

Abstract
Reaction of S-allyl cysteine derivatives, generated by the selenocysteine ligation, with rhodium carbenoids, stabilized and unstabilized, enables the attachment of diverse functionality onto cysteine residues. The reaction is successfully applied to the introduction of lipid-like residues, a fluorous alkyl chain, and mono and disaccharides.
Introduction
Recently, we have described methods for the permanent ligation of thiols involving the coupling of either Se-allyl Bunte salts (Se-allyl selenosulfonates), or S-allyl S′-heteroaryl disulfides, with thiols to give Se-allyl selenosulfides or S-allyl disulfides, respectively, followed by a dechalcogenative 2,3-sigmatropic rearrangement to give the ligated products (Scheme 1).1 These complementary reactions, for which all steps take place at room temperature in protic media, were illustrated by the introduction of a range of allyl and prenyl groups to cysteine and other thiols.
Scheme 1.

Dechalcogenative Allylic Selenosulfide and Disulfide Ligations
By virtue of the reaction mechanism these reactions afford allylic sulfides as products, thereby opening up avenues for further functionalization, one of which is the 2,3-sigmatropic rearrangement of allylic sulfur ylides as we describe here.
The 2,3-sigmatropic rearrangement of allylic sulfur ylides has been known for many years and has found widespread application in organic synthesis.2 With an eye to eventual applications in the modification of peptides, proteins and other bioconjugates, for our investigation we selected the modification of this reaction popularized by Kirmse and Doyle, in which the sulfur ylide is generated by transition metal catalyzed addition of a diazoalkane to an allylic sulfide (Scheme 2).3 Our choice of the Kirmse-Doyle reaction was further guided by current interest in the deployment of transition metal-catalyzed reactions in peptide chemistry,4 and more particularly by the recent publication of Francis on the reaction of a stabilized vinyl diazo acetate, catalyzed by dirhodium tetracetate, with tryptophan residues in horse heart myoglobin and substilin Carlsberg in aqueous ethylene glycol.5
Scheme 2.

Reaction of metal carbenoids with allyl thio ethers
Results and Discussion
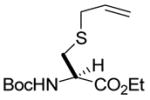
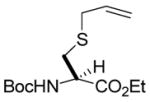
We began with a feasibility study in which a series of diazoalkanes were allowed to react with S-allyl or S-methylallyl cysteine derivatives, obtained by the selenosulfide ligation method, in the presence of catalytic Rh2(OAc)4 in dimethoxyethane at room temperature. From the results of these experiments (Table 1) it is clear that a variety of simple alkyl groups may be introduced into cysteine in this manner, with moderate yields consistent with earlier studies on simple allylic sulfides.3a–d,6 In each case the new stereogenic center formed as a result of the sigmatropic rearrangement was obtained as an approximately 1:1 mixture of isomers.
Table 1.
Reaction of carbenoids with cysteine derivatives.
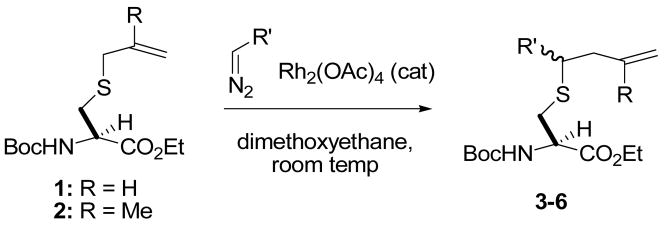 | |||
|---|---|---|---|
| Cmpd | Diazo Deriv | Product (% yield) | |
| 1 | 1 | ethyl diazoacetate |
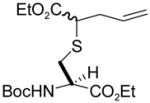
3 (53) |
| 2 | 2 | ethyl diazoacetate |
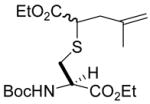
4 (42) |
| 3 | 1 | TMSdiazomethane |
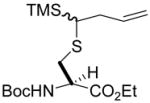
5 (57) |
| 4 | 2 | Me(CH2)14COCHN2 |
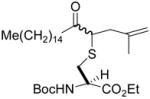
6 (52) |
Attention was turned to the functionalization of a tripeptide 7 (Table 2). Given the importance of the introduction of lipids onto cysteine in peptide and protein chemistry and biochemistry,7 entries 3 and 4 of Table 2 are especially noteworthy. In view of the recent interest in the fluorous tagging of peptides and proteins, attention is also called to entry 5 of Table 2.8
Table 2.
Reaction of carbenoids with S-allyl glutathione.
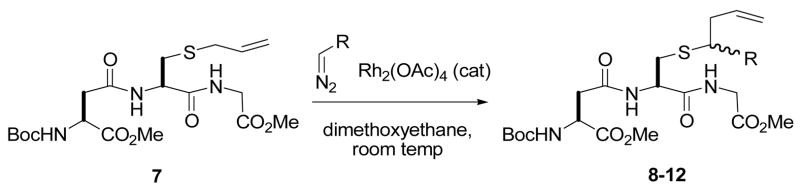 | ||
|---|---|---|
| R | Product (% Yield) | |
| 1 | CO2Et | 8 (45) |
| 2 | C6H5 | 9 (45) |
| 3 | CO(CH2)14Me | 10 (38) |
| 4 | (CH2)9Mea | 11 (35) |
| 5 | (CH2)2(CF2)5CF3a | 12 (32) |
Diazo precursors to 11 and 12 were prepared from the hydrazones with Pb(OAc)4 and were used immediately.
With the exception of the tryptophan case discussed below, the main byproducts from the chemistry presented here are those of dimerization of the intermediate metal carbenoids, as is typical of this type of reaction. Analysis of crude reaction mixtures by NMR spectroscopy indicates that the mass balance of the amino acid or peptide derivatives is made up largely by the unreacted substrate; insertion into the peptide or carbamate NH bond is not a major problem, as anticipated from the work of Francis.5
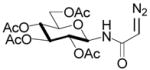
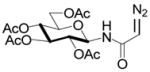
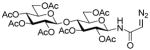
Interest in the glycosylation of cysteine residues as a means of peptide and protein glycosylation7a,9 led us to investigate carbohydrate-based diazoalkanes. To this end, peracetyl β-D-glucosyl and β-D-chitobiosyl diazo amides 13 and 14 were obtained from the glycosyl amines, via the tosyl hydrazones. An important feature in the design of 13 and 14 was the use of the diazoamide function rather than the much more common diazoesters. This choice was made based on the trans-nature of the amide bond, with its high barrier to inversion relative to the ester bond, which it was anticipated would prevent the metal carbenoid intermediate from “biting back” on the carbohydrate moiety. This supposition was borne out in practice, as the only carbohydrate-based byproducts observed upon activation with Rh2(OAc)4 were those resulting from dimerization of the carbenoid, which is typical for this type of reaction.
