

Abstract
Hantaviruses such as Hantaan virus (HTNV) and Andes virus cause two human diseases, hemorrhagic fever with renal syndrome and hantavirus pulmonary syndrome, respectively. For both, disease pathogenesis is thought to be immunologically mediated and there have been numerous reports of patients with elevated levels of proinflammatory and inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), in their sera. Multiple viruses have developed evasion strategies to circumvent the host cell inflammatory process, with one of the most prevalent being the disruption of nuclear factor kappa B (NF-κB) activation. We hypothesized that hantaviruses might also moderate host inflammation by interfering with this pathway. We report here that the nucleocapsid (N) protein of HTNV was able to inhibit TNF-α-induced activation of NF-κB, as measured by a reporter assay, and the activation of endogenous p65, an NF-κB subunit. Surprisingly, there was no defect in the degradation of the inhibitor of NF-κB (IκB) protein, nor was there any alteration in the level of p65 expression in HTNV N-expressing cells. However, immunofluorescence antibody staining demonstrated that cells expressing HTNV N protein and a green fluorescent protein-p65 fusion had limited p65 nuclear translocation. Furthermore, we were able to detect an interaction between HTNV N protein and importin α, a nuclear import molecule responsible for shuttling NF-κB to the nucleus. Collectively, our data suggest that HTNV N protein can sequester NF-κB in the cytoplasm, thus inhibiting NF-κB activity. These findings, which were obtained using cells transfected with cDNA representing the HTNV N gene, were confirmed using HTNV-infected cells.
Tumor necrosis factor alpha (TNF-α) is a major proinflammatory cytokine produced by a variety of cell types that include macrophages, endothelial cells, and epithelial cells, and the receptor is constitutively expressed on most cell types (44). TNF-α is pleiotropic and can regulate the response of immune cells, as well as induce inflammation, differentiation, and apoptosis, and is involved in protecting the host from pathogen infections. Upon TNF-α ligand binding, TNF-associated death domain (TRADD) associates with the TNF receptors and serves as an adaptor molecule that recruits TNF receptor-associated factor 2 (TRAF-2) and receptor-interacting protein (14). This complex of proteins leads to the activation of kinases that phosphorylate the inhibitor of κB (IκB) (14). The ubiquitin-proteosome pathway initiates the degradation of IκB, allowing for NF-κB dimers to translocate to the nucleus and regulate the transcription of NF-κB target genes (12).
NF-κB transcription factors are dimers composed of five subunits belonging to the Rel family (reviewed in reference 12). The five subunits, p65 (Rel A), Rel B, c-Rel, p50, and p52, can form various dimers (2, 42, 43). The p50/p65 heterodimers are the best-characterized and are the most abundant form of the NF-κB transcription factors in most cell types (12). IκB is responsible for sequestering NF-κB in the cytoplasm by masking its nuclear localization signal (NLS) (3, 10, 13). Proteins that are too large to diffuse through the nuclear pore require a transport system consisting of importin α and importin β (28). Proteins utilizing this transport system to translocate to the nucleus contain an NLS that is recognized and bound by importin α (11, 21, 28). To date, seven importin α proteins (importin α1 to α7), all of which carry various cargo, including signal transducers and activators of transcription (STATs) and NF-κB (6, 7, 27), have been identified. Importantly, Fagerlund et al. recently reported that TNF-α-induced nuclear localization of p50/p65 heterodimers is mediated by importin α3 and importin α4 (6). However, there have also been reports of importin α1's interacting with NF-κB, but the role of importin α1 in transport and activation remains unclear (5, 6). Because of the multifaceted nature of TNF-α and NF-κB, the NF-κB subunits and the importin α proteins have become prime targets of viruses to evade the outcome of inflammatory pathways.
