

Abstract
Increased histone acetylation has been correlated with increased transcription, and regions of heterochromatin are generally hypoacetylated. In investigating the cause-and-effect relationship between histone acetylation and gene activity, we have characterized two yeast histone deacetylase complexes. Histone deacetylase-A (HDA) is an ≈350-kDa complex that is highly sensitive to the deacetylase inhibitor trichostatin A. Histone deacetylase-B (HDB) is an ≈600-kDa complex that is much less sensitive to trichostatin A. The HDA1 protein (a subunit of the HDA activity) shares sequence similarity to RPD3, a factor required for optimal transcription of certain yeast genes. RPD3 is associated with the HDB activity. HDA1 also shares similarity to three new open reading frames in yeast, designated HOS1, HOS2, and HOS3. We find that both hda1 and rpd3 deletions increase acetylation levels in vivo at all sites examined in both core histones H3 and H4, with rpd3 deletions having a greater impact on histone H4 lysine positions 5 and 12. Surprisingly, both hda1 and rpd3 deletions increase repression at telomeric loci, which resemble heterochromatin with rpd3 having a greater effect. In addition, rpd3 deletions retard full induction of the PHO5 promoter fused to the reporter lacZ. These data demonstrate that histone acetylation state has a role in regulating both heterochromatic silencing and regulated gene expression.
Keywords: chromatin, acetylation, telomeres, PHO5, CUP1
The acetylation of the ɛ-amino group of specific lysines present in the amino termini of histones has been correlated with increased gene activity. Chromosomal regions that are actively transcribed often contain hyperacetylated histones, whereas silent heterochromatic regions are hypoacetylated (1, 2). Examples of this correlation have been demonstrated for hyperacetylated H4 in chromatin near the active chicken globin gene (3), the human platelet-derived growth factor gene (4), and the Drosophila male X chromosome (5). In contrast, histone H4 of human (6) and yeast (7) heterochromatin is distinctly hypoacetylated at H4 lysines 5, 8, and 16. However, in Drosophila (8) and in yeast (9), the fourth site, lysine 12, is acetylated even in heterochromatin.
Since both inactive (but “poised”) and active genes are highly enriched in an acetylated nucleosome fraction (3, 4, 10), it is unlikely that acetylation is merely a consequence of gene activity. Rather, histone acetylation may be a general means of preparing a gene for transcription. This view is supported by in vitro data using episomal DNA, which demonstrates that histone acetylation allows a fraction of nucleosomal DNA to be released from the repressive confines of the core particle into the internucleosomal linker region (11). Moreover, increased histone acetylation enhances the ability of the USF, GAL4 (12), and TFIIA (13) transcription factors to bind in vitro to DNA when contained in a nucleosome.
In a manner that is not understood, certain sites of acetylation have special significance. For example, acetylation of H4 lysines 5 and 12 is associated with nucleosome assembly in a number of diverse eukaryotes (14), and acetylation of H4 lysine 16 is found preferentially in the transcriptionally hyperactive X chromosome of Drosophila male larvae (5, 8). Deletions and mutations of H4 lysines 5, 8, and 12 have little, if any, effect on heterochromatic silencing in yeast. However, a mutation at position 16 can strongly derepress silencing (15–18). Lysine 16 has been shown to be involved in mediating the interaction between the histone H4 N terminus and silencing information regulator (SIR) repressors of yeast heterochromatin in vitro and in whole cell extracts (19, 20). However, modification of lysine 16 may occur independently of H4-SIR interactions and we cannot assume a priori that acetylation is a means of regulating silencing. Similar considerations are also important in euchromatin, where histones may interact with other regulators such as TUP1 (21).
One approach to the study of histone acetylation in vivo has involved the use of sodium butyrate and trichostatin A (TSA) as inhibitors of the deacetylase enzymes (1, 22). However, we do not know how many different histone deacetylases exist in a eukaryotic cell nor whether they are all equally sensitive to such inhibitors. In addition, we do not know the extent to which these inhibitors are specific only for deacetylases. Results using these inhibitors must therefore be interpreted with caution.
To study the cause-and-effect relationship between histone acetylation and transcription more directly in vivo, we have purified histone deacetylase enzyme complexes in the yeast Saccharomyces cerevisiae and identified protein components of these enzymes. These studies have led to the identification of at least two activities [histone deacetylase-A (HDA) and -B (HDB)] that possess different sensitivities to the histone deacetylase inhibitor TSA. HDA (≈350 kDa) is highly sensitive to TSA with over 80% of its activity inhibited in the presence of 10 nM TSA, while HDB (≈600 kDa) is much less sensitive with less than 20% inhibition (23).
We have now cloned and sequenced a component of HDA (which we have designated HDA1). HDA1 shares significant sequence similarity to a factor, RPD3, required for optimal transcription of certain genes in yeast. We further demonstrate that RPD3 is associated with HDB. Disruption of HDA1 and RPD3 affects histone H3 and H4 acetylation, silencing by telomeric heterochromatin and regulated gene activity.
MATERIALS AND METHODS
Cloning and Plasmid Construction.