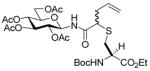
Glucosyl diazoamide 13 was attached to allyl hexadecyl sulfide (15) to establish the validity of the method (Table 3) before couplings to amino acid and peptide-based sulfides were undertaken (Table 3, entries 2–5), providing access to a new class of neoglycoconjugates.10 It is especially noteworthy that, although glycoamino acids and peptides 16–20 are cysteine derivatives, the amide linkage employed opens up the possibility of the application of this chemistry, coupled with native peptide ligation11 and our dechalcogenative allylation protocols,1 as mimics of the N-linked glycoproteins,12 for which new methods are constantly being sought.9d,e,13
Table 3.
Rh2(OAc)4 Catalyzed Glycosylation
| Substrate | Diazo Compound | Product (%yield) | |
|---|---|---|---|
| 1 |
15 |
13 |
16(52) |
| 2 |
1 |
13 |
17(48) |
| 3 |
1 |
14 |
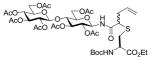
18(54) |
| 4 |
7 |
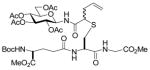
13 |
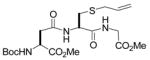
19(34) |
| 5 |
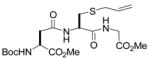
7 |
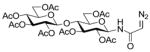
14 |
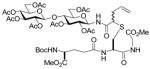
20(41) |
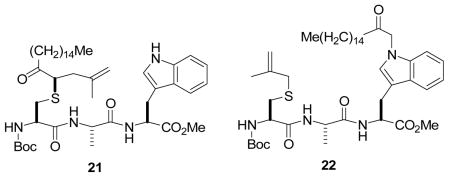
Finally, in view of the work of Francis,5 we briefly investigated chemoselectivity with S-methallyl Boc-L-Cys-L-Ala-L-Trp-OMe1 with a diazoketone. Literature work on the addition of Rh carbenoids to sulfides in the presence of indoles provided grounds for optimism that our chemistry would be applicable in the presence of tryptophan,14 however, complex reaction mixtures were obtained from which only two products, 21 and 22, were obtained pure in low yield (18 and 9%, respectively). The insertion of stabilized rhodium carbenoids into the indole N-H bond, as in the formation of 22, is a known reaction pathway,15 and is consistent with the structures proposed by Francis for reaction with protein-based tryptophan residues.5 At least for the present, it appears that the application of the Doyle-Kirmse reaction to S-allylated peptides and proteins is not compatible with the presence of tryptophan.16
Experimental Section
Phenyl diazomethane was prepared following the procedure reported by Creary.17
1-Diazo-2-heptadecanone was prepared following the procedure reported by Scott and Sumpter.18 A freshly prepared solution of diazomethane (30 mmol) in anhydrous ether (60 mL) was cooled to 0 °C and stirred at high speed. To this cooled solution, hexadecanoyl chloride (2.75 g, 10 mmol) in anhydrous ether (20 mL) was added dropwise over 20 min. The resulting reaction mixture was stirred cold for an additional 30 min and then at room temperature for 60 min. After this period of time the reaction was complete, and excess diazomethane was removed by evacuating the flask with a water aspirator pump in the hood. After the diazomethane has been removed, the remaining ethereal solution was concentrated by rotary evaporation to give crude compound. Pure 1-diazo-2-heptadecanone was obtained as yellow solid (2.61 g, 93% yield) by chromatography on silica gel using 10% ethyl acetate/hexane as an eluent. 1H NMR: δ 5.24 (s, 1H), 2.27–2.38 (m, 2H), 1.58–1.64 (m, 2H), 1.23 (br. s, 24H), 0.86 (t, J = 7.0, 3H). 13C NMR: δ 195.4, 54.2, 41.1, 31.9, 29.7 (3C), 29.6 (2C), 29.5 (2C), 29.4 (2C), 29.2, 25.3, 22.7, 14.1. ν 2120, 2100, 1620 cm−1. EIHRMS Calcd for C17H32N2O [M]+: 280.2515, found: 280.2520.
1-Diazoundecane
1-Diazoundecane was prepared following the procedure reported by Shechter and Holton.19 Undecanal (1.36 g, 8.0 mmol) was added to stirred anhydrous hydrazine (2.56g, 80.0 mmol) at 55°C. The reaction was continued 45 min at 55–65 °C. After the mixture had been cooled to room temperature, methylene chloride (25 mL) was added. The solution was washed with saturated aqueous sodium chloride (3 × 10 mL), dried over potassium carbonate, and concentrated under reduced pressure to a volume of 5 mL. Dimethylformamide (10 mL) was added, and remaining methylene chloride was removed by vacuum evaporation. The solution of undecanal hydrazone in dimethylformamide was cooled to −78 °C (15 min) and diluted with cold tetramethylguanidine (4 mL). Lead tetraacetate (3.90 g, 8.8 mmol) was added in 5 min, and the mixture was stirred 60 min at −78 °C. The reaction solution was diluted with cold hexane (3 × 20 mL) and extracted at −78 °C. The combined cold hexane extracts were washed with cold (−30 °C) 30% aqueous potassium hydroxide (2 × 10 mL), small pieces of dry ice were added, and the solution was then filtered to give a rose-red solution of 1-diazoundecane in hexane which was used directly in further reaction.
9-Diazo-1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluorononane
4,4,5,5,6,6,7,7,8,8,9,9,9-Tridecafluorononanal (8.0 mmol) was added to stirred anhydrous hydrazine (2.56g, 80.0 mmol) at 55 °C. The reaction was continued 45 min at 55–65 °C. After the mixture had been cooled to room temperature, methylene chloride (25 mL) was added. The solution was washed with saturated aqueous sodium chloride (3 × 10 mL), dried over potassium carbonate, and concentrated under reduced pressure to a volume of 5 mL. Dimethylformamide (10 mL) was added, and remaining methylene chloride was removed by vacuum volatization. The solution of undecylic aldehyde hydrazone in dimethylformamide was cooled to −78 °C (15 min) and diluted with cold tetramethylguanidine (4 mL). Lead tetraacetate (3.90 g, 8.8 mmol) was added in 5 min, and the mixture was stirred 60 min at −78 °C. The reaction solution was diluted with cold hexane (3 × 20 mL) and extracted at −78 °C. The combined cold hexane extracts were washed with cold (−30 °C) 30% aqueous potassium hydroxide (2 × 10 mL), small pieces of dry ice were added, and the solution was then filtered to give a rose-red solution of 1-diazo-undecane in hexane which was used directly.