Hantaan virus (HTNV) is a member of the Bunyaviridae family, which consists of segmented negative-sense or ambisense enveloped viruses. The trisegmented RNA genome (comprising the S [small], M [medium], and L [large] segments) of HTNV encodes the N protein, glycoproteins (Gn and Gc), and the transcriptional polymerase (L polymerase), respectively, in the virus cRNA. The Hantavirus genus is unique among the genera in this family because it is the only one with viruses that are rodent borne rather than arthropod borne. Hantaviruses establish prolonged and persistent infections in their rodent reservoirs and cause no overt signs of illness. However, some of the hantaviruses can cause two distinct types of human disease: hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary syndrome (HPS) (34). Hantavirus-associated diseases are thought to be immunologically mediated, and there have been numerous reports of patients with elevated levels of TNF-α in plasma during the acute phase of HFRS (20, 24). TNF-α-positive cells can also be found in kidney and lung biopsy specimens from HFRS and HPS patients (29, 41). Clinically, hypotension and patient outcomes correlate with the levels of TNF-α (24).
Recently, studies have begun to focus on the ability of hantaviruses to antagonize the innate immune response. The Gc proteins of the HPS viruses, Andes virus (ANDV) and NY-1 virus, can inhibit the activation of two important innate immune pathways, those of double-stranded RNA and interferon (IFN). NY-1 virus Gc has been shown to antagonize the activation of RIG-I, a protein involved in the induction of IFN (1). ANDV Gc can inhibit the phosphorylation of transcription factors STAT1 and STAT2, and this inhibition has been demonstrated previously in ANDV-infected cells (39). Furthermore, Sin Nombre virus is unable to induce the production of IFN-α/β during infection (32). These results show that hantaviruses have evolved multiple strategies to circumvent innate immune responses. Considering the clinical relevance of TNF-α during infection, we hypothesized that HTNV might be able to inhibit this inflammatory pathway. Here, we examined the ability of HTNV proteins to prevent downstream effects of TNF-α-induced activation of NF-κB.
MATERIALS AND METHODS
Cells, plasmids, reagents, and virus.
The human embryonic kidney cell line 293T was maintained in Dulbecco's minimum essential medium that was supplemented with 10% fetal bovine serum (HyClone), 250 μg/ml of amphotericin B (Gibco), and 100 IU of penicillin-streptomycin (Gibco). The human lung epithelial cell line A549 was maintained in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum, 0.5 μg/ml of amphotericin B, and 100 IU of penicillin-streptomycin. All cells were incubated at 37°C in 5% CO2 for the periods of time indicated below.
The pNF-κB-hrGFP reporter plasmid (Stratagene) expresses green fluorescent protein (GFP) from a promoter containing only a TATA box and five sequential κB binding elements. The construction of the expression vector pWRG7077 was described earlier (35). The cDNA encoding the full-length HTNV M segment was cloned into the pWRG7077 vector as described previously (15), yielding pWRG7077-HTNV-M. The cDNA encoding the HTNV S segment (36) was excised by digestion with BamHI and cloned into the BglII site of pWRG7077, generating pWRG7077-HTNV-S. All importin α constructs contained N-terminal FLAG and were cloned as described previously (38, 45). The pCAGGS-GFP-p65 plasmid was generated by cloning the p65 open reading frame into the EcoRV and NheI sites of the pCAGGS-GFP expression vector. The pCAGGS-GFP plasmid is a variant that contains multiple cloning sites flanking the GFP coding sequence, allowing for the expression of amino-terminal GFP fusion proteins.
For the transfection of 293T cells in 2-cm2 wells, 3 μl of FuGENE6 (Roche) was diluted in 100 μl of serum-free medium and 1 μg of DNA was added. The transfection mixture was allowed to incubate at room temperature for 20 min and added to the cells for 24 h. For the transfection of A549 cells in 2-cm2 wells, 0.5 μg of DNA was added to 100 μl of serum-free medium, after which 0.5 μl of PLUS reagent (Invitrogen) was added and the mixture was incubated for 5 min at room temperature. LTX reagent (1.5 μl; Invitrogen) was subsequently added to the mixture, and the mixture was incubated for an additional 25 min at room temperature. For larger wells, reagent amounts were scaled up proportionately to the size.
HTNV strain 76-118 (22) was propagated in roller bottles containing Vero E6 cells and used at a low multiplicity of infection (MOI). Fourteen days posttransfection, supernatants were harvested and clarified by centrifugation. The titers of aliquots were determined by a plaque assay. In all experiments, A549 cells were infected at an MOI of 5 to ensure that most of the cells were expressing viral protein. A549 cells were used for experiments because they are permissive for HTNV and transfectable. Complete EMEM was used as a mock-infected control and as the diluting agent in virus preparations before adsorption. Virus was allowed to adsorb at 37°C in 5% CO2 for 2 h. Cells were subsequently washed, and medium was replaced with fresh complete EMEM. Mock-infected and infected cells were then returned to the same incubation conditions for 5 days.
