

Abstract
Leishmania are parasites that survive within macrophages by mechanism(s) not entirely known. Depression of cellular immunity and diminished production of interleukin 1β (IL-1β) and tumor necrosis factor α are potential ways by which the parasite survives within macrophages. We examined the mechanism(s) by which lipophosphoglycan (LPG), a major glycolipid of Leishmania, perturbs cytokine gene expression. LPG treatment of THP-1 monocytes suppressed endotoxin induction of IL-1β steady-state mRNA by greater than 90%, while having no effect on the expression of a control gene. The addition of LPG 2 h before or 2 h after endotoxin challenge significantly suppressed steady-state IL-1β mRNA by 90% and 70%, respectively. LPG also inhibited tumor necrosis factor α and Staphylococcus induction of IL-1β gene expression. The inhibitory effect of LPG is agonist-specific because LPG did not suppress the induction of IL-1β mRNA by phorbol 12-myristate 13-acetate. A unique DNA sequence located within the −310 to −57 nucleotide region of the IL-1β promoter was found to mediate LPG’s inhibitory activity. The requirement for the −310 to −57 promoter gene sequence for LPG’s effect is demonstrated by the abrogation of LPG’s inhibitory activity by truncation or deletion of the −310 to −57 promoter gene sequence. Furthermore, the minimal IL-1β promoter (positions −310 to +15) mediated LPG’s inhibitory activity with dose and kinetic profiles that were similar to LPG’s suppression of steady-state IL-1β mRNA. These findings delineated a promoter gene sequence that responds to LPG to act as a “gene silencer,” a function, to our knowledge, not previously described. LPG’s inhibitory activity for several mediators of inflammation and the persistence of significant inhibitory activity 2 h after endotoxin challenge suggest that LPG has therapeutic potential and may be exploited for therapy of sepsis, acute respiratory distress syndrome, and autoimmune diseases.
Leishmania are obligate intracellular protozoan parasites of mammalian macrophages. The hallmark of leishmaniasis is macrophage deactivation. Studies of the immunopathogenesis of leishmaniasis have reported that macrophage antileishmanial activity is decreased by several cytokines (1–4). Deactivation by cytokines explains events during acute disease, but initial events of how Leishmania enters the macrophage without activating macrophages remain poorly defined.
Several lines of evidence suggest that lipophosphoglycan (LPG), a conserved major glycolipid molecule on the surface of all Leishmania species, is required for intracellular survival and may mediate macrophage deactivation (for reviews, see refs. 5 and 6–8). The most direct evidence is the rapid elimination of LPG-deficient leishmania and the protection from macrophage killing conferred by passive transfer of LPG to LPG-deficient parasite (6, 8). Moreover, LPG is rapidly transferred from the parasite to the surface of the macrophage after in vitro infection suggesting that LPG when shed from the leishmania may mediate deactivation of macrophage functions (8–10).
In studies of patients with acute visceral leishmaniasis, we observed diminished in vitro production of interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) in response to bacterial lipopolysaccharide (LPS, endotoxin) and heat-killed Listeria (11). Similarly, macrophages infected in vitro with Leishmania donovani amastigotes or treated with purified LPG produced lower amounts of IL-1β (10, 12–14). IL-1β is known to be an important mediator of immunity and inflammation (15–18). However, the mechanism(s) by which LPG down-regulates cell function and cytokine gene expression is not known (10). Herein, we report that LPG affects IL-1β gene expression by predominantly suppressing transcriptional activity, and a unique DNA sequence of the IL-1β promoter mediates LPG’s inhibitory effect. Furthermore, the kinetics of LPG’s inhibitory activity and suppression of several agonists (e.g., endotoxin, TNF-α, and Staphylococcus) relevant to clinical states of overt activation of macrophages were defined. The potential to exploit LPG as an antiinflammatory agent for clinical use in overt cell-activation states is discussed.
MATERIALS AND METHODS
Human Monocytes and Their Isolation, Cell Lines, and Reagents.
Human peripheral blood cells obtained by adherence to plastic were 99% monocytes (3, 4). Human monocytic cells THP-1 (TIB-202) and plasmid containing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA were obtained from American Type Culture Collection (ATCC no. 57090). Plasmids containing IL-1β cDNA were obtained from Steve Gillis, Immunex, Seattle, WA, or from ATCC (no. 39925) (19). Reagents and manufacturers were as follows: endotoxin from Escherichia coli serotype 055:B5 (Sigma) and TNF-α (gift from Genentech).
LPG.
LPG was isolated and purified from L. donovani promastigotes as described (20). The molecular weight of LPG was taken as 9.5 × 106, and 10 μg/ml is equal to 1 μM. Structurally, LPG consists of four distinct domains: (i) a saccharide cap, (ii) a repeating phosphodisaccharides galactosyl-mannose, (iii) a phosphosaccharide core, and (iv) a lyso-alkyl-phosphatidylinositol (5). Two structural domains of LPG used in these experiments were repeating phosphodisaccharides galactosyl-mannose, and lyso-alkyl-phosphatidylinositol. These LPG fragments were prepared as described elsewhere (21, 22). The methods for isolation and treatment of LPG and fragments removes or deactivates endotoxin. The LPG and LPG fragments used in these experiments was free of protein and endotoxin (<10 pg per 100 μg, as determined by the Limmulus amebocyte assay) (20).
Cell Culture and Treatment with LPG and Stimuli.
Human monocytes were cultured in endotoxin-free complete medium (3, 4). THP-1 monocytic cells were maintained in complete medium [RPMI 1640 medium/2 mM l-glutamine/penicillin (100 units/ml)/streptomycin (100 μg/ml), supplemented with 5 × 10−5 M 2-mercaptoethanol, and 10% heat-inactivated fetal bovine serum]. For experiments to determine the effect of LPG, THP-1 cells (10 × 106 cells per condition) in complete medium were treated with or without 0.01–2 μM LPG (0.1–20 μg/ml) for different times either before (− h), simultaneously with (0 h), or after (+ h) the addition of inducers of IL-1β including endotoxin, TNF-α, phorbol 12-myristate 13-acetate (PMA), or opsonized heat-killed Staphylococcus epidermidis. A single colony of S. epidermidis grown overnight was washed, diluted to 3 × 108 organisms per ml in normal saline, heated at 65°C for 1 h, irradiated with 3000 rads (1 rad = 0.01 Gy), and stored at −70°C. Bacteria opsonized by addition of 0.9 ml of a 1:10 dilution of the stock to 0.1 ml of serum for 2 h by tumbling rotation were diluted in medium and added to monocytes.
To exclude that LPG affects global cell function and viability, human cell lines (THP-1 and U937 monocytes and A3.01 T cells), peripheral blood mononuclear cells, and murine RAW 264.7 monocytic cells were cultured in medium or medium containing LPG (2 μM) at 37°C for 3 days or for 7 days in the presence of phytohemagglutinin (2 μg/ml). Cells cultured with LPG or medium alone were similar in cell viability (as measured by flow cytometry of propidium iodide-stained cells and trypan blue exclusion), proliferative response to mitogen (as measured by [3H]thymidine incorporation), and protein synthesis (as measured by protein content and lactate dehydrogenase activity per mg of protein in cell lysates).
Northern Blot Analysis.
THP-1 cells are functionally very similar to peripheral blood monocytes, in particular, the regulation of IL-1β gene (23–28). Cells treated with or without LPG and/or inducers of IL-1β were harvested by gentle scrapping with a rubber policeman and pelleted by centrifugation (500 × g, 5 min). Total RNA was extracted (RNA STAT-60 solution, Tel-Test Friendswood, TX) by a single-step method (29) and quantitated by spectrophotometry. Equal amount of total RNA was resolved on a 1.2% agarose/formaldehyde denaturing gel and transferred by capillary action onto nylon membrane (0.45 μm pore-size Nytran, Schleicher & Schuell). Membranes were prehybridized and hybridized with 32P-labeled cDNA (30). IL-1β or GAPDH cDNA fragment was excised with restriction enzymes and purified using standard techniques (30). Purified IL-1β or GAPDH cDNAs (0.1 μg) were labeled with [α-32P]dCTP using random-priming method (Boehringer Mannheim). After hybridization, washed membranes were exposed to film. Quantitative analysis was performed using PhosphorImager analyzer (Molecular Dynamics). In experiments using S. epidermidis, cells were washed twice with ice-cold medium and centrifugation at 160 × g and 250 × g, each for 5 min to remove excess S. epidermidis. The percent suppression of IL-1β mRNA was normalized by the amount of GAPDH in each condition, which varied <10%.
IL-1β mRNA Stability.
THP-1 cells (107 cells per condition) stimulated with endotoxin at 2 μg/ml or endotoxin at 2 μg/ml plus 2 μM LPG for 2 h were treated with actinomycin D (6 μg/ml) and cells were harvested for total RNA at the time indicated. IL-1β mRNA present 2 h after endotoxin or endotoxin plus LPG treatment was used as a reference to compare residual IL-1β mRNA. An equivalent amount of RNA for each condition separated and transferred to nylon membrane was probed with 32P-labeled IL-1β cDNA or GAPDH cDNA.
In Vitro Transcription by the Nuclear Run-Off Assay.
THP-1 cells (5 × 107 cells) treated with or without 2 μM LPG for 2 h were stimulated with endotoxin at 2 μg/ml for 1–2 h, at 37°C in 5% CO2/95% air, and nuclei were isolated as described (31). The nuclear run-off assay for in vitro transcription was performed as described (31).
Plasmid Constructs.
The pTK.CAT (4.5 kb), containing the thymidine kinase (TK) promoter, the chloramphenicol acetyltransferase (CAT) gene, the simian virus 40 polyadenylylated site, and parts of the pUC plasmid including ampicillin-resistance (AmpR) gene was deleted of the TK promoter and IL-1β genomic sequence (positions −1110 to +15) or truncated sequence was linked upstream to CAT gene (XhoI–HindIII) (23). 5′ truncated promoter sequences were created by restriction enzyme digestion of the isolated IL-1β sequence. The truncation mutants are: −1110(XbaI), −680 (Sau3AI), −310 (DraI), −131 (HindIII), and −57 (DdeI), with numbers indicating base pairs upstream relative to the transcription start site (24). We used human IL-1β promoter fragments containing position −3757 to +11, full-length (XT-CAT), and a deletion mutant (positions −3757 to −2729 linked to −131 to +11) derived from λBDC-454 and inserted into CAT gene plasmid vector pA10CAT (3M) (25, 26).
Transfection and CAT Assay.
THP-1 cells were transfected with supercoiled plasmid DNA containing IL-1β promoters (3–5 μg per 107 cells) using modified DEAE-dextran method and (23) incubated in RPMI 1640 medium supplemented with 2.5% fetal bovine serum and l-glutamine for 48 h were treated with LPG for 2 h (or not treated) and challenged with endotoxin. Harvested cells were disrupted in 150 μl of 0.25 M Tris·HCl, by three freeze–thaw cycles. Supernatants clarified by centrifugation (15,000 × g, 5 min) were collected and equal amounts of protein extracts (Commassie blue assay, Bio-Rad) varying from 50 to 75 μg between experiments were used for the CAT assay as described (30, 31) and quantitated by PhosphorImager for 2–16 h. In some experiments, cells cotransfected with 2 μg of control plasmid pSV β-galactosidase (Promega) was used to normalize transfection efficiency. Basal CAT activity in medium condition was subtracted and the percent inhibition was expressed as [1 − (relative units from LPG plus inducer/relative units from inducer)] × 100.
RESULTS
Effect of LPG on LPS-Induced IL-1β Gene Expression.
Fig. 1 shows that LPG from Leishmania suppressed the steady-state mRNA of IL-1β induced by endotoxin. Maximal suppression of IL-1β steady-state mRNA (>90%) was observed when LPG was added between 2 h before and 1 h after stimulation with endotoxin. Interestingly, LPG treatment 2 h after endotoxin challenge retained inhibitory activity, suppressing 70% of IL-1β steady-state mRNA. Control experiments showed that LPG had no detectable effect on expression of GAPDH, a constitutively expressed gene; in contrast LPG inhibited IL-1β mRNA in the same sample (Fig. 1 Inset). Moreover, LPG had no effect on cell viability (over 98% living), proliferative response to mitogen, and protein synthesis (data not shown). Increasing the time of LPG treatment before or after endotoxin challenge resulted in lower inhibitory activity of LPG. In fact, LPG’s inhibitory activity was undetected when LPG (1 or 2 μM) was added 4 h before endotoxin challenge (data not shown). Lack of LPG’s inhibitory activity in the latter case is not due to loss of membrane-bound LPG because the amounts of LPG bound to the cell surface detected by monoclonal antibody and flow cytometry (FACS) analysis were similar between cells pretreated with LPG for 2 h and 4 h (32, 33).
Figure 1.
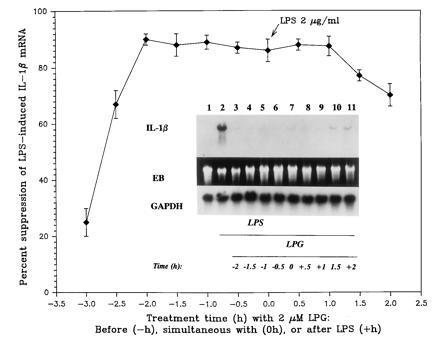
LPG-suppressed endotoxin (LPS)-induced IL-1β gene expression. Each data point is the mean ± SD of three to six experiments. (Inset) Autoradiograph of a representative Northern blot analysis of IL-1β mRNA (1.7 kb) and GAPDH mRNA (1.2 kb), and photograph of ethidium bromide-stained gel (EB). Lanes: 1, medium; 2–11, LPS at 2 μg/ml; 3–11, LPG at indicated times in relations to challenge with LPS.
To see whether the whole LPG molecule is required for the inhibitory effect, we tested two structural domains of LPG, namely, phosphodisaccharides galactosyl-mannose and lyso-alkyl-phosphatidylinositol (8, 10, 32). LPG (2 μM) pretreatment for 2 h resulted in ≈90% suppression of endotoxin-inducted of IL-1β mRNA; in contrast, treatment with phosphodisaccharides galactosyl-mannose or lyso-alkyl-phosphatidylinositol (2 μM for 2 h), followed by endotoxin (2 μg/ml) challenged had no effect on IL-1β mRNA level (n = 3, data not shown). We next examined the effects of varying concentration of LPG on endotoxin-induced IL-1β gene expression. Illustrated in Fig. 2 is the finding that treatment with increasing concentrations of LPG from 0.01 to 2.0 μM resulted in greater suppression of IL-1β mRNA level; maximum inhibition achieved was greater than 90%. Increasing the dose of endotoxin from 0.1 to 3 μg/ml shifted the dose–suppression curve to the right but did not overcome LPG’s inhibitory effect.
Figure 2.
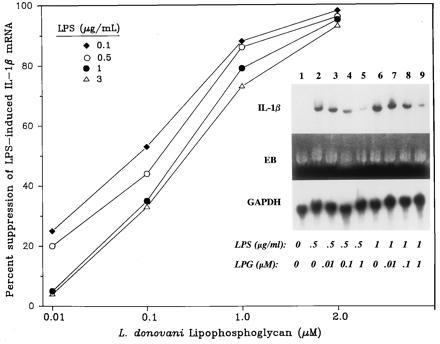
IL-1β mRNA LPG-suppressed in a dose-dependent manner. THP-1 cells treated with LPG (2 h) were challenged with LPS (mean of three or four experiments; SD <10% of the mean). (Inset) An autoradiograph of one representative Northern analysis. Lanes: 1, medium; 2–5, LPS at 0.5 μg/ml; 6–9, LPS at 1 μg/ml; 3 and 7, LPG at 0.01 μM; 4 and 8, LPG at 0.1 μM; 5 and 9, LPG at 1.0 μM.
LPG Suppression on IL-1β Gene Expression Is Not Reversible by Washing, Is Agonist-Specific, and Affects Primary Monocytes.
To examine the specificity of LPG’s inhibitory activity, (i) we found that washing cells pretreated with LPG (2 h) could not reverse the suppression of endotoxin-induced IL-1β steady-state mRNA (Fig. 31, lanes 3 and 4 compared with lanes 5 and 6). (ii) We observed that LPG pretreatment (2 h) inhibited the induction of IL-1β mRNA by cytokine TNF-α and serum-opsonized S. epidermidis but not by PMA. As shown in Fig. 32a, LPG inhibited TNF-α-induced IL-1β mRNA by 60 ± 10% (mean ± SD, n = 5). In contrast, LPG did not suppress steady-state IL-1β mRNA in response to PMA (Fig. 32b, PMA versus PMA plus LPG: lane 3 versus 4 and lane 5 versus 6, respectively, n = 3) while able to inhibit endotoxin-triggered IL-1β gene expression (Fig. 32b, lane 1 versus 2). We next examined the effect of varying the time of LPG treatment on S. epidermidis-induced IL-1β gene expression. LPG treatment of THP-1 cells 2 h before, simultaneously with, or 2 h after challenge with S. epidermidis suppressed IL-1β mRNA levels by 80 ± 19%, 40 ± 10%, and 20 ± 5%, respectively (Fig. 33 and data not shown). We also evaluated whether LPG had similar inhibitory effect on peripheral blood (primary) monocytes. Primary monocytes pretreated with LPG (2 μM, 2 h) and challenged with endotoxin (2 μg/ml) or S. epidermidis had suppressed levels of IL-1β mRNA by ≈80% (data not shown).
Figure 3.
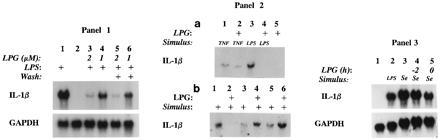
(1) Washing to removal LPG had no effect on LPG suppression of IL-1β gene expression. THP-1 cells pretreated or not with 1 or 2 μM LPG for 2 h were washed twice, resuspended in medium or LPS at 2 μg/ml, and cultured for 4 h. Lanes: 2, medium; 1 and 4–6, LPS at 2 μg/ml; 3 and 5, 2 μM LPG; 4 and 6, 1 μM LPG; 3 and 4, not washed; 5 and 6, washed after treatment with LPG. (2) LPG suppressed the induction of IL-1β mRNA by TNF-α but not by PMA. (a) THP-1 cells treated with LPG (2 μM, 2 h) were stimulated with TNF-α at 50 ng/ml (2 h) or with LPS at 2 μg/ml (4 h). Representative autoradiograph of four experiments. Lanes: 1, TNF-α; 2, TNF-α plus LPG; 3, LPS; 4, LPS/LPG; 5, LPG 2 μM. IL-1β was not detected in medium condition (data not shown). (b) Representative autoradiograph from three experiments showing IL-1β mRNA from THP-1 cells pretreated 2 μM LPG for 1 h (lanes 2, 4, and 6) and stimulated for 4 h with LPS at 2 μg/ml (lanes 1 and 2) or with PMA at 25 ng/ml (lanes 3 and 4) or at 50 ng/ml (lanes 5 and 6). (3) LPG suppressed S. epidermidis induction of IL-1β. Autoradiograph from THP-1 cells after the following treatments. Lanes: 4, 2 μM LPG for 2 h; 5, simultaneous treatment with 2 μM LPG and S. epidermidis (Se); 1, medium; 2, LPS at 2 μg/ml for 4 h; 3–5, opsonized heat-killed S. epidermidis.
Effect of LPG on IL-1β mRNA Stability and Transcription.
To characterize the suppression of mRNA levels, first, we examined whether LPG decreased mRNA stability. Although LPG enhanced the rate of IL-1β mRNA loss, as illustrated in Fig. 4, mRNA instability was increased by LPG by no more than 30%, which does not account for reduced steady-state IL-1β mRNA level of 90%. Therefore, we examined whether LPG’s inhibitory activity is mediated by suppressing IL-1β gene transcription in the nuclear-run off assay. Endotoxin induced high levels of IL-1β transcripts. LPG clearly inhibited the endotoxin-induced IL-1β mRNA transcription by ≈65% (Fig. 5, lane 2 versus 3). This inhibition is specific for IL-1β because in vitro transcription of GAPDH was similar all treatment conditions.
Figure 4.
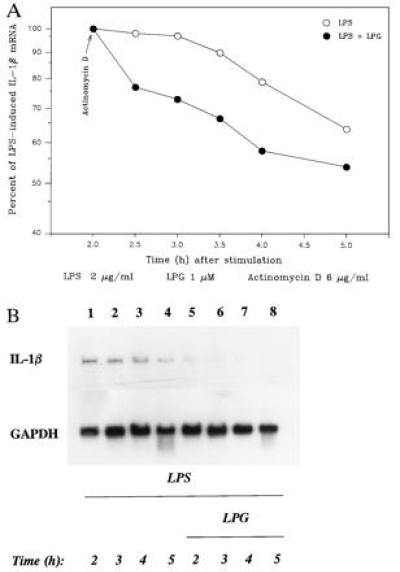
LPG lowers the stability of IL-1β mRNA. (A) IL-1β mRNA stability after LPS or LPS/LPG treatment, and the amount of IL-1β mRNA present at 2 h. Each condition was taken as the reference for comparison of residual IL-1β mRNA at the later times (mean of three or four experiments; SD < 10% the of mean). (B) Representative autoradiograph of the effect of LPG on mRNA levels of IL-1β and GAPDH. Lanes: 1–8, LPS at 2 μg/ml; 5–8, 2 μM LPG.
Figure 5.
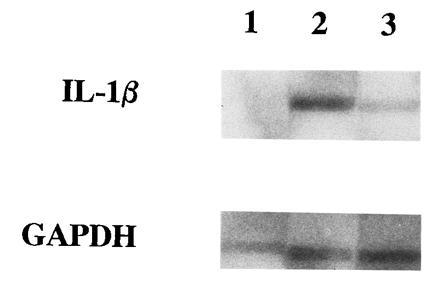
LPG suppresses transcription of IL-1β gene. Illustrated is an autoradiograph of one representative of three nuclear run-off assays. Lanes: 1, medium; 2, LPS; 3, LPG/LPS. IL-1β and GAPDH transcripts detected were indicated.
IL-1β Promoter Analysis and Identification of a LPG Inhibitory Response Gene Sequence.
