

Abstract
Recently, we identified a novel signaling pathway involving Epac, Rap, and phospholipase C (PLC)ε that plays a critical role in maximal β-adrenergic receptor (βAR) stimulation of Ca2+-induced Ca2+ release (CICR) in cardiac myocytes. Here we demonstrate that PLCε phosphatidylinositol 4,5-bisphosphate hydrolytic activity and PLCε-stimulated Rap1 GEF activity are both required for PLCε-mediated enhancement of sarcoplasmic reticulum Ca2+ release and that PLCε significantly enhances Rap activation in response to βAR stimulation in the heart. Downstream of PLCε hydrolytic activity, pharmacological inhibition of PKC significantly inhibited both βAR- and Epac-stimulated increases in CICR in PLCε+/+ myocytes but had no effect in PLCε–/– myocytes. βAR and Epac activation caused membrane translocation of PKCε in PLCε+/+ but not PLCε–/– myocytes and small interfering RNA-mediated PKCε knockdown significantly inhibited both βAR and Epac-mediated CICR enhancement. Further downstream, the Ca2+/calmodulin-dependent protein kinase II (CamKII) inhibitor, KN93, inhibited βAR- and Epac-mediated CICR in PLCε+/+ but not PLCε–/– myocytes. Epac activation increased CamKII Thr286 phosphorylation and enhanced phosphorylation at CamKII phosphorylation sites on the ryanodine receptor (RyR2) (Ser2815) and phospholamban (Thr17) in a PKC-dependent manner. Perforated patch clamp experiments revealed that basal and βAR-stimulated peak L-type current density are similar in PLCε+/+ and PLCε–/– myocytes suggesting that control of sarcoplasmic reticulum Ca2+ release, rather than Ca2+ influx through L-type Ca2+ channels, is the target of regulation of a novel signal transduction pathway involving sequential activation of Epac, PLCε, PKCε, and CamKII downstream of βAR activation.
Phospholipase C (PLC)3-mediated hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) results in inositol triphosphate (IP3)-mediated Ca2+ release from intracellular stores and diacylglycerol-mediated activation of protein kinase C. This ubiquitous signaling pathway plays an integral role in regulating many physiological processes, including those of the cardiovascular system. PLCε is a recently identified bifunctional PLC isoform that possesses both PIP2 hydrolytic and Rap guanine nucleotide exchange factor (GEF) activity (1–4). The activity of PLCε is uniquely regulated by direct binding of small G-proteins including Ras, Rap, and Rho (5, 6). PLCε activity is also stimulated by the heterotrimeric G-protein subunits Gαs, Gβγ, and Gα12/13 (5, 7, 8) but direct binding of these subunits to PLCε has not been demonstrated. In primary astrocytes isolated from PLCε+/+ and PLCε–/– mice, multiple G protein-dependent upstream signals rely critically on PLCε-dependent generation of IP3 and diacylglycerol (9).
We recently discovered a surprising role for PLCε regulation downstream of the β-adrenergic receptor (βAR) in cardiac myocytes (10). Compared with normal mice, PLCε–/– mice exhibit reduced left ventricular developed pressure in response to strong βAR stimulation (10). This deficit results from a decrease in isoproterenol (Iso)-dependent stimulation of electrically evoked Ca2+ release from the sarcoplasmic reticulum (SR) in single ventricular cardiac myocytes. βAR stimulation increases cardiac Ca2+ release in a cAMP/protein kinase A (PKA)-dependent mechanism through phosphorylation of multiple targets of the cardiac excitability and Ca2+ handling machinery (11). Recently, we identified a PKA-independent, PLCε-mediated pathway that contributes to maximal Iso-dependent enhancement of Ca2+-induced Ca2+ release (CICR) in cardiac myocytes (12) and explains the decreased βAR function in PLCε–/– mice. This novel pathway requires cAMP-dependent activation of the RapGEF, Epac (13), which subsequently stimulates Rap-dependent activation of PLCε.
Here, we establish a mechanistic link between PLCε activity and CICR by showing that Epac/Rap/PLCε-mediated enhancement of CICR in the heart requires both PLCε-PIP2 hydrolytic and PLCε-RapGEF activities and that downstream of PLCε, both PKCε and CamKII are required for Epac-dependent enhancement of Ca2+ release. In addition, voltage clamp experiments reveal that Iso-dependent activation of the Epac/PLCε pathway in the heart does not significantly alter Ca2+ influx through L-type calcium channels indicating that Ca2+ release from the sarcoplasmic reticulum is the ultimate target of this pathway.
EXPERIMENTAL PROCEDURES
Isolation of Cardiac Myocytes—Adult ventricular cardiomyocytes (AVM) were isolated from male, 4–6-month-old wild type or PLCε–/– mice (C57/B6 background) as previously described (10). Briefly, mice were anesthetized with ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight) by intraperitoneal injection. Hearts were excised and digested by Langendorff perfusion using either Liberase Blendzyme I (Roche) or a mixture of collagenase A and D (Roche). Cells were plated on laminin (BD Biosciences)-coated coverslips or 35-mm tissue culture dishes in minimum essential medium supplemented with 2 mm l-glutamine, 2.5% fetal bovine serum (Hyclone), 1% penicillin/streptomycin, and 2.5 μm blebbistatin (Sigma) to prevent contractile activity.
Transduction of AVM with Adenovirus—Adenoviruses were prepared using the AdEasy system (Stratagene) with the murine cytomegalovirus promoter used to drive expression of YFP, PLCε wild type, PLCεH1460L, PLCεΔCDC25, or PLCεK2150E. For wild type and domain mutant PLCε adenoviruses, a second murine cytomegalovirus promoter was used to drive the expression of YFP. Adult AVM were isolated and adhered to laminin-coated coverslips for 2-h pre-infection. Plating media were removed and replaced with fresh media containing 300 multiplicity of infection of YFP control, wild type PLCε, or PLCε domain mutant adenovirus. After 2 h, the virus was removed and fresh media were added to the cells. The appearance of YFP fluorescence was used to determine the percentage of cells transduced at 24 h post-infection. PLC message was measured by semiquantitative PCR and protein was detected by Western blotting.