NF-κB GFP reporter assay.
293T cells were seeded into 24-well plates and cotransfected with the pNF-κB-hrGFP plasmid (500 ng) and empty pWRG7077 vector (pWRG7077-Empty), pWRG7077-HTNV-S, or pWRG7077-HTNV-M (0 to 500 ng) by using the FuGENE6 transfection reagent. After 24 h, cells were overlaid with medium containing various concentrations of TNF-α (0 to 100 ng/ml; Endogen) for 4 h. As a control for inhibition in this assay, some cells were transfected with pNF-κB-hrGFP only and treated with 0 to 50 μM concentrations of the proteosomal inhibitor MG132 (Calbiochem) for 2 h before the addition of TNF-α and throughout TNF-α treatment. Cells were then washed, and the medium was replaced with complete Dulbecco's minimum essential medium without TNF-α for an additional 24 h.
As a control experiment, cells were cotransfected with 500 ng of pCAGGS-GFP and 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M. After 24 h, cells were incubated in medium with or without 10 ng/ml TNF-α for an additional 24 h. GFP-positive cells were examined with a Nikon Eclipse TS100 microscope.
NF-κB transcription factor assay.
A549 cells were seeded into 24-well plates and transfected with 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M for 24 h or treated with MG132. Cells were left untreated or treated with 50 ng/ml of TNF-α for 15 min. Cytoplasmic and nuclear cell extracts were isolated using the nuclear extract kit (Active Motif). Cytoplasmic portions were used for Western blotting to examine the total amount of lysate, and all nuclear extracts were loaded to measure transcription factor activation. The activation of NF-κB subunits NF-κB1 (p50) and Rel A (p65) was measured using a transcription factor assay kit from Active Motif. The assay is essentially an enzyme-linked immunosorbent assay in which an oligonucleotide containing the consensus NF-κB binding site (5′-GGGACTTTCC-3′) is immobilized onto a 96-well plate. Nuclear cell extracts that contain the active form of NF-κB specifically bind to the oligonucleotide, and the subunits p50 and p65 are detected with antibodies. The optical density at 450 nm was determined on a spectrophotometer. For internal controls to validate the specificity of our assay, no lysates, Raji cell extract, and competitive oligonucleotides were used. Raji cell extracts were used as a positive control for p50 and p65 activation. A wild-type consensus oligonucleotide was used to monitor the inhibition of p50 and p65, as this prevents binding to the probe immobilized on the plate. A mutated consensus oligonucleotide was also used and had no effect on binding.
Immunofluorescence microscopy.
For the translocation assay, 293T cells were seeded onto coverslips and cotransfected with 500 ng of pCAGGS-GFP-p65 and 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M. After 24 h, cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) and permeabilized with 0.2% Triton X-100 in PBS. After being blocked with 10% goat serum in PBS, cells were stained with antibodies against HTNV nucleocapsid (N) protein (rabbit immune sera) and HTNV Gc (mouse immune sera). Alexa Fluor 546 secondary antibody was used to detect HTNV proteins.
For infections, A549 cells were seeded onto coverslips and mock infected or infected with HTNV. After being infected for up to 5 days, cells were left untreated or treated with 50 ng/ml of TNF-α for 15 min. All samples were then fixed with 3.7% formaldehyde in PBS and permeabilized with 0.2% Triton X-100 in PBS. After being blocked with 10% goat serum in PBS, cells were sequentially stained, first with antibodies to p65 (Santa Cruz Biotechnologies) and then with antibodies to HTNV N (Abcam) and HTNV Gc (mouse immune sera). Alexa Fluor 488 secondary antibodies were used to detect p65, while Alexa Fluor 546 was used to detect HTNV proteins. All coverslips were washed, mounted onto slides by using Prolong gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen), and examined with a Nikon Eclipse E600 microscope.
Western blots and coimmunoprecipitations.