Having found that LPG predominantly suppressed gene transcription, we explored the possibility that a specific sequence within IL-1β gene promoter mediated inhibition of gene transcription. In THP-1 monocytes transiently transfected with plasmids containing the 5′ flanking regions of IL-1β gene (positions −1110 to +15, −680 to +15, −310 to +15, −131 to +15, −57 to +15) linked to CAT gene (23, 24), LPG suppressed endotoxin-induced CAT activity by ≈40% in plasmids containing the promoter sequences from positions −1110 to +15, −680 to +15, and −310 to +15 (Fig. 6). The major DNA sequence required for LPG suppression of endotoxin-induced CAT activity appears to reside within the region of positions −310 to −57, because truncation of IL-1β promoter nucleotide sequences to position −131 significantly reduced LPG’s inhibitory activity to only 7 ± 1% (suppression of endotoxin-induced CAT activity) and deletion to −57 nucleotide sequence upstream to the transcriptional start site completely abrogated LPG’s inhibitory activity.
Figure 6.
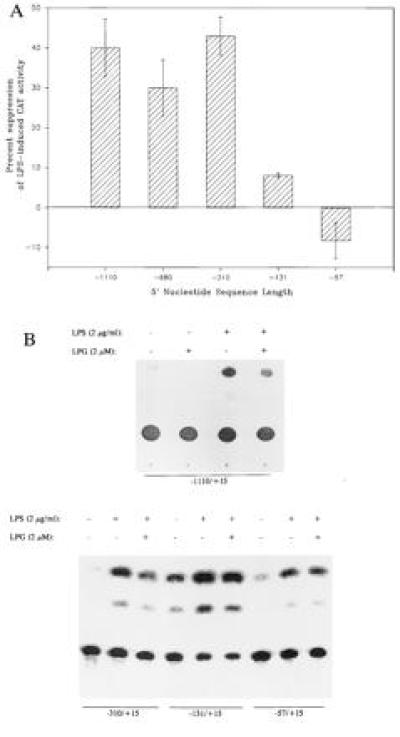
DNA nucleotide sequence (positions −310 to −57) of the IL-1β promoter is required for LPG’s inhibitory activity. THP-1 cells were transfected with plasmids containing the full-length (positions −1110 to +15) or 5′ truncation IL-1β promoter sequences. After 48 h of culture cells were treated or not with 2 μM LPG (2 h) and stimulated with LPS at 2 μg/ml (20–24 h). CAT activity of cell lysates was measured. LPS-induced CAT activity on average was 8-fold (6- to 18-fold) above medium control. Illustrated data are the mean of four to six experiments.
Previous reports have described that nucleotide sequence of positions −3757 to −2729 upstream to the region of positions −1110 to +15 of the IL-1β promoter contribute to maximal endotoxin-induced transcription (25, 26). To determine whether this region also mediated LPG’s inhibitory effect, we transiently transfected human THP-1 monocytes with a construct containing the full-length IL-1β construct, positions −3757 to +11, linked to CAT (XT-CAT, Fig. 7). Compared with basal CAT activity, endotoxin-induced CAT activity was increased by more than 20-fold. Pretreatment with LPG (2 μM) for 1 h suppressed endotoxin-induced CAT activity by 50 ± 5% (n = 5) (Fig. 7). We next tested the IL-1β promoter construct X2-HT-CAT (positions −3757 to −2729 directly linked to positions −131 to +11) in which the nucleotide sequence of positions −2729 to −131 has been deleted from the full-length IL-1β promoter. Fig. 7 showed that THP-1 monocytes transiently transfected with X2-HT-CAT responded to endotoxin (2 μg/ml) by increasing CAT activity by more than 30-fold from basal. In cells transfected with the deletional mutant construct X2-HT-CAT, pretreatment with LPG (2 μM, 1 h) suppressed endotoxin-induced CAT activity by only 8 ± 4%.
Figure 7.
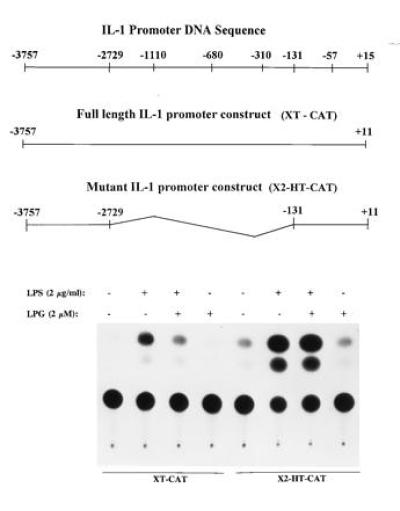
Positions −310 to −57 of the IL-1β promoter are required for LPG’s inhibitory activity. THP-1 cells transfected with plasmid containing positions −3757 to +11 (XT-CAT) or −3757 to −2729 linked to the promoter sequence positions, −131 to +11 (X2-HT-CAT), were treated with LPG (2 μM) (2 h) and challenge with LPS at 2 μg/ml (24 h). Cells were also cotransfected with 2 μg of control plasmid pSV β-galactosidase to normalize for transfection efficiency. After measurement and normalization for β-galactosidase expression, CAT activity were assayed and quantitated (n = 5).
Dose and Kinetic Characteristics of the LPG-Responsive IL-1β Promoter Sequence (Positions −310 to +15).
To provide additional support that the region of positions −310 to +15 of the IL-1β promoter mediates LPG’s inhibitory activity, we utilized this minimal IL-1β promoter sequence to characterize its response to LPG. In THP-1 monocytes transiently transfected with positions −310 to +15 of the IL-1β promoter sequence, LPG suppressed endotoxin-induced CAT activity in a dose-dependent manner similar to LPG’s inhibitory effect on steady-state mRNA (Fig. 8A). Moreover, maximal inhibitory activity of LPG was observed for LPG treatment from 2 h before endotoxin challenge to 1 h after endotoxin challenge, in a manner similar to LPG’s suppression of steady-state mRNA (Fig. 8B).
Figure 8.
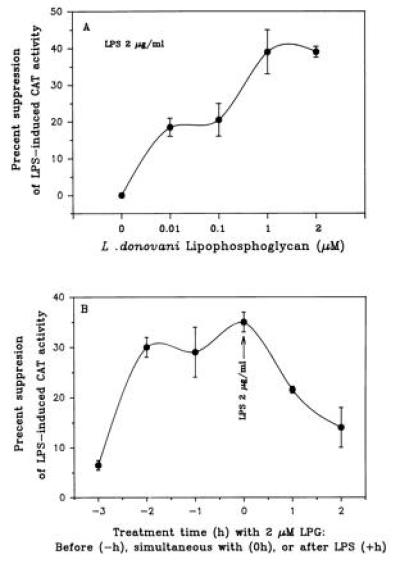
Time- and dose-dependent effects of LPG on transcription using the IL-1β promoter from positions −310 to +15. (A) Illustrated is the dose-dependent inhibitory activity of LPG on transcription activity (mean of four to eight experiments). (B) Kinetics of LPG treatment on LPS induction of CAT activity (n = 3 to 5 experiments). THP-1 cells transfected with the IL-1 β promoter plasmid were treated either with LPG (0–2 μM) for 2 h or with LPG (2 μM) for indicated time and challenged with LPS (2 μg/ml) for 24 h.
DISCUSSION
This report showed that LPG from Leishmania down-modulated endotoxin-induced IL-1β mRNA expression in a dose- and time-dependent manner. Its effect is agonist-specific: suppressing IL-1β gene expression induced by endotoxin, TNF-α, and S. epidermidis while having no effect on PMA-triggered IL-1β mRNA accumulation. This effect of LPG is gene-specific since the expression of a constitutive gene, GAPDH, was not affected by LPG. Moreover, removal of LPG did not abrogate this effect, the whole LPG molecules is required, and cell viability, proliferative response, and protein synthesis were not affected by LPG (data not shown and refs. 5–10). We have also found that LPG’s effect is not restricted to THP-1 cells and IL-1β gene. LPG inhibited endotoxin-induced expression of TNF-α genes in both THP-1 cells and (primary) blood monocytes (data not shown, unpublished results) and inhibited the release of immunoreactive IL-1 β from primary monocytes challenged with either endotoxin or S. epidermidis. Remarkably, despite stimulation with endotoxin or S. epidermidis for 72 h, LPG retained significant inhibitory activity on IL-1β protein production (data not shown).
LPG suppression of endotoxin-induced IL-1β mRNA is predominantly mediated by inhibiting transcription. LPG’s inhibition of gene transcription required a unique IL-1β promoter sequence located within the region of positions −310 to −57. This is evident by deletion and truncation analysis of the IL-1β promoters and the finding that deletion of the DNA sequence of positions −310 to −57 from the promoter (full length) abrogated LPG’s inhibitory activity. Furthermore, that the sequence from positions −310 to −57 of the IL-1β promoter mediates LPG’s effect is shown by its transcriptional activity having a dose and kinetic profiles that were similar to LPG’s suppression of steady-state level of IL-1β mRNA (Fig. 8 versus Figs. 1 and 2). These data strongly suggest that a unique DNA sequence located in the region of positions −310 to −57 of the IL-1β promoter mediates LPG’s inhibitory effect and argue that LPG-mediated response is not merely by antagonizing endotoxin-mediated response. The finding that a unique IL-1β promoter DNA sequence mediates LPG’s inhibitory effect is novel and not previously reported.
Our observation extends some findings by several investigators but are in distinct contrast to other findings. The discrepancy between our findings is likely due to treatment with LPG in our study while Reiner et al. (14) examined monocytes infected with amastigotes, which possess very low amounts of LPG (5). In a prior study, Descoteaux et al. (33) found that LPG alone triggered expression of c-fos by murine macrophages; in contrast LPG treatment suppressed endotoxin-induced c-fos mRNA while enhancing endotoxin-triggered TNF-α mRNA level. Our difference may be due to studies conducted in murine versus human mononuclear phagocytes.
We believe alternative mechanism(s) may play a more important role. (i) Multiple AP-1 (fos/jun) sites are present in the full length IL-1β promoter upstream to the region of positions −310 to −57 while LPG’s inhibitory activity is restricted to this region. (ii) The PU.1 sites (and possibly NF-IL6) within the nucleotide sequence from positions −310 to +15 are critical for IL-1β gene expression in response to endotoxin, TNF-α, and lipoarabinomannan of Mycobacterium tuberculosis (23–26). (iii) Additional DNA binding sites are also present within the nucleotide sequence of positions −310 to +15 of the IL-1β promoter gene (23–26, 34–39)]. Based on the finding of a LPG response promoter sequence, we speculate that (i) LPG diminishes IL-1β transcription by inhibiting binding and/or endotoxin activation of DNA binding protein(s) or (ii) LPG transiently induces an as yet unidentified nuclear binding protein(s) that acts as a transcription repressor.
LPG likely alters cell function via a membrane event because within 15 min after infection with L. donovani, LPG is transferred to macrophage’s cell surface (9, 32). LPG may interfer with endotoxin signaling by competing for endotoxin binding protein or CD14 or by insertion into the cell lipid bilayer (40–42). Alternatively, LPG may interact with an undefined receptor-signaling pathway leading to suppressed gene expression as suggested by the finding that (i) LPG treatment 2 h after endotoxin challenge blocked IL-1β gene expression and (ii) the inhibition of gene transcription is mediated by a LPG response DNA sequence (positions −310 to −57) in the IL-1β promoter, while response elements critical for maximal endotoxin effect are upstream to this region.
The finding that a parasite product modulates expression of a nonself gene has precedence. For example, sodium salicylate, a product of plants, has been reported to inhibit the activation of human NFkB; a nematode has been shown to direct the expression of root-specific gene that modifies root formation of the plant to supports nematode development; and lastly, lipoarabinomannan of M. tuberculosis-induced IL-1β gene expression via PU.1 (and possibly NF-IL6) DNA motifs (23–26, 37, 43, 44). However, most examples involve enhancing gene expression; therefore, LPG may be the first negative-regulator of gene expression that mediates its action by a unique promoter sequence acting as a “gene silencer”.
The suppression of IL-1β (and TNF-α, unpublished data) gene expression by LPG to multiple agonists provides an explanation for macrophage deactivation and defective T-cell response in patients with leishmaniasis (4, 45). Suppression of IL-1β and TNF-α gene expression may be Leishmania’s evolutionary strategy to block immune response because both cytokines activates macrophage anti-Leishmania activity (2, 3); and IL-1β is a costimulus for memory T-cell activation and triggers IL-2 production (15–18, 46). In addition to suppressing cytokine expression, we have shown that LPG binds avidly to endothelial cells and inhibits endotoxin-triggered endothelial cell adhesion to leukocytes (32).
In light of these findings it seems feasible that LPG, an evolutionary perfected molecule, represents a new class of compounds that may be exploited for clinical use as an antiinflammatory agent for overt vascular cell-activation states. Sepsis initiated by endotoxin is amplified by proinflammatory cytokines, such as, IL-1β and TNF-α. The organ damages involve overt activation of phagocytes (e.g., monocytes) and of vascular endothelium. Current strategies for treatment of sepsis have significant limitations, and thus far, have not demonstrated clinical benefit (47). The features of LPG found in this study and on endothelial cells (32) strongly suggest that LPG has therapeutic potential for treatment of sepsis, acute respiratory distress syndrome, and autoimmunity.
Acknowledgments
This work is dedicated to the memory of Dr. Sheldon M. Wolff. We thank Dr. W. D. Johnson for generous support, Drs. F. Laraque and S. K. Lo for critiques, Adair Russell for editorial comments, and S. He and Q. Zheng for computer assisted graphics. This work was supported in part by the Department of Medicine, the National Institutes of Health Grants PO1-AI-16282, R37-AI-22624, D43-TW00018, and AI-39606 (J.L.H.), MO1-00096 (W.N.R.), AI-20941 (S.J.T.), and P60 AR20613 and HL51957 (M.J.F.), the New York City Affiliate American Heart Association (J.L.H.), a Fogarty Training Grant (B.Z.), and “Stavros S. Niarchos” Endowment (D.E.H.).
Footnotes
Abbreviations: IL, interleukin; TNF-α, tumor necrosis factor α; LPG, lipophosphoglycan; PMA, phorbol 12-myristate 13-acetate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CAT, chloramphenicol acetyltransferase; LPS, lipopolysaccharide.
References
- 1.Bogdan C, Paik J, Vodovotz Y, Nathan C. J Biol Chem. 1992;267:23301–23308. [PubMed] [Google Scholar]
- 2.Hatzigeorgiou D H, He S H, Sobel J, Hafner A, Grabstein K, Ho J L. J Immunol. 1993;151:3682–3692. [PubMed] [Google Scholar]
- 3.Ho J L, He S H, Rios M J C, Wick E A. J Infect Dis. 1992;165:344–351. doi: 10.1093/infdis/165.2.344. [DOI] [PubMed] [Google Scholar]
- 4.Ho J L, Badaro R, Hatzigeorgiou D, Reed S G, Johnson W D., Jr Biother. 1994;7:223–235. doi: 10.1007/BF01878488. [DOI] [PubMed] [Google Scholar]
- 5.Turco S J, Descoteaux A. Annu Rev Microbiol. 1992;46:65–94. doi: 10.1146/annurev.mi.46.100192.000433. [DOI] [PubMed] [Google Scholar]
- 6.Handman E, Schnur L F, Spithill T W, Mitchell G F. J Immunol. 1986;137:3608–3613. [PubMed] [Google Scholar]
- 7.Elhay M, Kelleher M, Bacic A, McConville M J, Tolson D L, Pearson T W, Handman E. Biochem Parasitol. 1990;40:255–268. doi: 10.1016/0166-6851(90)90047-p. [DOI] [PubMed] [Google Scholar]
- 8.McNeely T B, Turco S. J Immunol. 1990;144:2745–2750. [PubMed] [Google Scholar]
- 9.Tolson D L, Turco S J, Pearson T W. Infect Immun. 1990;58:3500–3507. doi: 10.1128/iai.58.11.3500-3507.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frankenburg S, Leibovici V, Mansbach N, Turco S J, Rosen G. J Immunol. 1990;145:284–289. [PubMed] [Google Scholar]
- 11.Ho J L, Badaro R, Schwartz A, Dinarello C A, Gelfand J A, Sobel J, Barral A, Barral-Netto M, Carvalho E M, Reed S G, Johnson W D. J Infect Dis. 1992;165:1094–1102. doi: 10.1093/infdis/165.6.1094. [DOI] [PubMed] [Google Scholar]
- 12.Crawford G D, Wyler D J, Dinarello C A. J Infect Dis. 1985;152:315–322. doi: 10.1093/infdis/152.2.315. [DOI] [PubMed] [Google Scholar]
- 13.Reiner NE. J Immunol. 1987;138:1919–1925. [PubMed] [Google Scholar]
- 14.Reiner N E, Ng W, Wilson C B, McMaster R, Burchett S K. J Clin Invest. 1990;85:1914–1924. doi: 10.1172/JCI114654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luqman M, Greenbaum L, Lu D, Bottomly K. Eur J Immunol. 1992;22:95–100. doi: 10.1002/eji.1830220115. [DOI] [PubMed] [Google Scholar]
- 16.Stein P H, Singer A. Int Immunol. 1992;3:327–335. doi: 10.1093/intimm/4.3.327. [DOI] [PubMed] [Google Scholar]
- 17.Rogers H W, Sheehan K C F, Brunt L M, Dower S K, Unanue E R, Schreiber R D. Proc Natl Acad Sci USA. 1992;89:1011–1015. doi: 10.1073/pnas.89.3.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bancroft G J, Scheiber R D, Unanue E R. Immunol Rev. 1991;124:5–24. doi: 10.1111/j.1600-065x.1991.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 19.March C J, Mosley B, Larsen A, Cerretti D P, Braedt G, Price V, Gillis S, Henney C S, Kronheim S R, Grabstein K, Conlon P, Hopp T P, Cosman D. Nature (London) 1985;315:641–647. doi: 10.1038/315641a0. [DOI] [PubMed] [Google Scholar]
- 20.Orlandi P A, Turco S J. J Biol Chem. 1987;262:10384–10391. [PubMed] [Google Scholar]
- 21.Turco S J, Hull S R, Orlandi P A, Shepherd S D, Homans S W, Dwek R A, Rademacher T W. Biochemistry. 1987;26:6233–6238. doi: 10.1021/bi00393a042. [DOI] [PubMed] [Google Scholar]
- 22.Carver M A, Turco S J. Biochemistry. 1991;266:10974–10981. [PubMed] [Google Scholar]
- 23.Zhang Y, Doerflier M, Lee T C, Guillemin B, Rom W N. J Clin Invest. 1993;91:2076–2083. doi: 10.1172/JCI116430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Rom W N. Mol Cell Biol. 1993;13:3831–3837. doi: 10.1128/mcb.13.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirakawa F, Saito K, Bonagura C A, Galson D L, Fenton M J, Webb A C, Auron P E. Cell Biol. 1993;13:1332–1344. doi: 10.1128/mcb.13.3.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buras J A, Monks B G, Fenton M J. J Immunol. 1994;152:444–454. [PubMed] [Google Scholar]
- 27.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Int J Cancer. 1980;26:171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, Tada K. Cancer Res. 1982;42:1530–1536. [PubMed] [Google Scholar]
- 29.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 30.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., Struhl, K., Albright, L. M., Coen, D. M. & Varki, A. (1993) in Current Protocols in Molecular Biology, Vol. 1, Unit 4.9, pp. 4.9.1–4.9.14, and Unit 4.10, pp. 4.10.1–4.10.11.
- 32.Ho, J. L., Kim, H.-K., Sass, P. M., He, S., Geng, J., Xu, H., Zhu, B., Turco, S. J. & Lo, S. K. (1996) J. Immunol. 157, in press. [PubMed]
- 33.Descoteaux A, Turco S, Sacks D L, Matlashewski G. J Immunol. 1991;146:2747–2753. [PubMed] [Google Scholar]
- 34.Hunninghake G W, Monks B W, Geist L J, Monick M M, Monroy M A, Stinski M F, Webb A C, Monick J-M, Monroy M A, Stinski M F, Webb A C, Dayer J-M, Auron P E, Fenton M J. Mol Cell Biol. 1992;12:3439–3448. doi: 10.1128/mcb.12.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kominato Y, Galson D L, Waterman W R, Webb A C, Auron P E. Mol Cell Biol. 1995;15:58–68. [PMC free article] [PubMed] [Google Scholar]
- 36.Monks B G, Martell B A, Buras J A, Fenton M J. Mol Immunol. 1994;31:139–151. doi: 10.1016/0161-5890(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 37.Buras J A, Reenstra W R, Fenton M J. Mol Immunol. 1995;32:541–554. doi: 10.1016/0161-5890(95)00018-a. [DOI] [PubMed] [Google Scholar]
- 38.Wall L, deBoer E, Grosveld F. Genes Dev. 1988;2:1989–1100. doi: 10.1101/gad.2.9.1089. [DOI] [PubMed] [Google Scholar]
- 39.Ghazal P, Lubon H, Fleckenstein B, Hennighausen L. Proc Natl Acad Sci USA. 1987;84:3658–3662. doi: 10.1073/pnas.84.11.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wurfel M M, Kunitake S T, Lichenstein H, Kane J P, Wright S D. J Exp Med. 1994;180:1025–1035. doi: 10.1084/jem.180.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedron T, Girard R, Turco S J, Chaby R. J Biol Chem. 1994;269:2426–2432. [PubMed] [Google Scholar]
- 42.Miao L, Stafford A, Nir S, Turco S J, Flanagan T D, Epand R M. Biochemistry. 1995;34:4676–4683. doi: 10.1021/bi00014a022. [DOI] [PubMed] [Google Scholar]
- 43.Kopp E, Ghosh S. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 44.Opperman C H, Taylor C G, Conkling M A. Science. 1994;263:221–223. doi: 10.1126/science.263.5144.221. [DOI] [PubMed] [Google Scholar]
- 45.Karp C L, El-Saji S H, Wynn T A, Satti M M H, Kordojani H M, Hashim F A, Hag-Ali M, Neva F A, Nutman T B, Sacks D L. J Clin Invest. 1993;91:1644–1648. doi: 10.1172/JCI116372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bancroft G J, Sheehan K C F, Scheiber R D, Unanue E R. J Immunol. 1989;143:127–130. [PubMed] [Google Scholar]
- 47.Natanson C, Hoffman W D, Suffredini A F, Eichacker P Q, Danner R L. Ann Intern Med. 1994;120:771–783. doi: 10.7326/0003-4819-120-9-199405010-00009. [DOI] [PubMed] [Google Scholar]