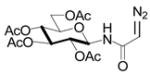
N-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)diazoacetamide
To a solution of tetra-O-acetyl-β-D-glucopyranosylamine (695 mg, 2.0 mmol, 1.0 equiv.) and glyoxylic acid p-toluenesulfonylhydrazone20 17,18 (533 mg, 2.2 mmol, 1.1 equiv.) in ice-cold THF (20 mL) was added, dropwise, DCC (454 mg, 2.2 mmol, 1.1 equiv.) in THF (10 mL). The mixture was allowed to warm to room temperature and stirring was continued over night. Then the solid was filtered off. After removal of the solvent, the filtrate was purified by flash chromatography (hexanes/EtOAc, 2:3) to afford a yellow solid (582 mg, 51% yield)
To a solution of the yellow solid (100 mg, 0.175 mmol, 1.0 equiv.) in methylene chloride (5.0 mL) was added triethylamine (0.35 mmol, 2.0 equiv.) in a nitrogen atmosphere. The mixture was stirred at room temperature over night. Then the solution was diluted with ethyl acetate (20 mL) and washed by water (2 × 10 mL), brine (2 × 10 mL), dried over sodium sulfate and concentrated. The remaining residue was then purified by flash chromatography (hexanes/EtOAc, 2:3) to afford the diazo compound (65 mg, 90% yield) as a viscous yellow oil. 1H NMR: δ 6.10 (d, J = 9.5 Hz, 1H), 5.26–5.31 (m, 2H), 5.03 (t, J = 9.5 Hz, 1H), 4.88 (t, J = 9.5, 1H), 4.81 (s, 1H), 4.29 (dd, J = 12.5, 4.5 Hz, 1H), 4.06 (dd, J = 12.5, 2.0 Hz, 1H), 3.80–3.94 (m, 1H), 2.00–2.06 (m, 12H). 13C NMR: δ 171.2, 170.7, 169.9, 169.7, 165.9, 78.5, 73.4, 72.7, 70.5, 68.2, 61.7, 48.1, 20.7 (4C). ν 2111, 1750, 1653 cm−1. ESIHRMS Calcd for C16H21N3O10Na [M+Na]+: 438.1125, found: 438.1128.
N-(2,3,6,2′,3′,4′,6′-Hepta-O-acetyl-β-cellobiosyl)-diazoacetamide
To a solution of hepta-O-acetyl-β-cellobiosylamine (857 mg, 1.35 mmol, 1.0 equiv.) and glyoxylic acid p-toluenesulfonylhydrazone5,6 (360 mg, 1.49 mmol, 1.1 equiv.) in ice-cold THF (10 mL) was added, dropwise, DCC (306 mg, 1.49 mmol, 1.1 equiv.) in THF (10 mL). The mixture was allowed to warm to room temperature and stirring was continued over night. Then the solid was filtered off. After removal of the solvent, the filtrate was purified by flash chromatography (hexanes/EtOAc, 2:3) to afford a yellow solid (735 mg, 63% yield)
To a solution of the yellow solid (739 mg, 0.86 mmol, 1.0 equiv.) in methylene chloride (10 mL) was added triethylamine (1.72 mmol, 2.0 equiv.) in a nitrogen atmosphere. The mixture was stirred at room temperature over night. Then the solution was diluted with ethyl acetate (20 mL) and washed by water (2 × 10 mL), brine (2 × 10 mL), dried over sodium sulfate and concentrated. The remaining residue was then purified by flash chromatography (hexanes/EtOAc, 2:3) to afford the diazo compound (560 mg, 93% yield) as a yellow solid. 1H NMR: δ 5.98 (d, J = 9.5 Hz, 1H), 5.22–5.26 (m, 2H), 5.12 (t, J = 9.5 Hz, 1H), 5.05 (t, J = 9.5 Hz, 1H), 4.89 (t, J = 9.5 Hz, 1H), 4.79 (t, J = 9.5 Hz, 1H), 4.78 (s, 1H), 4.48 (dd, J = 12.5, 4.5 Hz, 1H), 4.42 (dd, J = 12.5, 4.5 Hz, 1H), 4.34 (dd, J = 12.5, 4.5 Hz, 1H), 4.07–4.12 (m, 2H), 4.01 (dd, J = 12.5, 2.0 Hz, 1H), 3.72–3.76 (m, 2H), 3.62–3.65 (m, 1H), 1.95–2.12 (m, 21H). 13C NMR: δ 171.1, 170.5, 170.3, 170.2, 169.5, 169.3, 169.1, 166.1, 100.6, 78.3, 76.2, 74.5, 72.9, 72.5, 71.8, 71.5, 70.7, 67.8, 62.0, 61.5, 60.4, 47.9, 20.7. ν 2111, 1751, 1654 cm−1. ESIHRMS Calcd for C28H37N3O18Na [M+Na]+: 726.1970, found: 726.1981.
General Procedure for Ylide Formation and Rearrangement
To a solution of allyl sulfide compound (0.1 mmol, 1.0 equiv.) in 1,2-dimethoxyethane (5 mL), Rh2(OAc)4 (0.005 mmol, 0.05 equiv.) was added, followed by addition of diazo compound in a nitrogen atmosphere. The reaction mixture was stirred vigorously at room temperature for 10 h. Then another portion of diazo compound was added, and vigorous stirring was continued at room temperature for additional 12h. The solvent was removed by evaporation, and the remaining residue was purified by flash chromatography to afford the corresponding product.
N-(tert-Butoxycarbonyl)-S-(4-ethoxycarbonyl-1-buten-4-yl)-L-cysteine ethyl ester (3) was prepared according to the general procedure using 10 equiv. of ethyl diazoacetate. Colorless oil; 1H NMR: δ 5.72–5.77 (m, 2H), 5.37 (br. d, J = 9.0 Hz, 1H), 5.29 (br. d, J = 8.0 Hz, 1H), 5.06–5.13 (m, 4H), 4.54–4.55 (br. s, 2H), 4.16–4.23 (m, 8H), 3.40 (t, J = 7.0 Hz, 1H), 3.32 (dd, J = 8.5, 6.5 Hz, 1H), 3.03–3.14 (m, 3H), 2.91–2.95 (m, 1H), 2.56–2.59 (m, 2H), 2.38–2.43 (m, 2H), 1.44 (s, 9H), 1.43 (s, 9H), 1.27–1.29 (m, 12H). 13C NMR: δ171.8, 170.8, 155.1, 133.9, 133.8, 118.0, 80.1, 61.8, 61.7, 61.4, 61.3, 53.4, 53.0, 46.7, 46.2, 35.8, 35.5, 33.9, 28.3, 14.2. ESIHRMS Calcd for C17H29NO6S [M+Na]+: 398.1614, found: 398.1607.
N-(tert-Butoxycarbonyl)-S-(4-ethoxycarbonyl-2-methyl-1-buten-4-yl)-L-cysteine ethyl ester (4) was prepared according to the general procedure using 10 equiv. of ethyl diazoacetate. Colorless oil; 1H NMR: δ 5.35 (br. d, J = 8.1 Hz, 1H), 5.28 (br. d, J = 7.8 Hz, 1H), 4.81 (s, 2H), 4.74 (d, J = 7.5, 2H), 4.55 (br. s, 2H), 4.15–4.23 (m, 8H), 3.47–3.59 (m, 2H), 3.06–3.15 (m, 4H), 2.92–2.95 (m, 2H), 2.56–2.61 (m, 2H), 2.31–2.35 (m, 2H), 1.74 (s, 3H), 1.73 (s, 3H), 1.45 (s, 9H), 1.44 (s, 9H), 1.29–1.30 (m, 12H). 13C NMR: δ 172.0, 170.8, 155.2, 141.5, 141.4, 113.1, 80.1, 61.8, 61.4, 61.3, 53.4, 53.0, 45.3, 44.9, 39.6, 39.4, 33.8, 28.3, 22.3, 14.2. ESIHRMS Calcd for C18H31NO6S [M+H]+: 390.1950, found: 390.1944.