Transfected and infected A549 cell lysates were prepared with NuPAGE 2× lithium dodecyl sulfate sample buffer that contained NuPAGE reducing agent (Invitrogen) and complete protease inhibitor tablets (Roche). Proteins were separated on 4 to 12% gradient polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes by using the NuPAGE gel system (Invitrogen). Blots were blocked with 5% nonfat milk in Tris-buffered saline (TBS) and subsequently probed overnight at 4°C with antibodies directed against p50 (Santa Cruz Biotechnologies), p65 (Santa Cruz Biotechnologies), HTNV N (rabbit immune sera), IκBα (Santa Cruz Biotechnologies), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Cell Signaling Technology), histone H2B (Santa Cruz Biotechnologies), and FLAG (M2; Sigma-Aldrich). Blots were then washed with TBS containing Tween 20 and probed with the appropriate species-specific alkaline phosphatase-conjugated secondary antibody. Blots were developed using an alkaline phosphatase substrate detection system (Pierce Biotechnology) and exposed to X-ray film (GE Healthcare). For probes with multiple antibodies, some blots were stripped using Restore Plus Western blot stripping buffer according to the recommendations of the manufacturer (Pierce Biotechnology).
For the fractionation of lysates, A549 cells were mock infected or infected with HTNV for 5 days and left untreated or treated with 50 ng/ml of TNF-α for 15 min. Cells were washed, and cytoplasmic and nuclear fractions were isolated using extraction reagents from Pierce Biotechnology. Sample buffer was added, and the fractions were processed as described above for Western blotting. GAPDH antibody was used as a loading control for cytoplasmic fractions, and histone H2B was used as a control for nuclear fractions.
For coimmunoprecipitation experiments, A549 cells were seeded into six-well plates and transfected with 1.25 μg of FLAG-importin α1, FLAG-importin α2, FLAG-importin α3, or FLAG-importin α4 and 1.25 μg of pWRG7077-Empty or pWRG7077-HTNV-S plasmid. Twenty-four hours posttransfection, cells were left untreated or treated with 50 ng/ml of TNF-α for 15 min, washed with PBS, and lysed in 400 μl of lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, and protease inhibitors). Extracts were centrifuged at 13,000 rpm and 4°C for 10 min in an Eppendorf microcentrifuge, and the supernatant was precleared with a 50% slurry of protein A Sepharose beads in PBS. Anti-N protein antibody was subsequently added to the supernatant for 1 h at 4°C with shaking. Protein A Sepharose beads were used to precipitate the immune complex. The anti-N protein antibody-protein A beads were washed three times with 500 μl of TBS and boiled in 40 μl of NuPAGE lithium dodecyl sulfate sample buffer. Immunoprecipitated material and whole-cell lysates were analyzed in parallel by immunoblotting with antibodies directed against HTNV N protein and FLAG as described above.
RESULTS
HTNV N protein inhibits TNF-α-induced activation of NF-κB.
To date, there have been no reports of studies examining the ability of HTNV to antagonize the TNF-α signaling pathway. To determine if the HTNV N, Gn, or Gc protein could inhibit TNF-α-induced activation of NF-κB, a human NF-κB promoter that transcriptionally regulates the expression of GFP was used as a reporter. No GFP was detected in any of the samples that were left untreated, indicating that alone, the conditions of our experiment, including the presence of viral proteins, could not activate NF-κB (Fig. 1A). Transcriptional GFP expression from the NF-κB-responsive promoter was dose dependent, and HTNV N inhibited GFP expression at all concentrations of TNF-α compared to expression in the presence of the empty vector (Fig. 1A). HTNV glycoproteins had only little effect on GFP expression, suggesting that this phenomenon was N protein specific (Fig. 1A).
FIG. 1.

Effect of HTNV N protein on NF-κB gene expression. (A) 293T cells were cotransfected with 500 ng of pNF-κB-hrGFP and 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M. After 24 h, cells were incubated in medium with or without TNF-α (0 to 100 ng/ml) for 4 h. As a positive control for inhibition in our assay, cells transfected with only 500 ng of pNF-κB-hrGFP were pretreated with 50 μM MG132 for 2 h before the addition of TNF-α and throughout the experiment. (B) Cells were cotransfected with 500 ng of pNF-κB-hrGFP and 5 to 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M or transfected with 500 ng of pNF-κB-hrGFP and treated with 0 to 50 μM MG132. After 24 h, cells were incubated in medium with or without TNF-α (10 ng/ml) for 4 h. Following a 4-h treatment, medium was removed from all wells and replaced with medium lacking TNF-α for an additional 24 h. (C) Cells were cotransfected with 500 ng of pCAGGS-GFP and 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M. After 24 h, cells were incubated in medium with TNF-α (10 ng/ml) for an additional 24 h. GFP expression was examined by fluorescence microscopy.