The gene encoding HDA1 was obtained through probing a blot of the yeast genomic lambda library from American Type Culture Collection (ATCC) with the degenerate oligonucleotide ATCCCIGTIAGAGCTGCTACITC(C/T)GAAGA based on p75 peptide K16 (IPVRAATSEE) (23). After labeling with [γ-32P]ATP hybridization was carried in 6× standard saline citrate (SSC), 1× Denhardt’s solution (0.02% polyvinylpyrrolidone/0.02% Ficoll 400/0.02% bovine serum albumin), and 0.05% sodium pyrophosphate (NaPPi) at 42°C with 106 cpm/ml 32P-labeled oligonucleotide. After an overnight incubation, the membrane was washed 4× with 6× SSC 0.05% (NaPPi) at 23°C for 5 to 10 min each, and then once with 6× SSC 0.05% NaPPi at 42°C for 15 min.
The oligonucleotide showed strong hybridization to a 4.6-kb BamHI fragment from ATCC lambda clone 3194. This fragment was subcloned into pBluescript SK(+) (Stratagene), creating the plasmid pskB93. DNA sequencing confirmed that this gene was identical to a previously identified ORF (GenBank accession no. Z71297Z71297).
The hda1::TRP1 allele was made through deletion of the multiple cloning site of pskB93 through digestion with KpnI/HindIII, and then ExoIII using Erase-A-Base (Promega) resulting in the plasmid psB93. psB93 was digested with SacI/XbaI and then ExoIII digestion, resulting in pB93. An EcoRI/PstI TRP1 fragment from pYRP7 (24) was cloned into the EcoRI/PstI site of pB93, replacing the majority of the HDA1 coding region with TRP1 thus creating pB93TRP.
An rpd3::LEU allele was created through ligating a HindIII–BamHI fragment of the LEU2 gene prepared through PCR amplification of pRS415 (25) using the primers LEU2BAM (CGAGGGATCCCGAGGAGAACTTCTAGTATATC) and LEU2HIND (CGAGAAGCTTCTACGTCGTAAGGCCGTTTCTG) and was cloned into pMV34 (26) digested with HindIII/BamHI, resulting in the plasmid pMVL.
Yeast Strains.
Please see the strain list in Table 1. YEPD and other yeast media used were as described (27). HDA1 disruptions were performed in YDS2U, which is identical to YDS2 (MATa trp1-1 leu2-3 his3-11, 15 ura3-52 ade2-1 can1-100 (28) except that it carries the URA3 gene integrated at the ADH4 locus of chromosome VII L (29). Generation of hda1::TRP mutant allele was performed through PCR amplification from pB93TRP using T3 and D2.11 (ATAGCTCTGTATTATAC) primers before transformation and generation of the hda1Δ-1 mutation confirmed by Southern blotting. This strain was designated SRY2D2. Additionally, an independent hda1::TRP allele was created in YDS3 (28) and was designated SRY3D2.
Table 1.
Yeast strains used in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| YDS3 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | Ref. 27 |
| YDS2U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 URA3::Te1VIIL | This study |
| YDS21U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 URA3::Te1VR (2.1 kb from Te1) | This study |
| YDS35U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 URA3::Te1VR (3.5 kb from Te1) | This study |
| SRYD2U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 hda1::TRP1 URA3::Te1VIIL | This study |
| SRY21U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 hda1::TRP1 URA3::Te1VR (2.1 kb from Te1) | This study |
| SRY35U | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 hda1::TRP1 URA3::Te1VR(3.5 kb from Te1) | This study |
| SRY3D2 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 ura3-1 hda1::TRP1 | This study |
| SRYR34 | MATa ade2-1 can1-100 his3-11,15 12 trp1-1 rpd3:: LEU2 URA3::Te1VIIL | This study |
| SRYR39 | MATa ade2-1 can1-100 his3-11,15 12 trp1-1 rpd3:: LEU2 URA3::Te1VR(2.1 kb from Te1) | This study |
| SRYR40 | MATa ade2-1 can1-100 his3-11,15 12 trp1-1 rpd3:: LEU2 URA3::Te1VR(3.5 kb from Te1) | This study |
| SRYR3 | MATα ade2-1 can1-100 his3-11,15 12 trp1-1 ura3-1rpd3:: LEU2 | This study |
| SRYR38 | MATa ade2-1 can1-100 his3-11,15 12 hda1::TRP1 rpd3:: LEU2 URA3::Te1VIIL | This study |
| SRYR35 | MATα ade2-1 can1-100 his3-11,15 12 trp1-1 ura3-1hda1::TRP1 rpd3::LEU2 | This study |
RPD3 disruptions were performed through digestion of pMVL with AatII and BglII, and isogenic diploid strains heterozygous for hda1::TRP were transformed. After confirmation of the rpd3Δ-1 mutation by Southern blotting, cells were sporulated and rpd3Δ strains were designated either SRYR3 or SRYR34, and hda1Δ rpd3Δ strains were designated SRYR35 and SRYR38.
Generation of YDS21U and YDS35U was performed through integration of URA3 2.1 kb and 3.5 kb from telomere V-R using pVURAH2(+) and pVURAH3(+), respectively, (30) through digestion of these plasmids with NotI before transformation. After confirmation of faithful integration by Southern blotting, these strains were mated to SRYR35. The YDS21U-derived tetrad was then dissected to obtain SRY21U (hda1Δ) and SRYR39 (rpd3Δ)with URA3 at a distance of 2.1 kb from the telomere. The YDS35U tetrad was also dissected to yield SRY35U (hda1Δ) and SRYR40 (rpd3Δ), which contain URA3 at a distance of 3.5 kb from the telomere.
Western Blots.
Unlabeled histones were prepared from cells grown at 30°C in YEPD to an OD600 = 3 through preparation of yeast nuclei using Percoll (31) followed by extraction of the histones for 2 hr in 0.25 M HCl. Extracted histones were then precipitated with 25% trichloroacetic acid (TCA), washed once with 99:1 acetone/5 M HCl, and then once with acetone before resuspending in 8 M urea. Samples containing equal concentrations of histones were subjected to electrophoresis in 18% SDS/PAGE gels (30:0.8) acrylamide/bisacrylamide, and then transferred to Immobilon-P (Millipore) in transfer buffer with 0.1% SDS (32). The blots were probed with rabbit polyclonal antisera recognizing H4 acetyl-lysine 5 (R40/5), H4 acetyl-lysine 8 (R12/8), H4 acetyl-lysine 12 (R20/12), H4 acetyl-lysine 16 (R14/16), H3 acetyl-lysine 9 and 18 (R48/9,18), or H3 acetyl-lysine 14 (R224/14). Detection was performed using ECL (Amersham).
Western blotting of Mono S fractions was performed through SDS/PAGE of proteins in 8% gels followed by transfer to Immobilon-P (Millipore). A RPD3–GST fusion protein was created in pGEX2T (33) through PCR amplification from pMV34 (26) using the primers RPD3.5 (GATCGGATCCACAAAGTATGCCCCTAGTGTTCAG) and RPD3.3 (CGATGAATTCGTCATGCTCAACATGTAGGTCCCTCGC), encoding amino acids 383–433, and cloning into EcoRI–BamHI-digested pGEX2T (33). Rabbit polyclonal antibodies were generated using standard immunological techniques.
Immunoprecipitations of Histone Deacetylase Activity.
Each immunoprecipitation was performed from nuclear extracts prepared from 5 g of yeast cells as described previously (23). Antibodies directed against either HDA1 (23) or RPD3 (described above) were incubated overnight at 0°C with 5 μl of G-Sepharose purified antibody. Approximately 4 mg of protein A-Sepharose beads (equilibrated in PBS) was added to each reaction, and the beads were washed and assayed as described (23). Percoll, Ficoll, and other chromatographic materials were purchased from Pharmacia.
RESULTS
Cloning of HDA1.
To study the influence of histone acetylation on transcription, we have characterized two distinct histone deacetylase activities from S. cerevisiae termed HDA and HDB (23). The HDA activity is very sensitive to TSA (Ki = 9.7 nM), a noncompetitive inhibitor of histone deacetylases (23). SDS/PAGE analysis of purified HDA reveals that four proteins with relative molecular weights of 75, 73, 72, and 71 kDa reproducibly copurify with enzymatic activity.
To characterize the HDA complex, approximately 25 pmol of each protein was obtained, digested “in gel” using Achromobacter protease I (34), and subjected to sequence analysis. Of these, the p73 and p72 peptide sequences identified to date are identical, suggesting that they represent posttranslationally modified forms of each other (23). The sequence data for p75 were used to design degenerate primers that were used in the identification of an open reading frame (ORF) encoding the sequenced peptides (see Materials and Methods). We designate this gene HDA1.
HDA1 Shares Similarity to RPD3 and Three ORFs (HOS1, HOS2, and HOS3).
The encoded HDA1 protein sequence was compared with the sequences of GenBank. Of yeast sequences, the best scores were obtained for a group of sequences that include the RPD3 protein (26), a putative transcriptional regulator, and the gene products of several newly sequenced ORFs that we have termed HDA One Similar (HOS1, HOS2, and HOS3). These sequences were aligned using the pileup program (35) (Fig. 1). Independently, Taunton and colleagues (36) have found that the putative histone deacetylase HD1 from human cells also has similarity to RPD3. The similarity between HD1 and RPD3 (≈60% identity and 78% similarity over 450 amino acids) is greater than that between HDA1 and any of the yeast proteins or ORFs (in the range of 21–28% identity and 46–53% similarity over ≈498–743 amino acids). Therefore, it is possible that HD1 is the human RPD3 homolog. Interestingly, the Mycoplana bullata protein acetylpolyamine amidohydrolase (APH) shares a high degree of similarity to these proteins in this same region (30.9% identity, 55.4% similarity over 371 amino acids). Since deacetylation of acetyl-lysine residues involves recognition and cleavage of the amide bond, the similarity to this prokaryotic protein is particularly interesting. However, since APH is unlikely to be a histone deacetylase (bacteria do not contain histones), the level of similarity between HDA1 and the other yeast protein sequences is clearly insufficient to establish histone deacetylase function. Therefore, we confirmed histone deacetylase function both genetically and biochemically.
Figure 1.

Yeast sequences having similarity to HDA1 histone deacetylase. Comparison of HDA1 (GenBank accession no. Z71297Z71297), RPD3 (GenBank accession no. P32561P32561), HOS1 (GenBank accession nos. Z49219Z49219 23), HOS2 (GenBank accession no. X91837X91837), and HOS3 (GenBank accession no. U43503U43503). The sequences were aligned using the pileup program (Genetics Computer Group, Madison, WI). Regions of identity are blocked and shaded using the bestfit program. HDA1 and RPD3 show approximately 24.5% identity and 48.6% similarity over a region of approximately 498 amino acids. By comparison, the human HD1 protein and RPD3 show 60.3% identity and 78.4% similarity over a region of 450 amino acids, suggesting that HD1 may be the human RPD3 homolog. HDA1 and HOS1 show 21.1% identity, 52.9% similarity over 571 amino acids; HDA1 and HOS2, 28% identity, 51.3% similarity over 522 amino acids; HDA1 and HOS3, 24.9% identity, 45.7% similarity over 743 amino acids.
HDA1 and RPD3 Are Members of Distinct Histone Deacetylase Complexes.
To determine the relationship between HDA1 and RPD3, we created null alleles hda1Δ-1 and rpd3Δ-1 using gene replacement (see Materials and Methods). Histone deacetylase activities from wild-type, hda1Δ, rpd3Δ, and hda1Δ rpd3Δ mutant strains were then obtained through purifications performed in parallel (Fig. 2). When comparing the enzymatic profile from wild-type cells to that of the deacetylase mutants, it is apparent that an hda1 deletion leads to a strong reduction of the major enzyme peak (HDA) (Fig. 2A). Conversely, an rpd3 deletion leads to a decrease of HDB activity (Fig. 2B). These data suggest that while HDA1 is a member of the HDA complex, RPD3 is associated with the HDB complex. Interestingly, hda1Δ rpd3Δ double mutant cells still possess residual deacetylase activity (Fig. 2C). Whether this residual activity is that of a defective HDA or HDB complex or an independent copurifying activity remains to be determined.
Figure 2.
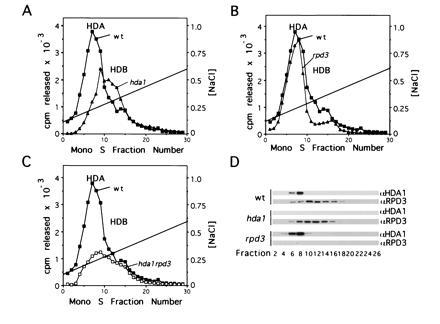
HDA1 and RPD3 are found in different histone deacetylase complexes. hda1 and rpd3 refer to hda1Δ and rpd3Δ, respectively. Mono S profiles of deacetylase activity in (A) YDS2U (wild-type) (▪), SRYD2U (hda1) (▴), (B) YDS2U (wild-type) (▪), SRYR34 (rpd3) (▴), and (C) YDS2U (wild-type) (▪), SRYR38 (hda1rpd3) (□). Nuclear extracts from 100 g cells were prepared and assayed as described (23), and proteins eluting between 150 mM and 350 mM NH4Cl from DEAE–Sepharose FF were dialyzed and chromatographed on Mono S HR 5/5 using identical parameters. Activity (3H-acetate released) is shown as a function of fraction number. (D) Western blots of Mono S fractions from YDS2U (wild-type), SRYD2U (hda1), and SRYR34 (rpd3) were probed with either anti-HDA1 (23) or anti-RPD3, and detected with ECL (Amersham).
We confirmed the presence of HDA1 and RPD3 in HDA and HDB respectively using Western blots of protein present in the Mono S fractions (Fig. 2D). Analysis of proteins present in wild-type preparations shows that HDA1 coelutes with HDA activity while RPD3 coelutes with HDB activity. Western blots of Mono S fractions from the deacetylase mutant strains extend these conclusions by demonstrating that the elution of HDA1 is not dependent on RPD3 nor is RPD3 elution dependent on HDA1. Additionally, immunoprecipitation of HDA histone deacetylase activity using an anti-HDA1 antibody is disrupted by an hda1 but not by an rpd3 deletion. Conversely, HDB histone deacetylase activity immunoprecipitated using an anti-RPD3 antibody is only disrupted by the rpd3 deletion and not by an hda1 deletion (Table 2). These data strongly argue that HDA1 is a component of HDA while RPD3 is a member of an independent activity associated with HDB.
Table 2.