N-(tert-Butoxycarbonyl)-S-(4-trimethylsilanyl-1-buten-4-yl)-L-cysteine ethyl ester (5) was prepared according to the general procedure using 5.0 equiv. of trimethylsilyl diazomethane. Colorless oil; 1H NMR: δ 5.89–5.91 (m, 2H), 5.35 (br. d, J = 8.0 Hz, 1H), 5.29 (br. s, 1H), 5.02–5.11 (m, 4H), 4.48 (br. s, 2H), 4.17–4.22 (m, 4H), 2.86–3.03 (m, 6H), 2.48–2.50 (m, 2H), 2.29–2.32 (m, 2H), 1.92–1.93 (m, 2H), 1.46 (s, 9H), 1.44 (s, 9H), 1.26–1.29 (m, 6H), 0.15 (s, 9H), 0.14 (s, 9H). 13C NMR: δ 171.1, 155.2, 137.3, 116.4, 116.3, 80.0, 61.7, 61.6, 53.3, 53.2, 38.5, 36.2, 32.6, 32.0, 28.3, 14.2, −2.3. ESIHRMS Calcd for C17H33NO4SSi [M+Na]+: 398.1798, found: 398.1793.
N-(tert-Butoxycarbonyl)-S-(2-methyl-5-oxo-4-icosyl)-L-cysteine ethyl ester (6) was prepared according to the general procedure using 5.0 equiv. of 1-diazo-2-heptadecanone. Pale yellow oil; 1H NMR: δ 5.27–5.30 (m, 2H) 4.81 (s, 2H), 4.69 (s, 2H), 4.49 (br. s, 2H), 4.21 (q, J = 7.0 Hz, 4H), 3.45–3.48 (m, 2H), 2.81–2.97 (m, 4H), 2.52–2.58 (m, 6H), 2.32–2.36 (m, 2H), 1.71 (s, 6H), 1.44 (s, 18H), 1.25–1.37 (m, 58 H), 0.87 (t, J = 11.0 Hz, 6H). 13C NMR: δ 206.3, 206.2, 170.8, 155.1, 141.6, 113.1, 80.2, 61.9, 53.2, 53.0, 51.0, 38.8, 38.1, 32.5, 29.7, 29.6, 29.4, 29.3, 29.2, 28.3, 23.9, 22.7, 22.5, 14.2. ESIHRMS Calcd for C31H57NO5S [M+H]+: 556.4036, found: 556.4027.
N-(tert-Butoxycarbonyl)-S-(4-ethoxycarbonyl-1-buten-4-yl)-glutathione dimethyl ester (8) was prepared according to the general procedure using 10 equiv. of ethyl diazoacetate. Pale yellow oil; 1H NMR: δ 7.16 (br. d, J = 5.0 Hz, 2H), 6.99 (br. d, J = 6.5 Hz, 1H), 6.89 (br. d, J = 6.0 Hz, 1H), 5.72–5.75 (m, 2H), 5.35 (br. d, J = 5.5 Hz, 2H), 5.09–5.14 (m, 4H), 4.62–4.67 (m, 2H), 4.32–4.38 (m, 2H), 4.18–4.24 (m, 4H), 4.01–4.09 (m, 4H), 3.74 (s, 6H), 3.73 (s, 6H), 3.52 (t, J = 7.3 Hz, 1H), 3.44 (t, J = 7.5 Hz, 1H), 3.19 (dd, J = 14.3, 6.3 Hz, 2H), 2.94–3.02 (m, 2H), 2.59–2.65 (m,2H), 2.44–2.48 (m, 2H), 2.32–2.38 (m, 4H), 2.16–2.21 (m, 2H), 1.93–2.01 (m, 2H), 1.43 (s, 18H), 1.24–1.29 (m, 6H). 13C NMR: δ 172.9, 172.6, 172.3, 170.4, 169.9, 155.7, 133.8, 118.1, 80.2, 61.7, 52.6, 47.2, 41.3, 35.7, 33.5, 33.0, 32.1, 28.6, 14.2. ESIHRMS Calcd for C24H39N3O10S [M+Na]+: 584.2254, found: 584.2255.
N-(tert-Butoxycarbonyl)-S-(4-phenyl-1-buten-4-yl)-glutathione dimethyl ester (9) was prepared according to the general procedure using 5.0 equiv. of phenyl diazomethane. Pale yellow oil; 1H NMR: δ7.31–7.32 (m, 4H), 7.22–7.25 (m, 6H), 7.02 (br. s, 1 H), 6.82 (br. s, 1H), 6.67 (br. d, J = 6.5 Hz, 1H) 6.60 (br. d, J = 7.0 Hz, 1H), 5.63–5.70 (m, 2H), 5.37 (br. s, 2H), 4.95–5.04 (m, 4H), 4.32–4.45 (m, 4H), 3.86–3.98 (m, 8H), 3.73 (s, 6H), 3.72 (s, 6H), 2.83 (dd, J = 12.0, 5.8 Hz, 1H), 2.69–2.71 (m, 2H), 2.58–2.64 (m, 5H), 2.27–2.31 (m, 2H), 2.21–2.24 (m, 2H), 2.11–2.18 (m, 2H), 1.89–1.96 (m, 2H), 1.42 (s, 9H), 1.41 (s, 9H). 13C NMR (125 MHz, CDCl3) δ172.9, 172.2, 172.1, 170.6, 170.0,169.9, 155.7, 142.0, 141.9, 135.1, 135.0, 128.7, 127.9, 127.5, 117.3, 80.2, 52.8, 52.5, 52.4, 52.2, 50.2, 50.0, 49.9, 41.3, 40.7, 40.6, 32.8, 32.2, 28.7, 28.5, 28.3. ESIHRMS Calcd for C27H39N3O8S [M+Na]+: 588.2356, found: 588.2364.
N-(tert-Butoxycarbonyl)-S-(5-oxo-4-icosyl)-glutathione dimeth-yl ester (10) was prepared according to the general procedure using 5.0 equiv. of 1-diazo-2-heptadecanone. Pale yellow oil; 1H NMR: δ 7.11–7.13 (br. s, 2H), 6.81 (br. d, J = 6.0 Hz, 1H), 6.80 (br. d, J = 6.5 Hz, 1H), 5.69–5.77 (m, 2H), 5.34–5.35 (br. d, J = 7.0 Hz, 2H), 5.07–5.09 (m, 4H), 4.55–4.59 (m, 2H), 4.36 (br. s, 2H), 3.95–4.09 (m, 4H), 3.74 (s, 12H), 3.44–3.49 (m, 2H), 2.80–2.90 (m, 4H), 2.55–2.62 (m, 6H), 2.32–2.46 (m, 6H), 2.14–2.17 (m, 2H), 1.91–1.97 (m, 2H), 1.51–1.57 (m, 4H), 1.42 (s, 18H), 1.16–1.28 (m, 48H), 0.85–0.88 (t, J = 7.0 Hz, 6H). 13C NMR: δ 207.7, 207.3, 172.9, 172.3, 172.2, 170.4, 170.3, 169.9, 155.7, 134.2, 134.1, 118.1, 118.0, 80.2, 52.8, 52.7, 52.6, 52.5, 52.4, 52.2, 41.3, 39.7, 39.6, 34.5, 32.4, 32.2, 32.1,31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 28.7, 28.5, 28.3, 23.9, 22.7, 14.1. ESIHRMS Calcd for C37H65N3O9S [M+Na]+: 750.4340, found: 750.4322.