Proteosomal degradation of IκB is required to activate NF-κB in the TNF-α pathway. In order to validate our assay, we included the proteosomal inhibitor MG132 as a control because it is a known inhibitor of this pathway. MG132 was also able to dramatically decrease the amount of GFP present in TNF-α-treated cells, and this decrease was comparable to the effect we observed in cells transfected with HTNV S segment cDNA (Fig. 1A).
We also examined whether TNF-α-induced activation of our reporter system correlated with the amount of viral protein or MG132 present in cells. As expected, increasing amounts of empty vector and HTNV glycoproteins had no effect on the expression of the NF-κB-GFP reporter (Fig. 1B). However, the inhibition of NF-κB-GFP was directly proportional to the total amount of HTNV N protein or MG132 in cells; hence, the greatest inhibition was observed with the maximal amount of N protein or MG132 present (Fig. 1B). It was noteworthy that the expression of the HTNV N and glycoprotein genes was not adversely affected by the amount of TNF-α or the time that cells were treated with TNF-α (data not shown).
Lastly, as a control for the specificity of action of HTNV N on the NF-κB promoter, cells were also cotransfected with a plasmid constitutively expressing GFP and the plasmids used in the experiments described above, as indicated in the legend to Fig. 1C. Twenty-four hours posttransfection, we observed no difference in the levels of expression of GFP in cells that also contained HTNV N or glycoprotein, indicating that HTNV protein expression alone was not enough to inhibit constitutive levels of GFP (Fig. 1C). Cells were then treated with TNF-α for an additional 24 h to determine if treatment alone or a combination of TNF-α and HTNV proteins were nonspecifically inhibiting GFP or causing cytotoxicity in our assay. Again, we could not discern any differences in the levels of GFP present in our cells. In addition, there was an increase in the amount of GFP-positive cells over the 24-h period, also suggesting that there were no cytotoxic effects (Fig. 1C).
HTNV N protein inhibits the activation of endogenous NF-κB p50 and p65.
In order to validate the NF-κB reporter assay described above, we next examined whether HTNV N protein was able to inhibit the activation of endogenous p50 and p65 subunits, the most abundant of the heterodimers used as an NF-κB transcription factor. To measure NF-κB transcription factor activation, we employed an enzyme-linked immunosorbent assay that measures binding to NF-κB consensus oligonucleotide sequences and then detects p50 and p65 subunits with antibodies. As expected, there was little activation in transfected and untreated cells, indicating that viral gene expression and transfection alone could not activate NF-κB proteins (Fig. 2A). In contrast, similar levels of activated p50 and p65 were detected in nuclear extracts from TNF-α-stimulated cells that were transfected with empty vector or HTNV M cDNA (Fig. 2A). Transfecting with HTNV S cDNA significantly inhibited the activation of p50 and p65, as determined with extracts of TNF-α-stimulated cells. This result was similar to what we observed in comparing MG132-treated cells and cells transfected with the empty vector (Fig. 2A). Although there was a small increase in the activation of p50 and p65 in HTNV S-expressing cells treated with TNF-α compared to that in untreated cells, this finding can most likely be explained by the fact that we were measuring the activation of endogenous NF-κB in a total cell population but not every cell was expressing the HTNV genes during the assay.
FIG. 2.
Endogenous NF-κB transcription activation in cells expressing HTNV N protein. A549 cells were transfected with 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M for 24 h. After treatment of the cells with 50 ng/ml of TNF-α for 15 min, cytoplasmic and nuclear extracts from lysates were prepared. (A) Nuclear extracts were allowed to bind to NF-κB consensus sequence oligonucleotides on 96-well plates and then probed with antibodies specific for NF-κB p65 or NF-κB p50. The absorbance reading for each sample was determined using a spectrophotometer. OD, optical density; MUT oligo, mutated consensus oligonucleotide; WT oligo, wild-type consensus oligonucleotide. (B) Cytoplasmic extracts were used for immunoblotting to detect HTNV N protein and GAPDH. Each point represents an average ± standard deviation of results for six samples. The statistical significance of results for HTNV S with TNF and MG132 with TNF was determined by comparing the results to those for the empty vector with TNF. Asterisks indicate significant differences (P < 0.05) as determined by Student's t test. +, with; −, without.