Immunoprecipitation of histone deacetylase activity using anti-HDA1 and anti-RPD3 antibodies
| Antibody | Wild type | hda1 | rpd3 |
|---|---|---|---|
| α-HDA1 | 1683 | 78 | 1521 |
| α-RPD3 | 362 | 346 | 89 |
| Preimmune-HDA1 | 86 | 79 | 83 |
| Preimmune-RPD3 | 79 | 79 | 90 |
| None | 79 | 85 | 82 |
Immunoprecipitations were performed as described (23) with antibodies directed against HDA1 or RPD3 or their preimmune sera. Nuclear extracts were prepared from YDS2U (wild type), SRYD2U (hda1Δ), or SRYR34 (rpd3Δ) as described (23). Each immunoprecipitation used nuclear extracts from approximately 5 g of yeast cells. Data are shown as cpm released in 2 h from 10 μg of a 3H-acetyl-histone substrate with a specific activity of 2.5 × 106 cpm/mg protein.
HDA1 and RPD3 Disruptions Result in Histone Hyperacetylation.
Since histone acetylation is maintained through competing histone acetyltransferase and deacetylase activities, it is expected that a loss of histone deacetylase activity would shift this equilibrium toward increased histone acetylation. To compare the steady-state levels of histone acetylation in wild-type, hda1Δ, and rpd3Δ mutants, we examined the acetylation state of H4 and H3 using acetylation site-specific antibodies. Equal concentrations of histones purified from hda1Δ, rpd3Δ, and hda1Δ rpd3Δ strains were serially diluted and subjected to SDS/PAGE before Western blotting. Identical blots were probed in parallel with antibodies recognizing either H4 acetylated at lysine 5, 8, 12, or 16 (37), or H3 acetylated at lysine 9/18 or 14 (38) (Fig. 3). We observed that histones prepared from hda1Δ cells react with antibodies recognizing each of the sites more strongly than with histones prepared from wild-type cells, suggesting hyperacetylation at all these sites (Fig. 3, compare lanes 1 and 3 containing 1 μg histones and also lanes 2 and 4 containing 3 μg histones). Although it is not possible to determine the exact increase in acetylation using this approach, from the comparison of lane 2 (wild type, 3 μg histone) and lane 3 (hda1Δ, 1 μg histone) we estimate approximately 3-fold or greater acetylation at all positions except H4 positions 8 and 16 (which have 1.5- to 2.0-fold increase). Deletion of RPD3 also leads to increased histone acetylation, but causes greater increases in the acetylation of H4 lysine 5 and 12 and H3 lysine 9/18 compared with hda1Δ (Fig. 3, compare lanes 3 and 5). Since deposition of newly synthesized H4 is associated with acetylation of lysine 5 and 12 (14), this finding raises the possibility that RPD3 may have a role in the deacetylation of H4 after its import into the nucleus.
Figure 3.
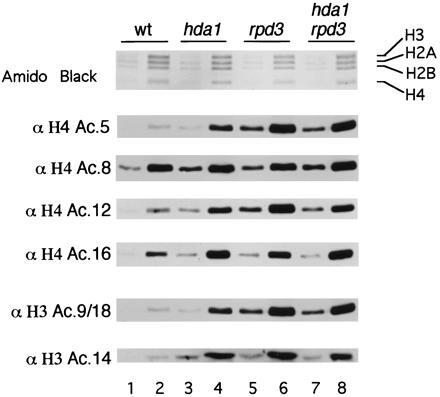
HDA1 and RPD3 disruptions results in histone H4 and H3 hyperacetylation. Relative histone acetylation levels in YDS2U (wild-type) (lanes 1 and 2), SRYD2U (hda1) (lanes 3 and 4), SRYR34 (rpd3) (lanes 5 and 6), and SRYR38 (hda1 rpd3) (lanes 7 and 8) were determined by loading 1 μg (lanes 1, 3, 5, and 7) or 3 μg (lanes 2, 4, 6, and 8) of histones. After transfer, blots were probed separately with antibodies specific for H4 acetyl-lysine 5, 8, 12, or 16. Additonally, blots were probed with antibodies specific for H3 acetyl-lysine 9 and 18 (9/18) or 14. Antibody binding was then detected using ECL.
HDA1 and RPD3 Disruptions Increase Repression at Telomeric Heterochromatin.
We next investigated whether increased histone acetylation levels affect the repression of the silent mating loci and a subtelomeric region. Both of these are in hypoacetylated chromatin and have features associated with heterochromatin (7). Since increased acetylation might be expected to derepress heterochromatin, we examined the mating ability of MATa cells (as a measure of HMLα repression) but found no significant change in mating ability caused by hda1Δ or rpd3Δ (data not shown). The extent of repression in subtelomeric regions can be measured by integrating the URA3 gene near the telomere (in this case, right arm of chromosome V) and monitoring expression of URA3 by sensitivity of cells to the drug 5-fluoroorotic acid (5-FOA) (29). This assay is extremely sensitive in that a several-fold increase in URA3 expression can result in an increase of 6–7 orders of magnitude in 5-FOA sensitivity (18, 29). Contrary to the predicted effect of increased histone acetylation, we find that both hda1Δ and rpd3Δ increased repression of URA3 integrated 2.1 kb (Fig. 4A) and 3.5 kb (Fig. 4B) from the telomere when comparing serial dilutions of mutant and wild-type strains. Quantitation of this effect shows that at 2.1 kb from the telomere, hda1Δ and rpd3Δ cells are 3.6-fold and 24.6-fold, respectively, more resistant to 5-FOA as compared with wild-type cells. When URA3 is integrated at 3.5 kb, hda1Δ and rpd3Δ cells are > 1 × 104 and > 1 × 105 more resistant, respectively. The effect of hda1Δ and rpd3Δ is repression specific in that the growth of these cells on media lacking uracil is indistinguishable from wild type (data not shown).