PCR—For detection of PLCε mRNA in PLCε–/– AVMs transduced with either wild type or domain mutant PLCε adenovirus constructs, total RNA was isolated using the RNAeasy mini kit (Qiagen, Inc., Valencia, CA) following the manufacturers recommendations. The Superscript III RT-PCR kit (Invitrogen) was used with 100 ng of total RNA template for reverse transcriptase-PCR with mouse PLCε primers 5′-ACCCTGCGGTAAATGTTCTG-3′ and 5′-ATGTGAATTCCGTGCTACCC-3′ to yield a 300-bp product. Glyceraldehyde-3-phosphate dehydrogenase primers 5′-CAACGGGAAGCCCATCACCAT-3′ and 5′-CCTTGGCAGCACCAGTGGATGC-3′ yielding a 350-bp product was used as control. Reverse transcriptase was performed for 30 min at 42 °C followed by incubation at 94 °C for 2 min. The PCR parameters were denaturation at 94 °C for 30 s, annealing at 45 °C for 45 s, and extension at 72 °C for 30 s. The number of PCR cycles was 30 for glyceraldehyde-3-phosphate dehydrogenase and 35 for PLCε.
Electrically Evoked Ca2+ Transients—Electrically evoked Ca2+ transients were measured as previously described (10). For each experiment, data were collected in the absence of agonist for 5–15 cells to determine naïve Ca2+ transient amplitude. 1 μm Isoproterenol and 10 μm cpTOME were prepared in control Ringer solution (145 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2 and 10 mm Hepes, pH 7.4) and locally perfused for 20 s followed by 60 s of electrical stimulation (20 ms, 8 V, 0.5–1 Hz, 60 s) in the continued presence of agonist.
Pharmacological Inhibition of PKC, IP3 Receptors, and CamKII—To determine the effect of PKC inhibition on electrically evoked Ca2+ transient amplitude, cells were pre-treated for 5 min with 1 μm bisindolylmaleimide-1 (BIM) (Calbiochem), a broad specificity PKC inhibitor, followed by constant perfusion of BIM in the presence of 1 μm Iso or 10 μm 8-4-(chlorophenylthio)-2′-O-methyladenosine-3′, 5′-monophosphate (cpTOME). For IP3 receptor inhibition, cells were pretreated with 20 μm 2-aminoethoxydiphenyl borate (2-APB) for 5 min followed by constant perfusion of 2-APB in the presence of 1 μm Iso. For CamKII inhibition, cells were pretreated with 1 μm KN93 (or inactive KN92) for 30 min followed by constant perfusion of KN93 in the presence of 1 μm Iso or 10 μm cpTOME.
PKC Translocation—AVM were isolated as described and plated at a density of 50,000 cells/60-mm tissue culture dish in serum-free minimal essential medium culture supplemented with 2 mm l-glutamine, 1% penicillin/streptomycin, and 2.5 μm blebbistatin. After 2 h, the media were changed to remove dead cells and debris. Cells were treated with 1 μm Iso for 30 s or 10 μm cpTOME for 3 min in a 37 °C incubator. Following treatment, dishes were placed immediately on ice and media and agonist were removed. Cells were washed two times with ice-cold phosphate-buffered saline supplemented with protease inhibitors. Cells were scraped into a lysis buffer (50 mm Hepes, pH 8.0, 3 mm MgCl2, 100 μm EDTA, 100 mm NaCl, 50 μm NaVO4, and protease inhibitors) and probe sonicated. Samples were then centrifuged at 100,000 × g at 4 °C for 15 min to pellet the membrane fraction. The membrane pellet was washed two times with ice-cold lysis buffer, and then re-suspended in sample buffer (125 mm Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% β-mercaptoethanol (1.42 m), and 0.25% bromphenol blue). Samples were resolved by 10% SDS-PAGE and Western blotted for specific PKC isoforms. PKCε and PKCα antibodies (Santa Cruz) were used at a 1:1000 dilution. Horseradish peroxidase-conjugated anti-rabbit IgG (Bio-Rad) was used at 1:1000.
PKC siRNA—PKCε-specific and CY3-labeled negative control siRNAs (Ambion) were reconstituted at 100 pm. Wild type AVM were isolated as before and media were changed prior to transfection. For each siRNA, 600 pmol was added to 600 μl of Opti-MEM. In a separate tube, 6 μl of Lipofectamine 2000 was added to 600 μl of Opti-MEM. After 5 min, siRNA and Lipofectamine tubes were mixed and incubated at room temperature for 20 min. The 200 pmol of the siRNA mixture was then added to each 35 mm dish of AVM. Efficiency of transfection was determined by fluorescence microscopy of AVM transfected with CY3-labeled negative control siRNA. Cells transfected with PKCε-specific siRNA or negative control siRNA were harvested in sample buffer at 24, 36, or 48 h post-transfection. Knockdown of PKCε protein was determined by quantitative Western blotting.
Western Blotting Phosphorylation Analysis—Phospho-Thr286 CamKII (1:1000) and total CamKII specific (1:1000) antibodies were from Santa Cruz, Phospho-Thr17 PLB (1:6000), Phospho-Ser16 PLB (1:6000), and total PLB (1:6000) antibodies were from Badrilla. Phospho-Ser2815 RyR2 antibody (1:2000) was kindly provided by Xander Wherens, Baylor College of Medicine, and total RyR2 antibody (1:2000) was from Affinity Bioreagents.
Perforated Patch Clamp—Briefly, individual AVM adhered to laminin-coated dishes were preloaded with 5 μm fluo-4 AM for 30 min at 37 °C in a Ringer solution. Myocytes were then washed 2 times with an external Ca2+ current recording solution containing: 140 mm tetraethylammonium-Cl, 2 mm MgCl2, 1.8 mm CaCl2, 0.005 mm blebbistatin, 10 mm glucose, and 10 mm Hepes, pH 7.4. Patch clamp electrodes had a pipette resistance of 1–2 megohms when backfilled with internal solution containing: 135 mm CsCl, 1 mm MgCl2, and 10 mm Hepes, pH 7.2. Ca2+ currents and transients were elicited using 200-ms test pulses from –50 to +70 mV in 10-mV increments delivered at 10-s intervals (0.1 Hz). Peak L-type Ca2+ current magnitude was normalized to total cell capacitance (pA/pF), plotted as a function of membrane potential (Vm), and fitted according to Equation 1,
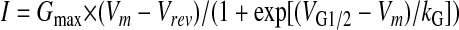 |
(Eq. 1) |
where Gmax is the maximal L-channel conductance, Vm is the test potential, VG1/2 is the voltage of half-maximal activation of Gmax, Vrev is the extrapolated reversal potential, and kG is a slope factor. The kinetics of Ca2+ current inactivation was described by fitting the inactivation phase to the following single exponential function using Equation 2,
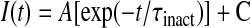 |
(Eq. 2) |
where I(t) is the current at time t after the depolarization, A is the amplitude of the inactivating component of current, τinact is the time constant of inactivation, and C represents the steady-state non-inactivating component of current. Ca2+ transients recorded during each test pulse were expressed as ΔF/F, where F represents baseline fluorescence and ΔF represents the fluorescence change from baseline.
Rap and Ras Activation—Hearts were excised from 4–6-month-old PLCε+/+ or PLCε–/– mice, cannulated through the aorta, perfused in the presence or absence of 1 μm isoproterenol for 10 min, and snap frozen in liquid nitrogen. Heart lysates were prepared by Polytron in a buffer containing 50 mm Tris-HCl, pH 7.4, 500 mm NaCl, 2.5 mm MgCl2, 1% Nonidet P-40 in 10% glycerol, and protease inhibitors. 1 mg of heart lysate was incubated with GST-tagged fusion protein corresponding to amino acids 788–884 of human RalGDS-Rap-GTP binding domain or 1–149 of human Raf-1-Ras GTP binding domain bound to glutathione-agarose in heart lysis buffer. Following incubation, beads were harvested by centrifugation, the supernatant removed, and beads were extensively washed with lysis buffer. After washing, beads were pelleted by centrifugation, resuspended in 4× SDS-sample buffer, resolved on a 15% polyacrylamide gel, and transferred to nitrocellulose for Western blotting.
Statistics—Data are given as mean ± S.E. Statistical significance was determined using unpaired Student's t test and a one-way analysis of variance (ANOVA) for multiple comparisons. Differences were considered statistically significant at p < 0.05.
RESULTS
To determine the relative roles of PLCε-RapGEF and PLC activities in the regulation of cardiac Ca2+ handling, we transduced freshly isolated PLCε–/– myocytes with adenoviruses directing expression of either wild type PLCε or mutants of PLCε previously shown to eliminate PLC hydrolytic activity (PLCεH1460L) (3), RapGEF activity (PLCεΔCDC25), or Rap (and other small GTPases) binding to the RA2 domain (PLCεK2150E) (1) (Fig. 1A). Based on PCR analysis of PLCε transcripts, all constructs were expressed to similar levels 24 h after transduction (Fig. 1B). Western blots of extracts from AVM infected with the PLCε mutant viruses indicate that the mutations do not affect expression of PLCε (Fig. 1C). Electrically evoked Ca2+ transients in transduced myocytes were then assessed in the presence or absence of either the βAR agonist isoproterenol (1 μm) or the direct Epac activator cpTOME (10 μm). As previously observed (12), isoproterenol-dependent enhancement of electrically evoked Ca2+ release was significantly increased 24 h after transduction of wild type PLCε in PLCε–/– AVM (Fig. 1D). Wild type PLCε expression also restored the 2-fold increase in evoked Ca2+ transient amplitude in response to Epac activation with cpTOME (Fig. 1E). In contrast, PLCε–/– AVM transduced with either PLCεH1460L or PLCεK2150E failed to respond to cpTOME and showed no increase in isoproterenol responsiveness compared with YFP control (Fig. 1, C–E) (supplemental Fig. S1). These results confirm that direct stimulation of PLCε hydrolytic activity by binding of a Ras family GTPase (likely Rap1) to the PLCε RA2 domain is required for maximal βAR-mediated increases in electrically evoked SR Ca2+ release. PLCε–/– AVM transduced with PLCεΔCDC25, the RapGEF-deficient PLCε mutant, failed to exhibit increased responsiveness to either isoproterenol or cpTOME (Fig. 1, D and E) (supplemental Fig. S1), indicating that PLCε-RapGEF activity is also required for the proper execution of the Epac/PLCε pathway during βAR-mediated regulation of SR Ca2+ release. We have previously shown that the Rap GEF deletion, PLCεΔCDC25, does not significantly affect intrinsic PIP2 hydrolysis activity (9).
FIGURE 1.

Epac-mediated enhancement of CICR requires both PLCε hydrolytic and RapGEF activities. A, domain structure of PLCε (CDC25 GEF, Ras family small GTPase guanine nucleotide exchange factor domain; PH, pleckstrin homology; EF, EF-hand Ca2+-binding domain, X and Y, PIP2 hydrolysis catalytic domain; C2, Ca2+-dependent lipid-binding domain; RA1 and RA2, Ras association domains). ΔCDC25(677–772), GEF deletion mutant, no RapGEF activity; H1460L, catalytic domain point mutation, lacks PIP2 hydrolysis activity; K2150E, RA2 domain point mutation, eliminates stimulation of PLC activity by Ras and Rap1. B, PLCε–/– cardiac myocytes were transduced with YFP, PLCε wild type, or PLCε domain mutant adenoviruses. 24 h post-transduction, equal expression of PLCε mRNA was demonstrated by reverse transcriptase-PCR (lower). C, cardiac myocytes were transduced with wild type (WT) and mutant PLCε viruses and protein expression was measured after 48 h by Western blotting. D and E, average (±S.E.) peak Ca2+ transient amplitude (Δ405/485) for naïve PLCε–/– myocytes and 24 h post-transduction with YFP control, PLCε wild type, or PLCε domain mutant adenovirus (300 multiplicity of infection) with or without 1 μm isoproterenol (D) or 10 μm cpTOME (E). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The inability of a RapGEF-deficient mutant of PLCε to rescue cpTOME and maximal isoproterenol-stimulated enhancement of CICR in PLCε–/– AVM suggests that Rap activation downstream of βAR stimulation is at least partially dependent on PLCε. Lysates prepared from control and Iso-perfused hearts of PLCε–/– or PLCε+/+ mice were analyzed for activated Rap (RapGTP) by pulldown with GST-RalGDS and activated Ras (RasGTP) by pulldown with GST-Raf-1-RBD. A significant increase in active Rap over basal levels was observed in lysates from Iso-treated PLCε+/+ myocytes (Fig. 2, left). In lysates prepared from PLCε–/– mice, detectable Rap activation was not observed under either basal or Iso-treated conditions (Fig. 2, right). On the other hand, similar levels of Ras activation were observed under basal and Iso-treated conditions in hearts from both PLCε+/+ and PLCε–/– mice. Total Rap and Ras levels were identical between PLCε–/– and wild type heart lysates. Together, these results are consistent with previous in vitro findings that PLCε acts specifically as a GEF for Rap, but not Ras (2).
FIGURE 2.

PLCε is required for βAR-mediated Rap activation in cardiac myocytes. Hearts from PLCε+/+ or PLCε–/– mice were cannulated through the aorta and perfused with or without 1 μm isoproterenol for 10 min. 1 mg of heart lysate was incubated with either GST-tagged RalGDS-RBD for assaying activated Rap or GST-tagged Raf1-RBD for activated Ras. Gβ1 was used as a gel loading control. Data are representative of 3 experiments showing similar results.
PLC-mediated hydrolysis of PIP2 results in generation of IP3 and diacylglycerol and the subsequent activation of Ca2+ release through IP3 receptors in the sarcoplasmic reticulum and/or PKC activation, respectively. A definitive role for IP3-mediated Ca2+ release in EC coupling in cardiac myocytes has not been identified despite intensive investigation (14). Type 2 IP3 receptors are the predominant IP3R isoform present in the heart. Analysis of type 2 IP3R knock-out mice indicates that the type 2 IP3 receptor is not required for βAR enhancement of Ca2+ release in atrial cardiac myocytes, but is important for endothelin-dependent regulation of Ca2+ release (15). To assess the potential role of IP3 in our cells, we compared electrically evoked Ca2+ after treatment with the IP3 receptor inhibitor, 2-APB (20 μm). 2-APB treatment did not alter either the Iso (Fig. 3A) or cpTOME (data not shown) responsiveness, supporting conclusions of previous studies that IP3 receptors do not directly contribute to Iso-dependent enhancement of CICR.
FIGURE 3.

Pharmacological inhibition of PKC attenuates βAR enhancement of CICR. A, IP3R inhibition with 2-APB (20 μm, 5 min pre-treatment) does not inhibit βAR-dependent increases in Ca2+ transient amplitude (Δ405/485). Ca2+ transients were measured in the absence and presence of 1 μm isoproterenol. Data are pooled from 10 to 15 cells per treatment condition. Results are average (±S.E.). B and C, PKC inhibition (1 μm BIM, 5 min pre-treatment) significantly blunts (B) isoproterenol- and (C) cpTOME-induced increases in Ca2+-transient amplitude in PLCε+/+, but not PLCε–/– cardiac myocytes. BIM did not affect naïve Ca2+ transient amplitude. Data were pooled from 20–40 cells for each treatment condition from n = 3 PLCε+/+ and n = 2 PLCε–/– mice. Results are average (±S.E.); ***, p < 0.001; #, p < 0.001 for PLCε–/– response compared with PLCε+/+; ns is not significant; one-way ANOVA, Bonferroni post test.
To test for PKC involvement in the βAR and Epac responses, electrically evoked Ca2+ transients in the presence or absence of isoproterenol were compared in AVM pretreated with 1 μm BIM, a broad specificity PKC inhibitor. BIM treatment significantly inhibited isoproterenol-stimulated enhancement of SR-Ca2+ release in wild type AVM (Fig. 3B, left). In addition, BIM treatment abolished the increase in electrically evoked Ca2+ transient amplitude observed following direct activation of Epac with cpTOME (Fig. 3C). However, BIM treatment did not alter Iso stimulation of electrically evoked Ca2+ transients in PLCε–/– myocytes (Fig. 3B, right) or baseline evoked transients (data not shown). These results indicate that the effects of BIM are specific to the PLCε-dependent pathway downstream of βAR stimulation and implicate PKC activation in this pathway.
There are 11 distinct isoforms of PKC, four of which are consistently detected in cardiac myocytes: α, βII, δ, and ε (16). To determine whether a specific isoform of PKC is activated downstream of PLCε, we monitored translocation of specific PKC isoforms to the particulate fraction following treatment of freshly isolated PLCε+/+ and PLCε–/– cardiac myocytes with either isoproterenol or cpTOME. Western blot analysis of the particulate fraction of wild type cardiac myocytes revealed a specific increase in PKCε in the membrane fraction in response to both isoproterenol and cpTOME treatment relative to nontreated control (Fig. 4A). In contrast, PKCα did not translocate to the membrane fraction in response to isoproterenol. In PLCε–/– AVM, neither isoproterenol nor cpTOME triggered translocation of PKCε to the membrane (if anything a small decrease in PKCε at the membrane was observed), placing PKCε downstream of the βAR/Epac/Rap/PLCε pathway.
FIGURE 4.

PLCε-dependent enhancement of CICR requires specific activation of PKCε. A, left, PKCε translocates to the membrane fraction following treatment with 1 μm isoproterenol (30 s) or 10 μm cpTOME (3 min) in PLCε+/+ but not PLCε–/– cardiac myocytes. PKCα does not translocate to the membrane in response to βAR stimulation. PMA treatment (500 nm, 10 min) was used as a positive control for PKC translocation. 3 μg of cardiac myocyte membrane fractions was analyzed for PKC isoform translocation. Gβ subunit was used as a loading control. Right, densitometric quantitation of PKCε membrane translocation from cells isolated from 5 PLCε+/+ and 3 PLCε–/– mice. Data are represented as a percentage of maximal translocation evoked by PMA treatment. **, p < 0.01; ***, p < 0.001; ns, not significant as compared with nontreated PLCε+/+ cells. One-way ANOVA, Bonferroni post-test. B, left, PLCε+/+ cardiac myocytes were transfected with PKCε-specific siRNA or a CY3-labeled negative control siRNA. PKCε protein levels are nearly completely knocked down in cardiac myocytes transfected with PKCε-specific siRNA relative to negative control siRNA at 36 h post-transfection. Lower left, PKCα protein levels are not significantly affected by PKCε siRNA 36 h post-transfection. Right, densitometric quantitation of PKCε protein expression from myocytes transfected with either PKCε siRNA or Cy3-labeled negative control siRNA pooled from three separate experiments. C, knockdown of PKCε significantly decreases isoproterenol-induced enhancement of CICR and completely eliminates cpTOME responsiveness in PLCε+/+ cardiac myocytes. Data are pooled Ca2+ transient amplitudes (Δ404/485) from 20 to 40 cells per condition, n = 3 mice. Results are average (±S.E.); ***, p < 0.001, one-way ANOVA, Bonferroni post-test.
To further test the role of PKCε downstream of the Epac/Rap/PLCε pathway, wild type AVM were transfected with either PKCε-specific siRNA or a CY3-labeled negative control siRNA. Transfection efficiency was nearly 100% (supplemental Fig. S2) and Western blot analysis revealed that PKCε (but not PKCα) protein levels were knocked down by at least 95% at 36 h post-transfection (Fig. 4B). PKCε protein levels were not substantially decreased in cells transfected with the negative control siRNA at all time points monitored (24, 36, and 48 h). Electrically evoked Ca2+ transients were monitored 36 h after transfection of wild type AVM with either PKCε or negative control siRNAs. Baseline electrically evoked Ca2+ transients were not different between control and PKCε siRNA-treated AVM. On the other hand, peak Ca2+ transient amplitude in the presence of isoproterenol was significantly inhibited and cpTOME responses abolished (Fig. 4C) in AVM treated with PKCε siRNA. These data are consistent with results obtained following pharmacological PKC inhibition with BIM (Fig. 3, B and C) and Iso/cpTOME-dependent PKCε membrane translocation (Fig. 4A), indicating that PKC acts downstream of PLCε and specifically implicates PKCε as the relevant PKC isoform involved.
A recent report demonstrated that CamKII is activated following Epac stimulation with cpTOME in rat cardiac myocytes, however, the mechanism for CamKII activation by Epac was not determined (17). PKC has also been shown to activate CamKII in rat ventricular myocytes (18) and CamKII is directly phosphorylated at Thr286 by PKC in vitro (19). Therefore, we determined if CamKII activation is required for Iso- and Epac-dependent enhancement of CICR, and if it is downstream of PKC. Iso- and cpTOME-induced enhancement of electrically evoked Ca2+ transients in wild type AVM were determined in the absence and presence of KN93, a specific CamKII inhibitor. KN93, but not the control compound KN92, attenuated Iso-induced enhancement of evoked release (Fig. 5A) and completely blocked cpTOME-induced enhancement (Fig. 5B). Co-treatment with BIM and KN93 did not further diminish the response to isoproterenol relative to treatment with either compound alone (data not shown). Additionally, KN93 had no effect on the Iso response in PLCε–/– AVM (Fig. 5A) or on baseline evoked transients (data not shown), supporting specific involvement of CamKII in the PLCε-dependent pathway.
FIGURE 5.

PKC-dependent activation of CamKII is required for PLCε-mediated enhancement of CICR. CamKII inhibition (1 μm KN-93, 30 min pretreatment) significantly blunts (A) Iso and (B) cpTOME (10 μm) enhancement of electrically evoked Ca2+ transient amplitude in AVM from PLCε+/+, but not PLCε–/– mice. Pretreatment with KN92, an inactive analogue of KN-93, does not affect Iso responsiveness. Data were pooled from 5 to 15 cells for each treatment condition from 3 PLCε+/+ and 3 PLCε–/– mice. Results are average (±S.E.); ***, p < 0.001; ns, not significant; one-way ANOVA, Bonferroni post test. C, phosphorylation of CamKII at Thr286 was measured by Western blotting of extracts of AVM treated with Iso (1 μm), cpTOME (10 μm), and BIM (1 μm) as indicated (n = 5 animals). Right panel, pooled data from densitometric quantitation. Results are average (±S.E.); *, p < 0.05; one-way ANOVA, Bonferroni post test. D, phosphorylation of RyR2 Ser2815 and PLB Thr17 was measured by Western blotting of extracts of AVM treated with cpTOME (10 μm) or BIM (1 μm) as indicated (n = 3 animals each). Right panel, pooled data from densitometric quantitation and normalized relative to untreated cells. Results are average (±S.E.); **, p < 0.01; ***, p < 0.001; one-way ANOVA, Bonferroni post test.
To determine whether CamKII activation was dependent on PKC, wild type cardiac myocytes were treated with either isoproterenol or cpTOME alone or in the presence of BIM and CamKII phosphorylation at Thr286 was measured by Western blotting (Fig. 5C). Both isoproterenol and cpTOME treatment increased CamKII phosphorylation at Thr286 relative to nontreated control. The cpTOME-dependent increase was blocked in the presence of BIM and the Iso-dependent increase was partially blocked by BIM. These data support the conclusion that the Epac/PLCε pathway can control CamKII activation in a PKCε-dependent, Ca2+-independent manner. That the Iso-dependent increase in phosphorylation was only partially blocked suggests that there are multiple mechanisms for CamKII activation downstream of Iso, one of which includes the Ca2+-independent Epac and PKC pathway, but may also result from changes in Ca2+ that occur with Iso-dependent regulation of PKA.
CamKII phosphorylates numerous Ca2+ handling proteins, including the L-type Ca2+ channel, RyR2 and PLB, involved in precisely controlling dynamic changes in intracellular calcium levels during the cardiac cycle (20). CamKII-dependent modulation of RyR function by phosphorylation at Ser2815 has been implicated as a means for positive regulation of SR-Ca2+ release downstream of βAR stimulation. CamKII also phosphorylates PLB at Thr17 to stimulate SR Ca2+ reuptake to increase content available for release. To determine whether Epac/PLCε stimulation results in a PKC-dependent, CamKII-mediated, phosphorylation of RyR2 and/or PLB, AVM were treated with cpTOME in the presence or absence of the PKC inhibitor BIM. Phosphorylation at the CamKII-specific sites was measured by Western blotting (Fig. 5D). cpTOME treatment significantly enhanced phosphorylation of RyR2 at Ser2815 and PLB at Thr17 in wild type AVM. These increases were ablated in AVM pretreated with BIM. These data identify at least two effector targets (RyR2 and PLB) of the Epac/PLCε pathway that could be involved in regulating the magnitude of CICR.
To examine the relationship between Ca2+ influx and release during EC coupling we conducted voltage clamp experiments to determine whether the Epac/PLCε pathway alters the properties of depolarization-induced L-type Ca+ channel function and RyR2-mediated Ca2+ release. Perforated patch clamp experiments in fluo-4-loaded AVM were conducted to simultaneously compare the voltage dependence, magnitude, and kinetics of L-type Ca2+ currents and global intracellular Ca2+ transients in wild type and PLCε–/– myocytes before and after βAR activation (Fig. 6 and supplemental Fig. S3). L-type Ca2+ currents and intracellular Ca2+ transients were elicited by 200-ms test pulses from –50 to +70 mV at 10-mV increments. No differences in the magnitude or kinetics of L-type Ca2+ currents (Fig. 6A) or global intracellular Ca2+ transients (Fig. 6C) were observed between PLCε–/– and PLCε+/+ AVM under basal conditions (closed symbols) (Table 1). Application of 1 μm Iso (open symbols) caused a similar 1.5–2-fold increase in peak L-type Ca2+ current density (Fig. 6A) and maximum channel conductance (Fig. 6B and Table 1) in both PLCε–/– and PLCε+/+ AVM. However, peak Iso-stimulated Ca2+ transient amplitude was significantly attenuated in PLCε–/– cardiac myocytes (Fig. 6, C and D, and Table 1), consistent with results observed in intact myocytes (Figs. 3B and 5A). In addition, the kinetics of Ca2+ current inactivation was significantly slower in myocytes from PLCε–/– mice (supplemental Fig. S3). These data indicate that alterations in action potential or L-type channel activity are not necessary PLCε-dependent regulation of CICR.
FIGURE 6.

PLCε ablation significantly reduces isoproterenol stimulation of depolarization-induced intracellular calcium transients without altering L-type Ca2+ current density. Perforated patch clamp experiments were used to simultaneously monitor depolarization-induced L-type Ca2+ currents (A and B) and intracellular Ca2+ transients (C and D) in the absence (closed symbols) and presence (open symbols) of βAR-stimulation with 1 μm isoproterenol in AVM from PLCε+/+ (circles) and PLCε–/– (squares) mice. A, average (±S.E.) voltage dependence of L-type Ca2+ current density. B, average (±S.E.) fold stimulation of maximal L-type Ca2+ conductance in AVM from 5 PLCε–/– and 5 PLCε+/+ mice. C, average (±S.E.) voltage dependence of intracellular Ca2+ transients. D, average (±S.E.) fold stimulation of peak intracellular Ca2+ transient (measured at –30 mV) in AVM from 5 PLCε–/– and 5 PLCε+/+ mice. *, p < 0.05, t test.
TABLE 1.
Parameters of IV and (ΔF/F)max data
Values represent mean (±S.E.) for n number of mice (from a total of 32 PLC+/+ and 27 PLC-/- myocytes). Parameters for the voltage dependence of Ca2+ current (I-V) was obtained by fitting myocytes within each group separately to the appropriate equation (I-V, Equation 1) as described under “Experimental Procedures,” (ΔF/F)max values are the mean (±S.E.) values obtained at a test potential of -10 mV.
| Gmaxa | kv | VG1/2 | Vrev | (ΔF/F)max | |
|---|---|---|---|---|---|
| nS/nF | mV | ||||
| PLCε+/+ naïve (n = 5 mice) | 108 ± 14 | 7.4 ± 0.4 | -14.3 ± 0.4 | 62.9 ± 1.1 | 0.15 ± 0.04 |
| PLCε+/+ Iso (n = 5 mice) | 197 ± 14b | 6.1 ± 1.4 | -18.9 ± 1.7 | 65.0 ± 0.9 | 0.45 ± 0.04b,c |
| PLCε-/- naïve (n = 5 mice) | 103 ± 14 | 6.6 ± 0.3 | -16.0 ± 2.4 | 64.7 ± 1.6 | 0.15 ± 0.03 |
| PLCε-/- Iso (n = 5 mice) | 162 ± 18d | 6.0 ± 0.2 | -20.1 ± 2.0 | 66.2 ± 1.9 | 0.27 ± 0.04d |
Gmax, maximal l-channel conductance; Vrev, reversal potential; VG1/2, potential at which G is half-maximal, respectively; kG, slope factor for I-V.
p < 0.01 compared to PLCε+/+ naive.
p < 0.05 compared to PLCε-/- Iso.
p < 0.05 compared to PLCε-/- naive.
DISCUSSION
PLCε is unique among PLC enzymes in that it possesses both phospholipase C and RapGEF activities. Physiological roles for both catalytic functions of PLCε are beginning to emerge (9, 10, 21, 22). Here we demonstrate that both PLCε hydrolytic and RapGEF activities are required for maximal βAR-mediated (and Epac-dependent) enhancement of electrically evoked Ca2+ release in the heart. We hypothesize that the PLCε-RapGEF activity ensures sufficient Rap activation to maintain PLCε hydrolytic activity, and that PLCε is required for sustained Rap activation in the heart. Epac stimulation by cAMP may initiate a low level of Rap activation that (undetectable in the PLCε–/– myocytes), in turn, stimulates PLCε to significantly amplify Rap activation that feeds forward to further stimulate PLCε and subsequent regulation of CICR. This model is supported by previous studies in transfected cells demonstrating that PLCε potentiates its own activation by RapGTP (2) and in primary astrocytes where PLCε RapGEF activity is required for sustained Rap activation and downstream ERK signaling (9). In addition, it is important to note that Rap generated from PLCε may also regulate other enzymes in the heart such as ERK5 where Rap-mediated inhibition protects against the development of hypertrophy (23). This would be consistent with our findings that PLCε–/– mice exhibit increased susceptibility to stress-induced hypertrophy (10) and that PLCε RapGEF activity modulates ERK signaling in astrocytes (9).
Downstream of PLCε activity, we demonstrate that diacylglycerol-mediated PKCε activation is required for maximalβAR-dependent enhancement of CICR (Fig. 3). Several reports have suggested that PKC activity modulates CICR in cardiac myocytes. Treatment with norepinephrine or phorbol myristic acid causes PKCε translocation to cross-striated t-tubular regions of cardiac myocytes upon activation, strategically placing this enzyme in position to phosphorylate proteins involved in Ca2+ handling (24). PKCδ and PKCε have been shown to mediate positive inotropy that is dependent on subcellular localization (25). Our data are consistent with a positive ionotropic effect of PKCε and indicate that PLCε is an upstream regulator of PKCε in the heart. siRNA knockdown of PKCε strongly suppressed cpTOME-dependent increases in calcium transient amplitudes suggesting that PKCε is the major isoform of PKC involved in Epac/PLCε-dependent responses. On the other hand, the inhibition of Iso-dependent responses by PKCε siRNA appeared less than observed with BIM treatment (Figs. 3B versus 4C). One possibility is that another PKC isoform such as PKCδ is involved in the Iso-dependent response or that a more complete inhibition of PKCε that might be achievable with BIM may be required to fully inhibit the Iso-response.
We also identified PKC-dependent CamKII regulation as an essential downstream component of the Epac/PLCε pathway in AVM. βAR and cpTOME enhancement of electrically evoked Ca2+ release was suppressed by PKC inhibition with BIM (Fig. 3, B and C), PKCε knockdown (Fig. 4C), and CamKII inhibition with K93 (Fig. 5) in wild-type PLCε+/+ AVM. On the other hand, neither PKC inhibition, CamKII inhibition, nor the combination, had any significant effect on the already reduced Iso-dependent regulation of evoked release in PLCε–/– AVM, indicating that both PKCε and CamKII are downstream from PLCε activation. We also show that phosphorylation of CamKII at Thr286 is increased by cpTOME in a PKC-dependent manner. These data are consistent with a previous report demonstrating that activation of Epac stimulates CamKII Thr286 phosphorylation (17) but extend this result to show that Epac-dependent CamKII activation relies on PLCε-dependent PKCε activity. Although a mechanism for PKC-dependent activation of CamKII has not been clearly delineated, CamKII Thr286 has been shown to be directly phosphorylated by PKC in vitro (19). Thr286 autophosphorylation results in tight binding of calmodulin such that Ca2+ is no longer required for activation. If CamKII Thr286 is directly phosphorylated by PKC it should also result in Ca2+-independent regulation. Physiological evidence for PKC-dependent regulation of CamKII is sparse but two previous studies have shown that α-adrenergic receptor stimulation facilitates PKC-mediated activation of CamKII (18, 26). PKCε may not directly phosphorylate CamKII in cardiac myocytes in response to Epac/PLCε activation but we clearly demonstrate that PKC activation is upstream of CamKII Thr286 phosphorylation in this pathway. It is also possible that PKCε itself phosphorylates components of the Ca2+ handling machinery, but our data indicate that any such activity alone is insufficient because CamKII inhibition with KN93 completely blocks cpTOME-dependent enhancement of Ca2+ transient amplitudes (Fig. 5B). This indicates that PKC activation by this pathway is not sufficient to cause increases in Ca2+ transients. Nevertheless, it remains formally possible that local Ca2+ release, dependent on PKC activation, could lead to CamKII autophosphorylation. We propose that in cardiac myocytes, linear activation of Epac-PLCε-PKCε-CamKII mediates a component of Iso-dependent regulation of evoked Ca2+ release in cardiac myocytes.
Potential targets downstream of this pathway include the L-type Ca2+ channel, RyR2, and phospholamban. PLCε ablation did not markedly affect isoproterenol-stimulated increases in L-type Ca2+ channel current density in perforated patch clamp experiments (Fig. 6, A and B) and L-type Ca2+ channel activity was not significantly altered by 20 μm cpTOME (data not shown). In the same cells Iso-stimulated enhancement of depolarization-induced Ca2+ transients was significantly attenuated (Fig. 6, C and D). The fact that Ca2+ release elicited by a uniform voltage clamp pulse is reduced in PLCε–/– myocytes, suggests that changes in action potential waveform are not likely responsible for the reduction in Iso responsiveness. Together, these data indicate that the Epac/PLCε/PKCε/CamKII pathway contributes enhanced release of Ca2+ from the SR during βAR stimulation that is not dependent on changes in Ca2+ influx. Two proteins that control release of Ca2+ from the SR, RyR2 and PLB, are phosphorylated at CamKII-specific sites in response to Epac stimulation, supporting this idea. The observed increase in Ca2+ release in response to a uniform Ca2+ influx could arise from a combination of both an increase in RyR2 sensitivity to activation by Ca2+ influx (27, 28) and an increase in Ca2+ reuptake and load. The observed increase in Ca2+ release we report here differs from results of Pereira et al. (17) who report that Epac activation decreases evoked Ca2+ release due to CamKII-dependent SR store depletion. Apparent discrepancies between our results and Pereira et al. (17) could be due to differences in protocol or species used, but the overall conclusions that Epac activation results in CamKII activation and RyR(Ser2815) phosphorylation are in agreement.
Roles of PKC and CamKII in both normal and pathological cardiac function have been steadily emerging over the last several years, but our understanding of the physiological mechanisms that control activation of these pathways have lagged behind. Here, we have outlined a novel, PLCε/PKCε/CamKII-dependent regulatory mechanism for regulating cardiac CICR in adult ventricular cardiac myocytes. Previous studies have implicated CamKII-dependent phosphorylation of RyR2 as a means for regulating SR-Ca2+ release downstream of Epac or βAR stimulation (17, 28). We extend these findings by identifying key mechanistic links between Epac activation and regulation of CICR such that the majority of the components of the pathway are now defined. This pathway is clearly important for cardiac function because mice lacking PLCε exhibit a significantly impaired ability to respond to βAR stimulation. This impairment is manifested by both decreased cardiac function in response to isoproterenol administration in whole animals (10) and decreased βAR-dependent enhancement of CICR in AVM isolated from PLCε–/– mice (12). PLCε–/– mice also show an increased sensitivity to stress-induced cardiac hypertrophy (10) and it remains to be determined how components of the pathway could contribute to this pathology.
Supplementary Material
Acknowledgments
We thank Joan Heller-Brown for critical review of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GMR01053536 (to A. V. S.), AR044657 (to R. T. D.), and DK56294 (to G. G. K.). This work was also supported by American Heart Association Scientist Development Grant 045343T (to B. C. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4 and Methods.
Footnotes
The abbreviations used are: PLC, phosphoinositide-specific phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3, inositol 1,4,5-trisphosphate; GEF, guanine nucleotide exchange factor; βAR, β-adrenergic receptor; AVM, adult ventricular myocytes; PKA, protein kinase A; PKC, protein kinase C; CICR, calcium-induced calcium release; SR, sarcoplasmic reticulum; PLB, phospholamban; RyR, ryanodine receptor; BIM, bisindolylmaleimide-1; cpTOME, 8-4-(chlorophenylthio)-2′-O-methyladenosine-3′, 5′-monophosphate; GST, glutathione S-transferase; YFP, yellow fluorescent protein; Iso, isoproterenol; 2-APB, 2-aminoethoxydiphenyl borate; CamKII, calmodulin kinase II; siRNA, small interfering RNA; ANOVA, analysis of variance; ERK, extracellular signal-regulated kinase.
References
- 1.Kelley, G. G., Reks, S. E., Ondrako, J. M., and Smrcka, A. V. (2001) EMBO J. 20 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin, T. G., Satoh, T., Liao, Y., Song, C., Gao, X., Kariya, K., Hu, C. D., and Kataoka, T. (2001) J. Biol. Chem. 276 30301–30307 [DOI] [PubMed] [Google Scholar]
- 3.Lopez, I., Mak, E. C., Ding, J., Hamm, H. E., and Lomasney, J. W. (2001) J. Biol. Chem. 276 2758–2765 [DOI] [PubMed] [Google Scholar]
- 4.Shibatohge, M., Kariya, K., Liao, Y., Hu, C. D., Watari, Y., Goshima, M., Shima, F., and Kataoka, T. (1998) J. Biol. Chem. 273 6218–6222 [DOI] [PubMed] [Google Scholar]
- 5.Kelley, G. G., Reks, S. E., and Smrcka, A. V. (2004) Biochem. J. 378 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wing, M. R., Snyder, J. T., Sondek, J., and Harden, T. K. (2003) J. Biol. Chem. 278 41253–41258 [DOI] [PubMed] [Google Scholar]
- 7.Wing, M. R., Houston, D., Kelley, G. G., Der, C. J., Siderovski, D. P., and Harden, T. K. (2001) J. Biol. Chem. 276 48257–48261 [DOI] [PubMed] [Google Scholar]
- 8.Schmidt, M., Evellin, S., Weernink, P. A., von Dorp, F., Rehmann, H., Lomasney, J. W., and Jakobs, K. H. (2001) Nat. Cell Biol. 3 1020–1024 [DOI] [PubMed] [Google Scholar]
- 9.Citro, S., Malik, S., Oestreich, E. A., Radeff-Huang, J., Kelley, G. G., Smrcka, A. V., and Brown, J. H. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 15543–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, H., Oestreich, E. A., Maekawa, N., Bullard, T. A., Vikstrom, K. L., Dirksen, R. T., Kelley, G. G., Blaxall, B. C., and Smrcka, A. V. (2005) Circ. Res. 97 1305–1313 [DOI] [PubMed] [Google Scholar]
- 11.Bers, D. M. (2002) Nature 415 198–205 [DOI] [PubMed] [Google Scholar]
- 12.Oestreich, E. A., Wang, H., Malik, S., Kaproth-Joslin, K. A., Blaxall, B. C., Kelley, G. G., Dirksen, R. T., and Smrcka, A. V. (2007) J. Biol. Chem. 282 5488–5495 [DOI] [PubMed] [Google Scholar]
- 13.Bos, J. L. (2003) Nat. Rev. Mol. Cell Biol. 4 733–738 [DOI] [PubMed] [Google Scholar]
- 14.Woodcock, E. A., and Matkovich, S. J. (2005) Pharmacol. Ther. 107 240–251 [DOI] [PubMed] [Google Scholar]
- 15.Li, X., Zima, A. V., Sheikh, F., Blatter, L. A., and Chen, J. (2005) Circ. Res. 96 1274–1281 [DOI] [PubMed] [Google Scholar]
- 16.Dorn, G. W., II, and Force, T. (2005) J. Clin. Investig. 115 527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira, L., Metrich, M., Fernandez-Velasco, M., Lucas, A., Leroy, J., Perrier, R., Morel, E., Fischmeister, R., Richard, S., Benitah, J. P., Lezoualc'h, F., and Gomez, A. M. (2007) J. Physiol. 583 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O-Uchi, J., Komukai, K., Kusakari, Y., Obata, T., Hongo, K., Sasaki, H., and Kurihara, S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 9400–9405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waxham, M. N., and Aronowski, J. (1993) Biochemistry 32 2923–2930 [DOI] [PubMed] [Google Scholar]
- 20.Maier, L. S., and Bers, D. M. (2007) Cardiovasc. Res. 73 631–640 [DOI] [PubMed] [Google Scholar]
- 21.Ikuta, S., Edamatsu, H., Li, M., Hu, L., and Kataoka, T. (2008) Cancer Res. 68 64–72 [DOI] [PubMed] [Google Scholar]
- 22.Tadano, M., Edamatsu, H., Minamisawa, S., Yokoyama, U., Ishikawa, Y., Suzuki, N., Saito, H., Wu, D., Masago-Toda, M., Yamawaki-Kataoka, Y., Setsu, T., Terashima, T., Maeda, S., Satoh, T., and Kataoka, T. (2005) Mol. Cell. Biol. 25 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodge-Kafka, K. L., Soughayer, J., Pare, G. C., Carlisle Michel, J. J., Langeberg, L. K., Kapiloff, M. S., and Scott, J. D. (2005) Nature 437 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Disatnik, M. H., Buraggi, G., and Mochly-Rosen, D. (1994) Exp. Cell Res. 210 287–297 [DOI] [PubMed] [Google Scholar]
- 25.Kang, M., and Walker, J. W. (2005) J. Mol. Cell Cardiol. 38 753–764 [DOI] [PubMed] [Google Scholar]
- 26.O-Uchi, J., Sasaki, H., Morimoto, S., Kusakari, Y., Shinji, H., Obata, T., Hongo, K., Komukai, K., and Kurihara, S. (2008) Circ. Res. 102 1378–1388 [DOI] [PubMed] [Google Scholar]
- 27.Currie, S., Loughrey, C. M., Craig, M. A., and Smith, G. L. (2004) Biochem. J. 377 357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wehrens, X. H. T., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004) Circ. Res. 94 e61–e70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.