N-(tert-Butoxycarbonyl)-S-(1-tetradecen-4-yl)-glutathione dimethyl ester (11) was prepared according to the general procedure using 5.0 equiv. of 1-diazo-undecane. Pale yellow oil; 1H NMR: δ 7.16 (br. d, J = 5.0 Hz, 2H), 6.84 (br. d, J = 7.0 Hz, 2H), 5.81–5.84 (m, 2H), 5.33 (br. d, J = 6.5 Hz, 2H), 5.05–5.10 (m, 4H), 4.52–4.53 (m, 2H), 4.39 (br. d, J = 4.5 Hz, 2H), 3.98–4.05 (m, 4H), 3.74 (s, 12H), 2.97–2.98 (m, 2H), 2.76–2.79 (m, 4H), 2.34–2.38 (m, 8H), 2.11–2.19 (m, 2H), 1.85–1.96 (m, 2H), 1.51–1.54 (m, 2H), 1.43 (s, 18H), 1.24–1.39 (m, 34H), 0.85–0.88 (m, 6H). 13C NMR: δ 172.9, 172.1, 170.7, 169.9, 155.7, 135.6, 135.5, 117.3, 117.2, 80.2, 52.8, 52.7, 52.5, 52.4, 46.3, 45.9, 41.3, 39.1, 34.6, 34.2, 32.3, 32.1, 31.9, 29.6, 29.5, 29.4, 28.6, 28.3, 26.8, 26.7, 22.7, 14.1. ESIHRMS Calcd for C31H55N3O8S [M+H]+: 630.3788, found: 630.3773.
N-(tert-Butoxycarbonyl)-S-(1-tridecafluorododecen-4-yl)-glutathione dimethyl ester (12) was prepared according to the general procedure using 5.0 equiv. of 9-diazo-1,1,1,2,2,3,3,4,4,5,-5,6,6-tridecafluorononane. Pale yellow oil; 1H NMR: δ 7.11 (br. s, 2H), 6.88 (br. s, 2H), 5.74–5.77 (m, 2H), 5.45 (br. s, 2H), 5.33–5.36 (m, 2H), 5.12–5.18 (m, 4H), 4.57–4.58 (m, 1H), 4.31–4.42 (m, 2H), 3.98–4.13 (m, 4H), 3.75 (s, 12H), 3.29–3.32 (m, 2H), 3.04–3.23 (m, 4H), 2.74–2.95 (m, 4H), 2.33–2.51 (m, 4H), 2.17–2.20 (m, 2H), 1.89–2.16 (m, 4H), 1.44 (s, 9H), 1.45 (s, 9H). 13C NMR: δ 172.8, 171.9, 171.4, 169.9, 155.6, 133.4, 133.2, 118.4, 118.2, 80.7, 52.9, 52.7, 52.5, 45.8, 41.4, 39.5, 34.9, 34.3, 32.4, 32.2, 31.7, 29.7, 29.3, 28.8, 28.3,28.2, 27.8, 24.6. F19 NMR: δ −8.3, −41.7, −49.5, −50.5, −50.9, −53.7. ESIHRMS Calcd for C29H38F13N3O8S [M+H]+:836.2245, found: 836.2247.
N-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-(2-hexadecylsulfanyl)-4-pentenamide (16) was prepared according to the general procedure using 5.0 equiv. of N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-diazoacetamide. Colorless oil; 1H NMR: δ 7.21–7.30 (m, 2H), 5.66–5.79 (m, 2H), 5.30 (t, J = 9.5 Hz, 2H), 5.19–5.23 (m, 2H), 5.01–5.33 (m, 6H), 4.96–5.00 (m, 2H), 4.28–4.32 (m, 2H), 4.05–4.08 (m, 2H), 3.79–3.82 (m, 2H), 3.25–3.28 (m, 2H), 2.38–2.49 (m, 6H), 2.17 (s, 6H), 2.07 (s, 6H), 2.03 (s, 6H), 2.02 (s, 6H), 1.52–1.55 (m, 4H), 1.24–1.33 (m, 54H), 0.87 (t, J = 7.0 Hz, 6H). 13C NMR: δ 172.5, 172.4, 170.6, 170.4, 170.0, 169.6, 133.9, 133.8, 118.2, 117.9, 78.4, 73.6, 72.9, 72.8, 70.3, 68.2, 61.7, 49.5, 49.4, 36.5, 36.2, 31.9, 31.5, 31.4, 29.7, 29.6, 29.5, 29.4, 29.2, 29.0, 28.9, 22.7, 20.7, 20.6, 14.2. ESIHRMS Calcd for C35H59N3O10S [M+Na]+: 708.3758, found: 708.3732.
N-(tert-Butoxycarbonyl)-S-(4-N-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)aminocarbonyl-1-buten-4-yl)-L-cysteine ethyl ester (17) was prepared according to the general procedure using 5.0 equiv. of N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)diazoacetamide. Pale yellow oil; 1H NMR: δ 7.41 (br. d, J = 8.5 Hz, 1H), 7.27 (br. s, 1H), 5.55–5.74 (m, 2H), 5.64–5.74 (m, 1H), 5.50 (br. d, J = 7.5 Hz, 1H), 5.19–5.30 (m, 4H), 4.98–5.12 (m, 6H), 4.43 (br, 2H), 4.25–4.30 (m, 2H), 4.15–4.21 (m, 4H), 4.02–4.11 (m, 4H), 3.77–3.81 (m, 2H), 3.30–3.33 (m, 2H), 3.09 (br. d, J = 13.0 Hz, 1H), 2.94–2.97 (m, 1H), 2.68–2.74 (m, 2H), 2.60–2.65 (m, 1H), 2.48–2.54 (m, 1H), 2.33–2.40 (m, 2H), 1.98–2.05 (m, 24H), 1.47 (s, 9H), 1.46 (s, 9H), 1.21–1.27 (m, 6H). 13C NMR: δ 171.8, 171.6, 171.2, 170.7, 170.6, 170.4, 169.9, 169.5, 169.4, 155.5, 155.4, 133.9, 133.4, 118.2, 118.1, 80.5, 80.4, 73.6, 72.9, 72.7, 70.3, 68.1, 68.0, 62.0, 61.9, 61.7, 61.5, 60.4, 53.0, 49.8, 48.0, 36.2, 35.8, 34.2, 33.7, 28.4, 21.1, 20.7, 20.6, 14.2. ESIHRMS Calcd for C29H44N2O14S [M+Na]+: 699.2406, found: 699.2410.