Figure 4.
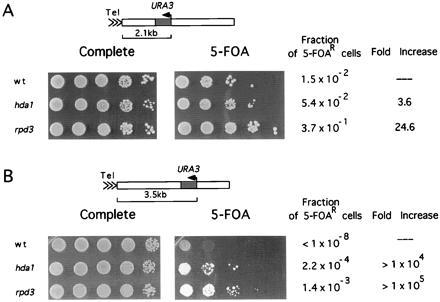
HDA1 and RPD3 disruptions increase telomeric silencing. The extent of URA3 gene repression was determined by measuring resistance to 5-FOA (29) (A) Strains YDS21U (wild-type), SRY21U (hda1), and SRYR39 (rpd3) containing URA3 integrated 2.1 kb from the telomere were grown on YEPD plates for 3 days at 30°C before 10-fold dilutions were spotted onto media with or without 5-FOA. To quantitate the fraction of cells capable of growing on 5-FOA-containing media, diluted cell suspensions were also spread and cell number was determined. (B) Strains YDS35U (wild-type), SRY35U (hda1), and SRYR40 (rpd3) containing URA3 integrated 3.5 kb from the telomere were grown on YEPD plates and assayed as in A.
RPD3 Disruption Retards PHO5 Promoter Activation.
To determine whether the deletion of HDA1 or RPD3 alters regulated gene expression, PHO5 promoter–lacZ fusions were assayed under noninducing and inducing conditions through β-galactosidase assays. In contrast to the effect in wild-type and hda1 cells, an rpd3 deletion retards PHO5–lacZ activation (Fig. 5A) This effect of rpd3Δ on PHO5 induction is consistent with previous data (26). However, the time course demonstrates that the rate of rate of β-galactosidase accumulation in rpd3Δ cells at later time points parallels that of wild-type cells, suggesting that rpd3Δ merely delays PHO5 induction. This delay is also seen for the genomic PHO5 gene whose transcript levels eventually surpass those of wild-type (data not shown). Interestingly, both hda1 and rpd3 deletions cause some hyperactivation at the CUP1–lacZ promoter construct (Fig. 5B).
Figure 5.
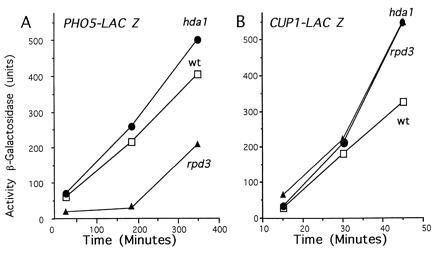
The effects of HDA1 and RPD3 disruptions on gene activity. Determination of noninduced and activated transcription in wild-type, hda1, and rpd3 cells was performed through β-galactosidase assays. YDS3 (wild-type) (□), SRY3D2 (hda1) (•), or SRYR3 (rpd3) (▴) was transformed with either (A) pMH313 (containing a 2.4-kb DraI fragment of the PHO5 promoter fused to lacZ) (39) or (B) pCLUC (containing the entire CUP1 promoter fused to lacZ) (40), and induced for various time periods (38) before harvesting and determining β-galactosidase activity in cell extracts by the following formula: units = OD420 × 103 mg−1·min−1. The values given represent an average of the data obtained from at least five independent transformants. The standard deviations for these data were <30% and <10%, respectively, for the PHO5 and CUP1 promoters.
DISCUSSION
HDA1 and RPD3 Histone Deacetylase Complexes.
Our results indicate that HDA1 and RPD3 are associated with distinct histone deacetylase complexes, HDA and HDB, respectively. The disruption of both HDA1 and RPD3 increase acetylation of histones H3 and H4. However, rpd3Δ causes stronger hyperacetylation at H4 lysines 5 and 12 as compared with hda1Δ. Acetylation of these sites is correlated with histone deposition and it is tempting to speculate that RPD3 may function to deacetylate histones soon after nucleosome assembly.
Interestingly, the hda1Δ rpd3Δ double mutant does not appear to increase acetylation more than either single mutant. This result suggests that the two enzymes may deacetylate the same chromatin domains, and that deletion of one is not compensated for by the other. Alternatively, other histone deacetylase activities may be up-regulated in the absence of HDA1 or RPD3. This latter possibility seems likely since there are three other open reading frames in the yeast genome (HOS1, HOS2, and HOS3) whose encoded proteins also have homology to HDA1 and RPD3 (Fig. 1). Additionally, deletion of HDA1 is accompanied by an increase in the size of the HDB peak of activity (Fig. 2A) and deletion of both HDA1 and RPD3 results in the presence of a significant amount of deacetylase activity (Fig. 2C). We may only be able to determine why the hda1 and rpd3 deletion effects are not additive when we have a better understanding of the chromosomal domains acetylated by these enzymes and the manner in which the enzymes are regulated.