To confirm that cells were expressing viral genes, we prepared cytoplasmic extracts and examined them by immunoblotting to detect N protein (Fig. 2B) or by immunofluorescent antibody staining to detect Gc (see Fig. 4). Levels of the housekeeping protein GAPDH were also measured to rule out the possibility that the decreased inhibition we observed for HTNV N protein and MG132 treatment was not due to differences in the amount of total lysate used in our assay (Fig. 2B).
FIG. 4.

Translocation of NF-κB p65 in HTNV N-expressing cells. 293T cells were cotransfected with 500 ng of pCAGGS-GFP-p65 and 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M for 24 h. After incubation, cells were fixed and stained with antibodies against HTNV N or Gc protein (red) and with DAPI to highlight nuclei (blue). Arrows indicate areas of protein accumulation.
HTNV N expression does not affect levels of p50 and p65 or the degradation of IκBα.
Because it was difficult to definitively determine by the above-described assays if the decreased NF-κB activation was due to retention in the cytoplasm or a decrease in the amount of total NF-κB present in cells expressing HTNV N, we performed Western blot analysis. No difference in the levels of p50 and p65 proteins were observed in any of the samples that were transfected and unstimulated or stimulated with TNF-α (Fig. 3). GAPDH was used as a control to ensure equal loading of samples.
FIG. 3.
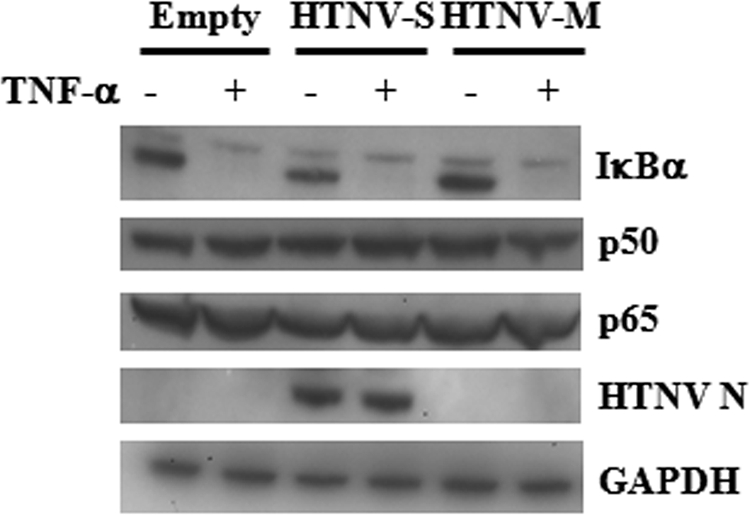
Examination of NF-κB p50 and p65 levels and TNF-α-induced IκBα degradation in HTNV N-expressing cells. A549 cells were transfected with 500 ng of pWRG7077-Empty, pWRG7077-HTNV-S, or pWRG7077-HTNV-M for 24 h. To induce the degradation of IκBα, cells were treated with 50 ng/ml of TNF-α for 15 min, and lysates were prepared for immunoblotting. For uninduced samples, cells were left untreated. Proteins were transferred onto PVDF membranes and probed with antibodies against IκBα, p50, p65, HTNV N protein, or GAPDH. +, with; −, without.
The TNF-α pathway requires the association of numerous signaling molecules such as TRADD, TRAF-2, and receptor-interacting protein, and the alteration of any of these molecules may inhibit the activation of NF-κB (44). The next question we wanted to address was if the NF-κB proteins were being targeted directly by HTNV N protein or if a signaling molecule further upstream was being inhibited, thus preventing the activation of NF-κB. Because IκBα serves as an inhibitor of NF-κB and its degradation is required for the translocation of activated NF-κB to the nucleus, we examined the ability of HTNV N protein to inhibit the degradation of IκBα in the presence of TNF-α. