

Abstract
It was previously shown that coexpression of the lactose permease of Escherichia coli in two contiguous fragments leads to functional complementation. We demonstrate here that site-directed thiol crosslinking of coexpressed permease fragments can be used to determine helix proximity in situ without the necessity of purifying the permease. After coexpression of the six N-terminal (N6) and six C-terminal (C6) transmembrane helices, each with a single Cys residue, crosslinking was carried out in native membranes and assessed by the mobility of anti-C-terminal-reactive polypeptides on immunoblots. A Cys residue at position 242 or 245 (helix VII) forms a disulfide with a Cys residue at either position 28 or 29 (helix I), but not with a Cys residue at position 27, which is on the opposite face of helix I, thereby indicating that the face of helix I containing Pro-28 and Phe-29 is close to helix VII. Similarly, a Cys residue at position 242 or 245 (helix VII) forms a disulfide with a Cys residue at either position 52 or 53 (helix II), but not with a Cys residue at position 54. Furthermore, low-efficiency crosslinking is observed between a Cys residue at position 52 or 53 and a Cys residue at position 361 (helix XI). The results indicate that helix VII lies in close proximity to both helices I and II and that helix II is also close to helix XI. The method should be applicable to a number of different polytopic membrane proteins.
Keywords: transport, bioenergetics, membrane protein structure, disulfide crosslinking, split permease
The lactose (lac) permease of Escherichia coli is a paradigm for secondary transport proteins that transduce free energy stored in an electrochemical ion gradient into a solute concentration gradient. This integral membrane protein that catalyzes the coupled stoichiometric translocation of β-galactosides and H+ has been solubilized, purified to homogeneity, reconstituted, and shown to be solely responsible for β-galactoside transport (1) as a monomer (see ref. 2). Based on circular dichroism and hydropathy analysis, a secondary-structure model was proposed (3) in which the permease contains 12 α-helical rods that traverse the membrane in zig-zag fashion connected by loops with the N and C termini on the cytoplasmic face. Evidence favoring many aspects of the model has been obtained from a variety of approaches (reviewed in ref. 4, in addition see refs. 5–7), and analysis of an extensive series of lac permease-alkaline phosphatase (lacY-phoA) fusions has provided unequivocal support for the topological predictions of the 12-helix model (8).
Site-directed and Cys-scanning mutagenesis have allowed delineation of amino acid residues in the permease that are important for active transport and/or substrate binding (reviewed in refs. 9 and 10). However, static and dynamic information at high resolution are required to understand the role of these residues in the transport mechanism. Because hydrophobic membrane proteins are notoriously difficult to crystallize, a high-resolution structure of lac permease is not available, and development of alternative methods for obtaining structural information is essential. In this respect, a functional permease molecule has been engineered in which all of the native Cys residues have been replaced (C-less permease), and single Cys residues have been placed at every position in the molecule. In addition to providing evidence that remarkably few amino acid residues are irreplaceable with regards to insertion, stability, or activity, the Cys-replacement mutants represent unique molecules that are being used to study permease structure–function relationships (reviewed in ref. 11).
A helix packing model of the C-terminal half of lac permease has been formulated (12) based on site-directed excimer fluorescence, which demonstrates that helix VIII (Glu-269) is close to helix X (His-322), helix IX (Arg-302) is close to helix X (Glu-325), and that helix X is in α-helical conformation (also see ref. 13). Furthermore, the presence of two pairs of charged residues that interact functionally—Asp-237 (helix VII) with Lys-358 (helix XI) (14–18) and Asp-240 (helix VII) with Lys-319 (helix X) (15, 17, 19)—indicates that helix VII is close to helices X and XI. The proximity relationships have been confirmed by engineering divalent metal-binding sites (bis- or tris-His residues) within the permease (20–22) and by the demonstration that monoclonal antibody 4B11 binds to the last two cytoplasmic loops (7). In addition, site-directed chemical cleavage studies support the positioning of helix X next to helices VII and XI and indicate further that helix V is in close proximity to helices VII and VIII (23). Finally, site-directed spin labeling and chemical crosslinking have provided direct support for the placement of helix V close to helices VII and VIII (24).
Yu et al. (25) have described a method for determining tertiary contacts between transmembrane domains in polytopic membrane proteins based on disulfide formation between paired Cys residues in purified segments of visual rhodopsin. Because coexpression of lac permease in two fragments leads to functional complementation (26, 27), we have extended this approach to show in situ that helices I and II are in close proximity to helices VII and XI in the folded tertiary structure of lac permease.
MATERIALS AND METHODS
Materials.
Protein A-conjugated horseradish peroxidase (HRP), enhanced chemiluminescence detection kits were obtained from Amersham. Avidin-conjugated HRP (avidin-HRP) was purchased from Pierce. Anti-C-terminal antibody was prepared as described (28). All other materials were reagent grade and obtained from commercial sources.
Construction of the N-Terminal Six Transmembrane Helices (N6) of Permease with Single Cys Replacements.
Construction of permease mutants containing single Cys replacements at position 27, 28, 29 (helix I) or 52, 53, 54 (helix II) in the Cys-less background has been described (29, 30). With each single Cys mutant, the biotin acceptor domain from the Klebsiella pneumoniae oxaloacetate decarboxylase (31) was inserted into the middle cytoplasmic loop as described (32). The 3′ half of the lacY gene in each construct was then removed by AflII digestion followed by intramolecular ligation yielding plasmid pN6, which encodes the N6 with a single Cys residue at a given position and the biotin acceptor domain at the C terminus (Fig. 1).
Figure 1.
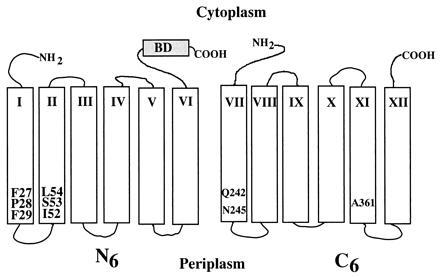
Secondary structure model of N6/C6 split permease. The lac permease is shown as the N-terminal six transmembrane helices (N6) and the C-terminal six transmembrane helices (C6). N6 has a biotin acceptor domain (BD) at the C terminus. Single Cys replacements in helix I (mutants F27C, P28C, and F29C), helix II (mutants I52C, S53C, and L54C), helix VII (mutants Q242C and N245C), and helix XI (mutant A361C) are numbered and highlighted.
Construction of the C-Terminal Six Transmembrane Helices (C6) of Permease with Single Cys Replacements.
Construction of plasmid pC6 encoding the C6 of the lac permease has been described (23). To place a single Cys residue at position 242, 245 (helix VII), or 361 (helix XI) in the C6 fragment, the BstXI–HindIII fragment of pC6 was replaced with the corresponding DNA fragment from a single Cys permease mutant.
Expression of Split Permeases and Membrane Preparation.
E. coli T184 (lacY−Z−) was transformed with both pN6 and pC6 each encoding a permease fragment with a given single Cys replacement. Cells (50 ml) were grown at 37°C to an OD600 of 1.0 and induced with isopropyl β-d-thiogalactoside (final concentration 1.0 mM) for 2 h. Cells were harvested by centrifugation, washed once with 20 mM Tris·HCl (pH 7.6)/2.0 mM EDTA and suspended in the same buffer. Lysozyme was added to a final concentration of 100 μg/ml, and the suspension was incubated on ice for 10 min. Membranes were prepared by sonification as described (33) and suspended in 20 mM Tris·HCl (pH 7.6).
Disulfide Crosslinking.
Crosslinking was carried out at 20°C for 10 min by adding aqueous iodine (final concentration of 0.5 mM) to membrane preparations. Reactions were terminated by adding 10 mM N-ethylmaleimide (NEM). Samples were mixed with sodium dodecylsulfate (NaDodSO4) loading buffer and subjected to electrophoresis in NaDodSO4/14% polyacrylamide gels. N6 with the biotin acceptor domain was detected with avidin-HRP (5); C6 was detected by immunoblotting with anti-C-terminal antibody (28).
Lac Transport.
Active transport was measured in E. coli T184 (lacY−Z−) transformed with given plasmids. Cell cultures grown overnight were diluted 20-fold, grown to OD600 of 0.8, induced with 1.0 mM isopropyl β-d-thiogalactoside for 2 h and harvested by centrifugation. Transport of [1-14C]lactose was assayed as described (34).
Protein Determinations.
Protein was assayed as described (35).
RESULTS
Functional Complementation of N6/C6 Fragments with Paired Cys Replacements.
Previous experiments show that coexpression of wild-type lacY fragments encoding contiguous polypeptides corresponding to the first and second halves of the permease (26, 27) leads to functional complementation. Consistently, active lac transport is observed in all 18 paired Cys N6/C6 permease fragments used in this study (Table 1). Thus, each pair accumulates lac to a steady state that is 30–60% of C-less permease.
Table 1.
Crosslinking efficiencies and transport activity of N6/C6 permease with paired Cys replacements
| Paired Cys positions (N6/C6) | Crosslinking efficiency* | Transport activity, nmol/mg† |
|---|---|---|
| 27/242 | − | 62 |
| 28/242 | +++ | 38 |
| 29/242 | +++ | 53 |
| 27/245 | − | 48 |
| 28/245 | ++ | 27 |
| 29/245 | + | 45 |
| 52/242 | ++ | 33 |
| 53/242 | +++ | 58 |
| 54/242 | + | 50 |
| 52/245 | +++ | 30 |
| 53/245 | +++ | 46 |
| 54/245 | − | 55 |
| 27/361 | − | 57 |
| 28/361 | − | 31 |
| 29/361 | − | 64 |
| 52/361 | + | 25 |
| 53/361 | + | 39 |
| 54/361 | − | 42 |
+++, >20% efficiency; ++, 10–20% efficiency; +, <10% efficiency; −, no detectable crosslinking.
Transport activity is shown as the steady-state level of lactose accumulation. C-less permease accumulates about 100 nmol lactose per mg protein.
Detection of Permease Fragments.
By using anti-C-terminal polyclonal antibody, it is apparent that C6 migrates at about 20 kDa (Fig. 2A). Furthermore, use of avidin-HRP shows that N6 with the biotin acceptor domain migrates at about 35 kDa (Fig. 2B).
Figure 2.

Disulfide crosslinking of N6/C6 permease fragments containing paired Cys residues in helices VII and I. Membranes were prepared from E. coli T184 expressing N6 and C6, each with a single Cys residue at a given position as indicated. Disulfide crosslinking was carried out at room temperature by incubating membranes (2 mg protein per ml) with 0.5 mM iodine for 10 min. Reactions were terminated by adding NEM to a final concentration of 10 mM. Samples containing approximately 20 μg of protein were subjected to NaDodSO4/PAGE and electroblotted. C6 was detected with anti-C-terminal antibody (A) and N6 with HRP-avidin (B).
Crosslinking Between Helices I and VII.
When membranes containing N6/C6 with paired Cys residues at position 242 (helix VII) and position 28 or 29 (helix I) are subjected to NaDodSO4/PAGE and probed with anti-C-terminal antibody, a single band migrating at a Mr of about 20 kDa is observed (Fig. 2A, lanes 1 and 3). Strikingly, however, when the membranes are oxidized with iodine prior to electrophoresis, high-efficiency crosslinking is observed (>20%), as evidenced by the appearance of a band migrating at the Mr of intact lac permease with the biotin acceptor domain (i.e., ≈52 kDa; lanes 2 and 4). In contrast, no crosslinking is detected between Cys residues at position 242 and 27, which is on the opposite face of helix I (Fig. 2A, lanes 5 and 6; see Fig. 6). In addition to anti-C-terminal antibody, both the N6 fragment, which migrates at ≈35 kDa, and the 52-kDa band react with avidin-HRP (Fig. 2B). Furthermore, when the oxidized samples are electrophoresed in the presence of dithiothreitol, the 52-kDa band is not evident when the blots are probed with either antibody or avidin-HRP (data not shown), demonstrating that the high molecular weight band represents disulfide crosslinked N6/C6. As determined by the same methodology, disulfide crosslinking is also observed between a Cys residue at position 245, which is on the same face of helix VII as position 242 (see Fig. 6) and a Cys residue at either position 28 or 29, but not with a Cys residue at position 27 (Table 1).
Figure 6.
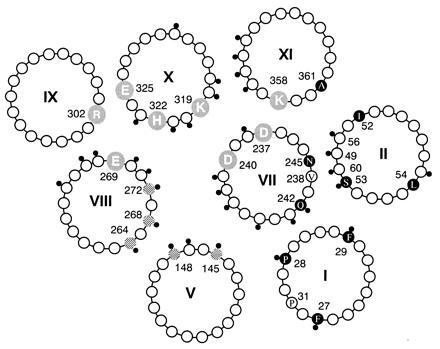
Packing of helices I, II, V, and VII–XI in the lac permease viewed from the periplasmic surface. The four essential residues (Glu-269, Arg-302, His-322, and Glu-325), two interacting pairs of Asp–Lys residues [Asp-237 (helix VII)/Lys-358 (helix XI), and Asp-240 (helix VII)/Lys-319 (helix X)] are highlighted. Positions of NEM-sensitive Cys replacements are indicated with a small black dot. Substrate protectable single Cys replacement mutants (145, 148, 264, 268, and 272) are crosshatched. Cys replacement mutants tested for disulfide crosslinking in this study are shown as solid circles. The arrangement of helices VII–XI has been described (reviewed in ref. 11). The placement of helix V has been established by site-directed chemical cleavage (23), site-directed electron spin resonance, and thiol crosslinking (24). The positioning of helices I and II is based on the results of this study.
Crosslinking Between Helices II and VII.
C6 with a Cys residue at position 245 (helix VII) was also coexpressed with N6 fragments containing a Cys residue at position 52, 53, or 54 (helix II). Oxidation of N6/C6 with paired Cys residues at positions 245 and 52 or 53 results in highly significant crosslinking (Fig. 3), which is reversed under reducing conditions (not shown). The crosslinked product reacts with both anti-C-terminal antibody (Fig. 3), as well as avidin-HRP (not shown). However, no crosslinking is detected with N6/C6 containing paired Cys residues at positions 245 and 54, which is on the opposite face of helix II from positions 52 and 53 (Table 1; see Fig. 6). Crosslinking of N6/C6 with paired Cys residues at positions 242 and 52 or 53, as well as low but significant crosslinking (<10%) between positions 242 and 54 is also observed (Table 1).
Figure 3.

Disulfide crosslinking of N6/C6 permease fragments containing paired Cys residues in helices VII and II. Membranes were prepared from E. coli T184 expressing N6 and C6 with paired Cys residue at position 245 in helix VII and 52 (A) or 53 (B) in helix II, and crosslinking was carried out as described in Fig. 2. Blots were probed with anti-C-terminal antibody.
Crosslinking Between Helices II and XI.
To examine further the relative positions of helices I and II in the tertiary structure of the permease, C6 containing a Cys residue at position 361 (helix XI) was coexpressed with N6 containing a Cys residue at position 27, 28, 29 (helix I) or position 52, 53, 54 (helix II). Among the six paired constructs tested (Table 1), low-efficiency N6/C6 crosslinking (<10%) is observed between Cys residues at position 361 and 52 or 53 (Fig. 4). However, no detectable crosslinking is observed between position 361 and 54 nor between position 361 and 27, 28, or 29 in helix I (Table 1).
Figure 4.
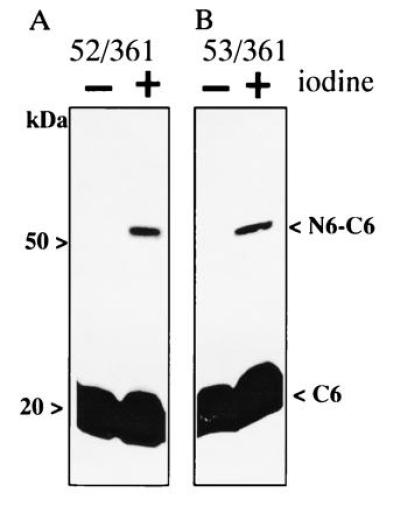
Disulfide crosslinking of N6/C6 permease fragments containing paired Cys residues in helices XI and II. Membranes were prepared from E. coli T184 expressing N6 and C6 with paired Cys residue at position 361 in helix XI and 52 (A) or 53 (B) in helix II, and crosslinking was carried out as described in Fig. 2. Blots were probed with anti-C-terminal antibody.
Chemical Crosslinking.
In addition to iodine-catalyzed disulfide bond formation, homobifunctional thiol crosslinking agents such as 1,6-bismaleimidohexane (BMH) and N,N′-p-phenylenedimaleimide have been used to crosslink each of the pairs described with results similar to those described for iodine. A representative example with BMH is shown in Fig. 5.
Figure 5.
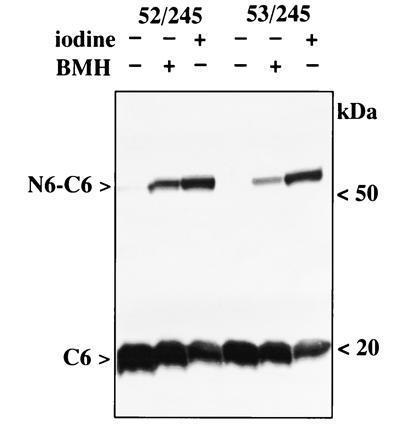
Chemical crosslinking of helices VII and II. Membranes were prepared from E. coli T184 expressing N6 and C6 with paired Cys residue at position 245 in helix VII and 52 (left) or 53 (right) in helix II. Iodine-catalyzed crosslinking was carried out as described in Fig. 2. For BMH crosslinking, membranes (2 mg protein per ml) were incubated with 0.5 mM BMH at room temperature for 10 min, and the reaction was terminated by adding dithiothreitol to a final concentration of 10 mM. Samples were probed with anti-C-terminal antibody. Although data are not shown, results similar to those observed with iodine were also obtained with BMH, as well as N,N′-p-phenylenedimaleimide, and each of the other paired Cys mutants described.
DISCUSSION
As part of a continuing effort to develop alternative methodologies for determining helix packing in membrane proteins that are resistant to crystallization (reviewed in ref. 11), we have now used site-directed disulfide crosslinking of coexpressed permease fragments, each with a single Cys residue at a defined position, a technique introduced to study tertiary interactions in visual rhodopsin (25). As shown, crosslinking is evident between a Cys residue at position 242 or 245 (helix VII) and a Cys residue at position 52 or 53 (helix II). In contrast, no crosslinking is observed between position 245 and position 54, which is on the opposite face of helix II from Ile-52 and Ser-53. Thus, the face of helix VII that contains Gln-242 and Asn-245 must be in close proximity to the face of helix II that contains Ile-52 and Ser-53 (Fig. 6). Moreover, weak crosslinking is observed between position 52 or 53 and position 361 in helix XI. Taken together, the data indicate that helix II is close to both helices VII and XI.
The placement of helix II (Fig. 6) is consistent with a number of other observations: (i) Asp-237 (helix VII) interacts functionally with Lys-358 (helix XI) (reviewed in ref. 11, and see ref. 18) and bis-His replacements at these positions form an Mn2+ binding site (21). (ii) Replacement of Asp-68 at the cytoplasmic end of helix II with Ser, Thr, or Glu abolishes permease activity (36), and several second-site revertants of D68S or D68T exhibit high activity (37). Interestingly, in 7 of 18 revertants, the inactive phenotype is suppressed by a mutation in the cytoplasmic half of helix VII in which Cys-234 is replaced with a bulkier residue (Phe or Trp). Moreover, 6 of 18 second-site suppressor mutations of D68S or D68T map at the periplasmic end of helix XI (Ser-366 → Phe, Val-367 → Glu, or Ala-369 → Pro). (iii) Substrate binding reduces the reactivity of single Cys mutant V238C (helix VII) with NEM (S. Frillingos, J.W., and H.R.K., unpublished results) and the emission spectrum of V238C permease labeled with 2-(4′-maleimidylanilino)-naphthalene-6-sulfonic acid is blue-shifted (J.W., S. Frillingos, and H.R.K., unpublished results), suggesting that this face of helix VII is conformationally active. (iv) Cys-scanning mutagenesis of helix II demonstrates that the NEM-sensitive Cys-replacement mutants lie on the same face as Ile-52 and Ser-53 (30). Therefore, helix II is positioned in such a manner that the NEM-sensitive face of helix II is in contact with the conformationally active face of helix VII (Fig. 6).
A Cys residue at position 28 or 29 (helix I) forms a disulfide with a Cys residue at position 242 or 245 (helix VII). Since no crosslinking is evident between position 242 or 245 and position 27, it is likely that the face of helix I containing Pro-28 and Phe-29 is in close proximity to the face of helix VII containing Gln-242 and Asn-245 (Fig. 6). It is noteworthy that the face of helix I containing Pro-28 and Pro-31 undergoes a ligand-induced conformational change (38). In addition, site-directed chemical cleavage (23), spin labeling, and chemical crosslinking (24) demonstrate close proximity between helices V, VII, and VIII. Because Cys-148 is clearly a component of a substrate binding site and Met-145 is in the vicinity (32, 39), helix I is not only in close proximity to helices VII and V, but may be oriented in such a manner that the conformationally active face containing Pro-28 and Pro-31 is in the vicinity of Cys-148 and Met-145 in helix V (Fig. 6).
Finally, the results presented here indicate that crosslinking of genetically engineered protein fragments in situ may represent a general approach for analyzing helix packing of polytopic membrane proteins without the necessity of protein purification. A similar strategy has been used to study the dimeric Tar receptor where both inter- and intra-subunit site-directed disulfide crosslinking have been observed (40–42). The split approach is particularly useful when intramolecular crosslinking does not alter the electrophoretic mobility of the protein, which is the case for most polytopic membrane proteins. Regarding functional complementation of split membrane proteins, the available evidence indicates that polytopic membrane proteins preserve function and tertiary structure when the peptide backbone is cleaved within the hydrophilic domains. Therefore, the forces between transmembrane domains in these proteins are apparently able to maintain tertiary organization in the membrane. For example, lac permease fragments with discontinuities in various periplasmic or cytoplasmic loops, but not within transmembrane domains, form functional complexes (26, 27, 43, 44). In addition, functional complementation of split membrane proteins has been observed with bacteriorhodopsin (45, 46), the β-adrenergic receptor (47), a voltage-dependent Na+ channel (48), the yeast a factor transporter (49), adenylate cyclase (50), the muscarinic acetylcholine receptor (51, 52), and the erythroid glucose transporter GLUT1 (53). Therefore, it is apparent that the approach described here may be applicable to a variety of integral membrane proteins.
Acknowledgments
This work was supported in part by National Institutes of Health Grant 1 R0 1 DK51131-01.
Footnotes
Abbreviations: lac, lactose; N6, the N-terminal six transmembrane helices of lac permease; C6, the C-terminal six transmembrane helices of lac permease; NEM, N-ethylmaleimide; BMH, 1,6-bismaleimidohexane; HRP, horseradish peroxidase.
References
- 1.Viitanen P, Newman M J, Foster D L, Wilson T H, Kaback H R. Methods Enzymol. 1986;125:429–452. doi: 10.1016/s0076-6879(86)25034-x. [DOI] [PubMed] [Google Scholar]
- 2.Sahin-Tóth M, Lawrence M C, Kaback H R. Proc Natl Acad Sci USA. 1994;91:5421–5425. doi: 10.1073/pnas.91.12.5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foster D L, Boublik M, Kaback H R. J Biol Chem. 1983;258:31–34. [PubMed] [Google Scholar]
- 4.Kaback H R. Int Rev Cytol. 1992;137:97–125. doi: 10.1016/s0074-7696(08)62674-1. [DOI] [PubMed] [Google Scholar]
- 5.Zen K, Consler T G, Kaback H R. Biochemistry. 1995;34:3430–3437. doi: 10.1021/bi00010a035. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Wu J, Carrasco N, Kaback H R. Biochemistry. 1996;35:990–998. doi: 10.1021/bi952166w. [DOI] [PubMed] [Google Scholar]
- 7.Sun, J., Frillingos, S. & Kaback, H. R. (1996) Biochemistry, in press.
- 8.Calamia J, Manoil C. Proc Natl Acad Sci USA. 1990;87:4937–4941. doi: 10.1073/pnas.87.13.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaback H R, Jung K, Jung H, Wu J, Privé G G, Zen K. J Bioenerg Biomembr. 1993;25:627–636. doi: 10.1007/BF00770250. [DOI] [PubMed] [Google Scholar]
- 10.Kaback H R, Frillingos S, Jung H, Jung K, Privé G G, Ujwal M L, Weitzman C, Wu J, Zen K. J Exp Biol. 1994;196:183–195. doi: 10.1242/jeb.196.1.183. [DOI] [PubMed] [Google Scholar]
- 11.Kaback H R. In: The Lactose Permease of Escherichia coli: Past, Present and Future. Konings W N, Kaback H R, Lolkema J S, editors. Vol. 2. Amsterdam: Elsevier; 1996. pp. 203–227. [Google Scholar]
- 12.Jung K, Jung H, Wu J, Privé G G, Kaback H R. Biochemistry. 1993;32:12273–12278. doi: 10.1021/bi00097a001. [DOI] [PubMed] [Google Scholar]
- 13.Lee J I, Varela M F, Wilson T H. Biochim Biophys Acta. 1996;1062:111–118. doi: 10.1016/0005-2736(95)00209-x. [DOI] [PubMed] [Google Scholar]
- 14.King S C, Hansen C L, Wilson T H. Biochim Biophys Acta. 1991;1062:177–186. doi: 10.1016/0005-2736(91)90390-t. [DOI] [PubMed] [Google Scholar]
- 15.Sahin-Tóth M, Dunten R L, Gonzalez A, Kaback H R. Proc Natl Acad Sci USA. 1992;89:10547–10551. doi: 10.1073/pnas.89.21.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunten R L, Sahin-Toth M, Kaback H R. Biochemistry. 1993;32:3139–3145. doi: 10.1021/bi00063a028. [DOI] [PubMed] [Google Scholar]
- 17.Sahin-Tóth M, Kaback H R. Biochemistry. 1993;32:10027–10035. doi: 10.1021/bi00089a019. [DOI] [PubMed] [Google Scholar]
- 18.Frillingos, S. & Kaback, H. R. (1996) Biochemistry, in press.
- 19.Lee J L, Hwang P P, Hansen C, Wilson T H. J Biol Chem. 1992;267:20758–20764. [PubMed] [Google Scholar]
- 20.Jung K, Voss J, He M, Hubbell W L, Kaback H R. Biochemistry. 1995;34:6272–6277. doi: 10.1021/bi00019a003. [DOI] [PubMed] [Google Scholar]
- 21.He M M, Voss J, Hubbell W L, Kaback H R. Biochemistry. 1995;34:15661–15666. doi: 10.1021/bi00048a009. [DOI] [PubMed] [Google Scholar]
- 22.He M M, Voss J, Hubbell W L, Kaback H R. Biochemistry. 1995;34:15667–15670. doi: 10.1021/bi00048a010. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Perrin D, Sigman D, Kaback H. Proc Natl Acad Sci USA. 1995;92:9186–9190. doi: 10.1073/pnas.92.20.9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Voss J, Hubbell W L, Kaback H R. Proc Natl Acad Sci USA. 1996;93:10123–10127. doi: 10.1073/pnas.93.19.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Kono M, McKee T D, Oprian D D. Biochemistry. 1995;34:14963–14969. doi: 10.1021/bi00046a002. [DOI] [PubMed] [Google Scholar]
- 26.Bibi E, Kaback H R. Proc Natl Acad Sci USA. 1990;87:4325–4329. doi: 10.1073/pnas.87.11.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zen K H, McKenna E, Bibi E, Hardy D, Kaback H R. Biochemistry. 1994;33:8198–8206. doi: 10.1021/bi00193a005. [DOI] [PubMed] [Google Scholar]
- 28.Carrasco N, Herzlinger D, Mitchell R, DeChiara S, Danho W, Gabriel T F, Kaback H R. Proc Natl Acad Sci USA. 1984;81:4672–4676. doi: 10.1073/pnas.81.15.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahin-Tóth M, Persson B, Schwieger J, Cohan M, Kaback H R. Protein Sci. 1994;3:240–247. doi: 10.1002/pro.5560030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frillingos, S., Sun, J., Gonzalez, A. & Kaback, H. R. (1996) Biochemistry, in press.
- 31.Cronan J E., Jr J Biol Chem. 1990;265:10327–10333. [PubMed] [Google Scholar]
- 32.Wu J, Kaback H R. Biochemistry. 1994;33:12166–12171. doi: 10.1021/bi00206a020. [DOI] [PubMed] [Google Scholar]
- 33.Frillingos S, Kaback H R. Biochemistry. 1996;35:5333–5338. doi: 10.1021/bi953068d. [DOI] [PubMed] [Google Scholar]
- 34.Consler T G, Tsolas O, Kaback H R. Biochemistry. 1991;30:1291–1298. doi: 10.1021/bi00219a019. [DOI] [PubMed] [Google Scholar]
- 35.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Jessen-Marshall A E, Paul N J, Brooker R J. J Biol Chem. 1995;270:16251–16257. doi: 10.1074/jbc.270.27.16251. [DOI] [PubMed] [Google Scholar]
- 37.Jessen-Marshall A E, Brooker R J. J Biol Chem. 1996;271:1400–1404. doi: 10.1074/jbc.271.3.1400. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Frillingos S, Kaback H R. Biochemistry. 1995;34:8257–8263. doi: 10.1021/bi00026a007. [DOI] [PubMed] [Google Scholar]
- 39.Jung H, Jung K, Kaback H R. Biochemistry. 1994;33:12160–12165. doi: 10.1021/bi00206a019. [DOI] [PubMed] [Google Scholar]
- 40.Falke J J, Koshland D E., Jr Science. 1987;237:1596–1600. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- 41.Pakula A, Simon M. Proc Natl Acad Sci USA. 1992;89:4144–4148. doi: 10.1073/pnas.89.9.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chervitz S A, Falke J J. Proc Natl Acad Sci USA. 1996;93:2545–2550. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wrubel W, Stochaj U, Sonnewald U, Theres C, Ehring R. J Bacteriol. 1990;172:5374–5381. doi: 10.1128/jb.172.9.5374-5381.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wrubel W, Stochaj U, Ehring R. FEBS Lett. 1994;349:433–438. doi: 10.1016/0014-5793(94)00719-5. [DOI] [PubMed] [Google Scholar]
- 45.Liao M J, Huang K S, Khorana H G. J Biol Chem. 1984;259:4200–4204. [PubMed] [Google Scholar]
- 46.Popot J-L, Gerchman S-E, Engelman D M. J Mol Biol. 1987;198:655–676. doi: 10.1016/0022-2836(87)90208-7. [DOI] [PubMed] [Google Scholar]
- 47.Kobilka B K, Kobilka T S, Daniel K, Regan J W, Caron M G, Lefkowitz R J. Science. 1988;240:1310–1316. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- 48.Stuhmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Nature (London) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 49.Berkower C, Michaelis S. EMBO J. 1991;10:3777–3785. doi: 10.1002/j.1460-2075.1991.tb04947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang W J, Krupinski J, Gilman A G. J Biol Chem. 1991;266:8595–8603. [PubMed] [Google Scholar]
- 51.Maggio R, Vogel Z, Wess J. FEBS Lett. 1993;319:195–200. doi: 10.1016/0014-5793(93)80066-4. [DOI] [PubMed] [Google Scholar]
- 52.Schoneberg T, Liu J, Wess J. J Biol Chem. 1995;270:18000–18006. doi: 10.1074/jbc.270.30.18000. [DOI] [PubMed] [Google Scholar]
- 53.Cope D L, Holman G D, Baldwin S A, Wolstenholme A J. Biochem J. 1994;300:291–294. doi: 10.1042/bj3000291. [DOI] [PMC free article] [PubMed] [Google Scholar]