HDA1 and RPD3 Affect Repression of Telomeric Heterochromatin.
The silent HM and telomeric loci, both of which resemble heterochromatin, contain histone H4, which is hypoacetylated at lysines 5, 8 and 16 when compared with active regions of the genome (9). It is expected that increased histone acetylation would cause enhance access of transcription factors to these domains causing derepression. However, the increases in chromosomal acetylation caused by both hda1Δ and rpd3Δ actually result in increased repression of a reporter gene (URA3) integrated at the telomeric end of chromosome V R. The mutation (rpd3Δ) has a larger effect on H4 acetylation and also has a stronger effect on hyper-repression. We did not observe increased repression at the HM locus (HMLα) examined. However, it has been reported that yeast cells made defective for HM silencing can have this phenotype suppressed by a number of extragenic (sds) mutations. One of the SDS genes, SDS6 is identical to RPD3 (41). Therefore, it is likely that defective RPD3 function increases repression at the silent mating loci and that this is seen only when repression of the HM loci is enfeebled.
It is possible that the correlation we observe between increased genomic acetylation and stronger repression of heterochromatin is due to increased acetylation of H4 lysine 12. This modification has been suggested to enhance the interaction between H4 and the silencing information regulators SIR3 and SIR4 (9). An alternative explanation is that the effects of increased acetylation on silencing are indirect through increased SIR3 expression as discussed below.
HDA1, RPD3, and Gene Regulation in Yeast.
Bulk yeast chromatin is highly acetylated in wild-type cells (42). Therefore, it is interesting that hda1Δ and rpd3Δ mutations alter gene expression in the ostensibly hyperacetylated euchromatic regions. Again, we find that rpd3Δ has the greatest effect on transcription, retarding full activation of PHO5-lacZ. Our analysis of genomic PHO5 by Northern blotting demonstrates similar retardation of PHO5 expression, except that PHO5 mRNA levels in rpd3Δ cells do eventually catch up with and even surpass those of wild-type cells (data not shown). Since hda1Δ and rpd3Δ lead to increases in CUP1 promoter activity, increased histone H3 and H4 acetylation levels may cause hyperactivation of a number of genes. Hyperactivation would be expected if increased acetylation allows enhanced transcription factor binding to chromatin (13). However, if genes involved in silencing heterochromatin are also transcribed at higher levels this could increase telomeric silencing. This possibility must be considered since even modest increases in SIR3 concentrations resulting from an additional SIR3 gene on a centromeric, single copy plasmid increases both silencing (30) and SIR3 spreading (20) considerably.
Genetically and biochemically, we have identified members of two histone deacetylases (HDA1 and RPD3) in yeast cells. The similarity of HDA1 and RPD3 and the finding that an RPD3 homolog (HD1) in humans interacts with the deacetylase inhibitors TSA and trapoxin suggests that these proteins may be the catalytic subunits of the histone deacetylase complexes (36). On the basis of sequence similarity, there may be a family of five histone deacetylases. Whether they are all in distinct histone deacetylase complexes (or even whether they are all histone deacetylases) is at this time uncertain. This must await a genetic and biochemical analyses of the HOS mutants. However, at this stage one must certainly entertain the possibility that several conserved histone deacetylases exist in yeast and that they are not totally redundant in function. The genetic and biochemical approaches described above should help us to understand how they mediate histone acetylation, heterochromatin, chromatin assembly, and gene activity states.
Acknowledgments
We thank Danny Rice for aid in sequence analysis, Thomas Sutherland for his assistance with yeast fermentation, and Sabine Strahl-Bolsinger for the use of yeast strain YDS2U. We are grateful to R. Mann, X. J. Ma, and other members of the Grunstein laboratory for helpful criticisms of this manuscript. We also thank Kevin Struhl for communicating unpublished data. This work was supported by Public Health Services National Institutes of Health Grants GM23674 and GM42421 (to M.G.), a National Research Service Award Postdoctoral Fellowship to S.E.R., and a National Cancer Institute National Research Service Award to A.A.C. Any inquiries regarding this work should be directed to M.G.
Footnotes
Abbreviations: HDA and HDB, histone deacetylase-A and -B, respectively; TSA, trichostatin A; SIR, silencing information regulator; 5-FOA, 5-fluoroorotic acid; ORF, open reading frame.