N-(tert-Butoxycarbonyl)-S-(4-N-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-cellobiosyl)aminocarbonyl-1-buten-4-yl)-L-cysteine ethyl ester (18) was prepared according to the general procedure using 5.0 equiv. of N-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-cellobiosyl)diazoacetamide. Pale yellow oil; 1H NMR: δ 7.26 (br. d, J = 9.0 Hz, 1H), 7.13 (br. d, J = 9.0 Hz, 1H), 5.59–5.61 (m, 2H), 5.46 (br. d, J = 6.6 Hz, 1H), 5.21–5.28 (m, 3H), 5.03–5.19 (m, 10H), 4.88–4.92 (m, 4H), 4.40–4.49 (m, 6H), 4.33–4.35 (m, 2H), 4.08–4.19 (m, 8H), 3.98–4.01 (m, 2H), 3.62–3.71 (m, 6H), 3.26–3.31 (m, 2H), 3.03–3.08 (m, 1H), 2.91–2.96 (m, 1H), 2.68–2.73 (m, 1H), 2.59–2.66 (m, 1H), 2.45–2.51 (m, 1H), 2.31–2.39 (m, 2H), 1.95–2.09 (m, 42H), 1.45 (s, 9H), 1.42 (s, 9H), 1.21–1.27 (m, 6H). 13C NMR: δ 171.6, 171.4, 170.8, 170.7, 170.6, 170.5, 170.2, 169.4, 169.3, 169.0, 155.3, 133.7, 133.4, 118.2, 118.1, 100.7, 80.378.3, 78.2, 76.2, 74.5, 72.9, 72.2, 72.1, 72.0, 71.5, 70.5, 67.8, 62.0, 61.9, 61.6, 60.4, 53.5, 53.1, 49.8, 48.4, 36.2, 35.8, 34.1, 33.5, 28.4, 21.1, 20.8, 20.7, 20.5, 14.2. ESIHRMS Calcd for C41H60N2O22S [M+H]+: 965.3431, found: 965.3424.
N-(tert-Butoxycarbonyl)-S-(4-N-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)aminocarbonyl-1-buten-4-yl)-glutathione dimethyl ester (19) was prepared according to the general procedure using 5.0 equiv. of N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-diazoacetamide. Pale yellow oil; 1H NMR: δ 7.69 (br. d, J = 8.5 Hz, 2H), 7.35 (br. s, 2H), 7.01 (br. d, J = 7.5 Hz, 2H), 5.73–5.76 (m, 2H), 5.35 (br. s, 2H), 5.26–5.31 (m, 4H), 4.99–5.14 (m, 6H), 4.82 (br. s, 2H), 4.32–4.40 (m, 4H), 4.03–4.14 (m, 4H), 3.82–3.87 (m, 2H), 3.73–3.79 (m, 12H), 3.63 (br. s, 2H), 2.96–3.31 (m, 4H), 2.62–2.64 (m, 2H), 2.52–2.54 (m, 2H), 2.35–2.37 (m, 4H), 2.00–2.19 (m, 28H), 1.43 (s, 9H), 1.42 (s, 9H). 13C NMR: δ 172.9, 172.3, 172.1, 170.8, 170.6, 170.3, 170.0, 169.6, 155.6, 133.9, 133.8, 118.2, 80.2, 78.3, 73.9, 73.1, 70.6, 70.5, 68.2, 68.1, 61.7, 60.4, 52.8, 52.6, 52.1, 52.0, 49.2, 45.4, 41.3, 41.2, 36.0, 35.2, 33.8, 32.6, 31.9, 28.3, 24.0, 21.0, 20.8. ESIHRMS Calcd for C36H54N4O18S [M+H]+: 885.3046, found: 885.3059.
N-(tert-Butoxycarbonyl)-S-(4-N-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-cellobiosyl)aminocarbonyl-1-buten-4-yl)-glutathione dimethyl ester (20) was prepared according to the general procedure using 5.0 equiv. of N-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-cellobiosyl)diazoacetamide. Pale yellow oil; 1H NMR: δ 7.55 (br. d, J = 9.1 Hz, 1H), 7.28 (br. d, J = 7.0 Hz, 1H), 6.98 (br. d, J = 8.1 Hz, 1H), 5.62–5.71 (m, 1H), 5.33 (br. d, J = 8.1 Hz, 1H), 5.20–5.27 (m, 4H), 5.10–5.14 (m, 3H), 5.03–5.07 (m, 4H), 4.89–4.92 (m, 4H), 4.61–4.68 (br. s, 1H), 4.48–4.52 (m, 5H), 4.34–4.37 (m, 2H), 4.25 (br. s, 1H), 4.03–4.12 (m, 8H), 3.71–3.76 (m, 10H), 3.64–3.66 (m, 3H), 3.12 (br. s, 1H), 3.01 (br. s, 1H), 2.52–2.61 (m, 1H), 2.50–2.51 (m, 1H), 2.35–2.37 (m, 2H), 1.98–2.13 (m, 21H), 1.42 (s, 9H). 13C NMR: δ 173.0, 172.3, 171.5, 170.6, 170.5, 170.3, 170.2, 169.9, 169.8, 169.3, 169.0, 155.6, 133.9, 133.8, 118.0, 100.6, 80.3, 78.1, 76.4, 74.9, 73.2, 73.1, 71.9, 71.8, 71.6, 70.5, 67.8, 61.8, 61.7, 61.5, 52.9, 52.6, 52.5, 52.4, 46.4, 41.4, 35.1, 32.8, 32.0, 28.3, 20.8. ESIHRMS Calcd for C48H70N4O26S [M+H]+: 1151.4072, found: 1151.4071.
N-(tert-Butoxycarbonyl)-S-(2-methyl-5-oxo-4-icosyl)-L-cysteinyl-L-alanyl-L-tryptophan methyl ester (21) was prepared according to the general procedure using 5.0 equiv. of 1-diazo-2-heptadecanone, which was contaminated by approximately 10% of 22. 1H NMR: (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.59 (s, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.34 (m, 2H) 7.16 (m, 2H), 7.09 (m, 2H), 7.00 (s, 1H), 6.98 (s, 1H), 6.80–6.67 (m, 4H), 5.28 (m, 1H), 5.15 (m, 1H), 4.89–4.80 (m, 4H), 4.73 (s, 1H), 4.70 (s, 1H), 4.47-4.37 (m, 2H), 4.20-4.10 (m, 2H), 3.694 (s, 3H), 3.692 (s, 3H), 3.53 (m, 2H), 3.37-3.23 (m, 4H), 2.74-2.73 (m, 4H), 2.62-2.50 (m, 6H), 2.38-2.31 (m, 2H), 1.74 (s, 3H), 1.73 (s, 3H,), 1.63-1.56 (m, 4H), 1.47 (s, 18H), 1.31 (d, J = 7.1 Hz, 6H), 1.30-1.20 (m, 52H), 0.88 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 207.6, 207.4, 172.04, 171.99, 171.1, 170.13, 170.07, 155.6, 141.6, 141.5, 136.3, 127.5, 127.4, 123.23, 123.19, 122.06, 122.04, 113.3, 111.38, 111.33, 109.5, 79.8, 54.0, 53.4, 53.1, 52.7, 52.4, 50.9, 50.8, 48.9, 39.1, 39.0, 38.0, 37.9, 32.4, 32.2, 31.9, 29.68, 29.65, 29.5, 29.4, 29.3, 29.2, 28.3, 27.4, 27.3, 23.9, 22.7, 22.4, 17.5, 17.4, 14.1. ESIHRMS Calcd for C44H71N4O7S [M+H]+: 799.5038, found: 799.5038; Calcd for C44H70N4O7SNa [M+Na]+: 821.4858, found: 821.4560.
N-(tert-Butoxycarbonyl)-S-(2-methylallyl)-L-cysteinyl-L-alanyl-L-Nω-(2-oxo-heptadecyl)-tryptophan methyl ester (22) was prepared according to the general procedure using 5.0 equiv. of 1-diazo-2-heptadecanone. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 7.7 Hz, 1H), 7.20 (m, 1H), 7.14-7.10 (m, 2H), 6.90 (s, 1H), 6.84 (br. d, J = 7.7 Hz, 1H), 6.59 (br. d, J = 7.6 Hz, 1H), 5.29 (m, 1H), 4.93-4.79 (m, 4H), 4.45 (quint, J = 7.4 Hz, 1H), 4.14 (m, 1H), 3.68 (s, 3H), 3.33 (d, J = 5.6 Hz, 2H), 3.03 (d, J = 13.4 Hz, 1H), 2.96 (d, J = 13.4 Hz, 1H), 2.72 (dd, J = 13.7, 5.9 Hz, 1H), 2.65 (dd, J = 13.7, 6.7 Hz, 1H), 2.35 (t, J = 7.4 Hz, 2H), 1.77 (s, 3H), 1.56 (m, 2H), 1.44 (s, 9H), 1.32 (d, J = 7.1 Hz, 3H), 1.30-1.20 (m, 26H), 0.88 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 206.4, 171.8, 171.3, 170.4, 140.7, 136.6, 128.1, 127.6, 122.4, 119.8, 119.0, 114.5, 109.6, 108.9, 80.4, 55.2, 53.8, 52.6, 52.5, 48.9, 39.4, 33.1, 31.9, 29.7, 29.5, 29.4, 29.1, 28.3, 27.3, 23.4, 22.7, 20.6, 18.0, 14.1. FABHRMS Calcd for C44H70N4O7SNa [M+Na]+: 821.4863, found: 821.4859.
Supplementary Material
Copies of NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.

Preparation of diazo amides 13 and 14
Acknowledgments
We thank the NIH (GM 62160) for partial support of this work.
References
- 1.(a) Crich D, Krishnamurthy V, Hutton TK. J Am Chem Soc. 2006;128:2544. doi: 10.1021/ja057521c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crich D, Brebion F, Krishnamurthy V. Org Lett. 2006;8:3593. doi: 10.1021/ol061381+. [DOI] [PubMed] [Google Scholar]
- 2.(a) Trost BM, Melvin SM. Sulfur Ylides. Academic Press; New York: 1975. [Google Scholar]; (b) Vedejs E. Acc Chem Res. 1984;17:358. [Google Scholar]
- 3.(a) Kirmse W, Kapps M. Chem Ber. 1968;101:994. [Google Scholar]; (b) Doyle MP, Tamblyn WH, Bagheri V. J Org Chem. 1981;46:4094. [Google Scholar]; (c) Doyle MP, Griffin JH, Chinn MS, van Leusen D. J Org Chem. 1984;49:1917. [Google Scholar]; (d) Doyle MP, Tamblyn WH, Bagheri V. J Org Chem. 1981;46:5094. [Google Scholar]; (e) Ando W, Yagihara T, Kondo S, Nakayama K, Yamato H, Nakaido S, Migita T. J Org Chem. 1971;36:1732. [Google Scholar]; (f) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo compounds. Wiley; New York: 1998. [Google Scholar]; (g) Aggarwal V, Richardson J. In: Science of Synthesis: Compounds with Two Carbon-Heteroatom Bonds. Padwa A, editor. Vol. 27. Thieme; Stuttgart: 2004. p. 21. [Google Scholar]; (h) Li AH, Dai LX, Aggarwal VK. Chem Rev. 1997;97:2341. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]; (i) Doyle MP, Forbes DC. Chem Rev. 1998;98:911. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; (j) Hodgson DM, Pierard FYTM, Stupple PA. Chem Soc Rev. 2001;30:50. [Google Scholar]
- 4.(a) McFarland JM, Francis MF. J Am Chem Soc. 2005;127:13490. doi: 10.1021/ja054686c. [DOI] [PubMed] [Google Scholar]; (b) Tilley DS, Francis MB. J Am Chem Soc. 2006;128:1080. doi: 10.1021/ja057106k. [DOI] [PubMed] [Google Scholar]; (c) van Maarseveen JH, Reek JNH, Back JW. Angew Chem Int Ed. 2006;45:1841. doi: 10.1002/anie.200504352. [DOI] [PubMed] [Google Scholar]; (d) Dibowski H, Schmidtchen FP. Angew Chem Int Ed. 1998;37:476. doi: 10.1002/(SICI)1521-3773(19980302)37:4<476::AID-ANIE476>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (e) Antos JM, Francis MB. Curr Op Chem Biol. 2006;10:253. doi: 10.1016/j.cbpa.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Antos JM, Francis MF. J Am Chem Soc. 2004;126:10256. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Carter DS, Van Vranken DL. Tetrahedron Lett. 1999;40:1617. [Google Scholar]; (b) Aggarwal VK, Ferrara M, Hainz R, Spey SE. Tetrahedron Lett. 1999;40:8923. [Google Scholar]
- 7.(a) Pachamuthu K, Schmidt RR. Chem Rev. 2006;106:160. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]; (b) Clarke S. Ann Rev Biochem. 1992;61:355. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]; (c) Schelhaas M, Nägele E, Kuder N, Bader B, Kuhlmann J, Wittinghofer A, Waldmann H. Chem Eur J. 1999;5:1239. [Google Scholar]; (d) Schelhaas M, Glomsda S, Hänsler M, Jakubke HD, Waldmann H. Angew Chem Int Ed. 1996;35:106. [Google Scholar]; (e) Brown MJ, Milano PD, Lever DC, Epstein WW, Poulter CD. J Am Chem Soc. 1991;113:3176. [Google Scholar]; (f) Pachamuthu K, Zhu X, Schmidt RR. J Org Chem. 2005;70:3720. doi: 10.1021/jo0482357. [DOI] [PubMed] [Google Scholar]
- 8.Brittain SM, Ficarro SB, Brock A, Peters EC. Nature Biotechnology. 2005;23:463. doi: 10.1038/nbt1076. [DOI] [PubMed] [Google Scholar]
