Am Chem Soc. 1996;118:9239. [Google Scholar]; (b) Gildersleeve J, Smith A, Sakurai k, Raghavan S, Kahne D. J Am Chem Soc. 1999;121:6176. [Google Scholar]
- 17.(a) Crich D, Mataka J, Zakharov L. J Am Chem Soc. 2002;124:6028. doi: 10.1021/ja0122694. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Mataka J, Sun S, Lam KC, Rheingold AR, Wink DJ. J Chem Soc, Chem Commun. 1998;24:2763. [Google Scholar]
- 18.Crich D, Sun S. J Am Chem Soc. 1997;119:11217. [Google Scholar]
- 19.Xia J, Matta KL, Abbas SA. Tetrahedron Lett. 2000;41:169. [Google Scholar]
- 20.Oikawa Y, Yoshioka T, Yonemitsu O. Tetrahedron Lett. 1982;23:889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.