References
- 1.Grunstein M. Annu Rev Cell Biol. 1990;6:643–678. doi: 10.1146/annurev.cb.06.110190.003235. [DOI] [PubMed] [Google Scholar]
- 2.Turner B M. Cell. 1993;75:5–8. [PubMed] [Google Scholar]
- 3.Hebbes T R, Thorne A W, Clayton A L, Crane-Robinson C. Nucleic Acids Res. 1992;20:1017–1022. doi: 10.1093/nar/20.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clayton A L, Hebbes T R, Thorne A W, Crane-Robinson C. FEBS Lett. 1993;336:23–26. doi: 10.1016/0014-5793(93)81601-u. [DOI] [PubMed] [Google Scholar]
- 5.Bone J R, Lavender J, Richman R, Palmer M J, Turner B M, Kuroda M I. Genes Dev. 1994;8:96–104. doi: 10.1101/gad.8.1.96. [DOI] [PubMed] [Google Scholar]
- 6.Jeppesen P, Turner B M. Cell. 1993;74:281–289. doi: 10.1016/0092-8674(93)90419-q. [DOI] [PubMed] [Google Scholar]
- 7.Braunstein M, Rose A B, Holmes A G, Allis C D, Broach J R. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- 8.Turner B T, Birley A J, Lavender J. Cell. 1992;69:375–384. doi: 10.1016/0092-8674(92)90417-b. [DOI] [PubMed] [Google Scholar]
- 9.Braunstein M, Sobel R R, Allis C D, Turner B, Broach J R. Mol Cell Biol. 1996;16:4349–4356. doi: 10.1128/mcb.16.8.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill L P, Turner B M. EMBO J. 1995;14:3946–3957. doi: 10.1002/j.1460-2075.1995.tb00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norton V G, Imai B S, Yau P, Bradbury E M. Cell. 1989;57:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- 12.Vettese-Dadey M, Grant P A, Hebbes T R, Crane-Robinson C, Allis C D, Workman J L. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- 13.Lee D Y, Hayes J J, Pruss D, Wolffe A P. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 14.Sobel R E, Cook R G, Perry C A, Annunziato A T, Allis C D. Proc Natl Acad Sci USA. 1995;92:1237–1241. doi: 10.1073/pnas.92.4.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayne P S, Kim U J, Han M, Mullen J R, Yoshizaki F, Grunstein M. Cell. 1988;55:27–39. doi: 10.1016/0092-8674(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 16.Johnson L M, Fisher-Adams G, Grunstein M. EMBO J. 1992;11:2201–2209. doi: 10.1002/j.1460-2075.1992.tb05279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Megee P C, Morgan B A, Mittman B A, Smith M M. Science. 1990;247:841–845. doi: 10.1126/science.2106160. [DOI] [PubMed] [Google Scholar]
- 18.Thompson J S, Ling X, Grunstein M. Nature (London) 1994;369:245–247. doi: 10.1038/369245a0. [DOI] [PubMed] [Google Scholar]
- 19.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser S M, Grunstein M. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- 20.Hecht A, Strahl-Bolsinger S, Grunstein M. Nature (London) 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- 21.Cooper J P, Roth S Y, Simpson R T. Genes Dev. 1994;8:1400–1410. doi: 10.1101/gad.8.12.1400. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida M, Horinouchi S, Beppu T. BioEssays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
- 23.Carmen A S, Rundlett S E, Grunstein M. J Biol Chem. 1996;271:15837–15844. doi: 10.1074/jbc.271.26.15837. [DOI] [PubMed] [Google Scholar]
- 24.Struhl K, Stinchcomb D T, Scherer S, Davis R W. Proc Natl Acad Sci USA. 1979;76:1035–1039. doi: 10.1073/pnas.76.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidal M, Gaber R F. Mol Cell Biol. 1991;11:6317–6327. doi: 10.1128/mcb.11.12.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 28.Laman H, Balderes D, Shore D. Mol Cell Biol. 1995;15:3608–3617. doi: 10.1128/mcb.15.7.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottschling D E, Aparicio O M, Billington B L, Zakian V A. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- 30.Renauld H, Aparicio O M, Zierath P D, Billington B L, Chhablani S K, Gottschling D E. Genes Dev. 1993;7:1133–1145. doi: 10.1101/gad.7.7a.1133. [DOI] [PubMed] [Google Scholar]
- 31.Ide G J, Saunders C A. Curr Genet. 1981;4:85–90. doi: 10.1007/BF00365686. [DOI] [PubMed] [Google Scholar]
- 32.Thiriet C, Albert P. Electrophoresis. 1995;16:357–361. doi: 10.1002/elps.1150160161. [DOI] [PubMed] [Google Scholar]
- 33.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 34.Kassavetis G A, Nguyen S T, Kobayashi R, Kumar A, Geiduschek E P, Pisano M. Proc Natl Acad Sci USA. 1995;92:9786–9790. doi: 10.1073/pnas.92.21.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taunton J, Hassig C A, Schreiber S L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 37.Clarke D J, O’Neill L P, Turner B. Biochem J. 1993;294:557–561. doi: 10.1042/bj2940557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balyaev N D, Keohane A M, Turner B M. Hum Genet. 1996;97:573–578. doi: 10.1007/BF02281863. [DOI] [PubMed] [Google Scholar]
- 39.Han M, Grunstein M. Cell. 1988;55:1137–1145. doi: 10.1016/0092-8674(88)90258-9. [DOI] [PubMed] [Google Scholar]
- 40.Hull M W, Erickson J, Johnston M, Engelke D R. Mol Cell Biol. 1994;14:1266–1277. doi: 10.1128/mcb.14.2.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sussel L, Vannier D, Shore D. Genetics. 1995;141:873–888. doi: 10.1093/genetics/141.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson D A. J Biol Chem. 1982;257:1565–1568. [PubMed] [Google Scholar]