- 9.(a) Gamblin DP, Garnier P, van Kasteren S, Oldham NJ, Fairbanks AJ, Davis BG. Angew Chem Int Ed. 2004;43:828. doi: 10.1002/anie.200352975. [DOI] [PubMed] [Google Scholar]; (b) Gamblin DP, Garnier SJ, Ward NJ, Oldham AJ, Fairbanks AJ, Davis BG. Org Biomol Chem. 2003;1:3642. doi: 10.1039/b306990g. [DOI] [PubMed] [Google Scholar]; (c) Doores KJ, Gamblin DP, Davis BG. Chem Eur J. 2006;12:656. doi: 10.1002/chem.200500557. [DOI] [PubMed] [Google Scholar]; (d) Davis BG. Chem Rev. 2002;102:579. doi: 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]; (e) Peri F, Nicotra F. Chem Comm. 2004:623. doi: 10.1039/b308907j. [DOI] [PubMed] [Google Scholar]; (f) Ohnishi Y, Ichikawa M, Ichikawa Y. Biorg Med Chem Lett. 2000;10:1289. doi: 10.1016/s0960-894x(00)00223-7. [DOI] [PubMed] [Google Scholar]; (g) Jobron L, Hummel G. Org Lett. 2000;2:2265. doi: 10.1021/ol006019o. [DOI] [PubMed] [Google Scholar]; (h) Cohen SB, Halcomb RL. Org Lett. 2001;3:405. doi: 10.1021/ol006908b. [DOI] [PubMed] [Google Scholar]; (i) Knapp S, Myers DS. J Org Chem. 2002;67:2995. doi: 10.1021/jo0110909. [DOI] [PubMed] [Google Scholar]; (j) Zhu XM, Pachamuthu K, Schmidt RR. Org Lett. 2004;6:1083. doi: 10.1021/ol036186z. [DOI] [PubMed] [Google Scholar]; (k) Zhu XM, Schmidt RR. Chem Eur J. 2004;10:875. doi: 10.1002/chem.200305163. [DOI] [PubMed] [Google Scholar]; (l) Thayer DA, Yu HN, Galan MC, Wong CH. Angew Chem Int Ed. 2005;44:4596. doi: 10.1002/anie.200500090. [DOI] [PubMed] [Google Scholar]; (m) Bertozzi CR, Kiessling LL. Science. 2001;291:2357. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]; (n) Galonic DP, Ide ND, van der Donk W, Gin DY. J Am Chem Soc. 2005;127:7359. doi: 10.1021/ja050304r. [DOI] [PubMed] [Google Scholar]
- 10.(a) Roy R. In: Carbohydrate Chemistry. Boons G-J, editor. Blackie; London: 1998. p. 243. [Google Scholar]; (b) Lee YT, Lee RC, editors. Neoglycoconjugates: Preparation and Applications. Academic; San Diego: 1994. [Google Scholar]; (c) Ichikawa Y, Matsukawa Y, Isobe M. J Am Chem Soc. 2006;128:3934. doi: 10.1021/ja056253f. [DOI] [PubMed] [Google Scholar]; (d) Ladmiral V, Mantovani G, Clarkson GJ, Cauet S, Irwin JL, Haddleton DM. J Am Chem Soc. 2006;128:4823. doi: 10.1021/ja058364k. [DOI] [PubMed] [Google Scholar]; (e) Hotha S, Kashyap S. J Org Chem. 2006;71:364. doi: 10.1021/jo051731q. [DOI] [PubMed] [Google Scholar]
- 11.Dawson PE, Kent SBH. Ann Rev Biochem. 2000;69:923. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 12.(a) Dwek RA, Butters TD. Chem Rev. 2002;102:283. [Google Scholar]; (b) Varki A. Glycobiology. 1993;3:97. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lehmann J. Carbohydrates: Structure and Biology. Thieme; Stuttgart: 1998. [Google Scholar]
- 13.(a) He Y, Hinklin RJ, Chang J, Kiessling L. Org Lett. 2004;6:4479. doi: 10.1021/ol048271s. [DOI] [PubMed] [Google Scholar]; (b) Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J Org Chem. 2005;70:7123. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]; (c) Wen S, Guo Z. Org Lett. 2001;3:3773. doi: 10.1021/ol0101988. [DOI] [PubMed] [Google Scholar]; (d) Mizuno M, Goto K, Miura T. Chem Lett. 2005;34:426. [Google Scholar]; (e) Kaneshiro CM, Michael K. Angew Chem Int Ed. 2006;45:1077. doi: 10.1002/anie.200502687. [DOI] [PubMed] [Google Scholar]; (f) Mandal M, Dudkin VY, Geng X, Danishefsky SJ. Angew Chem Int Ed. 2004;43:2557. doi: 10.1002/anie.200353625. [DOI] [PubMed] [Google Scholar]; (g) Lin H, Walsh CT. J Am Chem Soc. 2004;126:13998. doi: 10.1021/ja045147v. [DOI] [PubMed] [Google Scholar]; (h) Tanaka H, Iwata Y, Takahashi D, Adachi M, Takahashi T. J Am Chem Soc. 2005;127:1630. doi: 10.1021/ja0450298. [DOI] [PubMed] [Google Scholar]; (i) Doores KJ, Mimura Y, Dwek RA, Rudd PM, Elliott T, Davis BG. Chem Commun. 2006:1401. doi: 10.1039/b515472c. [DOI] [PubMed] [Google Scholar]; (j) Liu H, Wang L, Brock A, Wong CH, Schulz PG. J Am Chem Soc. 2003;125:1702. doi: 10.1021/ja029433n. [DOI] [PubMed] [Google Scholar]; (k) Brik A, Yang YY, Ficht S, Wong CH. J Am Chem Soc. 2006;128:5626. doi: 10.1021/ja061165w. [DOI] [PubMed] [Google Scholar]; (l) Hang HC, Bertozzi CR. Acc Chem Res. 2001;34:727. doi: 10.1021/ar9901570. [DOI] [PubMed] [Google Scholar]
- 14.Nyong AM, Rainier JD. J Org Chem. 2005;70:746. doi: 10.1021/jo0482413. [DOI] [PubMed] [Google Scholar]
- 15.(a) Salim M, Capretta A. Tetrahedron. 2000;56:8063. [Google Scholar]; (b) Gibe R, Kerr MA. J Org Chem. 2002;67:6247. doi: 10.1021/jo025851z. [DOI] [PubMed] [Google Scholar]
- 16.It can be similarly inferred that the reaction of rhodium carbenoids with protein-based tryptophan described by Francis is likely not compatible with the presence of nucleophilic sulfur as in cysteine and methionine.
- 17.Creary X. J Am Chem Soc. 1980;102:1502. [Google Scholar]
- 18.Scott LT, Sumpter CA. Org Synth. 1990;69:180. [Google Scholar]
- 19.Holton TL, Shechter H. J Org Chem. 1995;60:4725. [Google Scholar]
- 20.(a) House HO, Blankley CJ. J Org Chem. 1968;33:53. [Google Scholar]; (b) Ouihia A, Rene L, Badet B. Tetrahedron Lett. 1992;33:5509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.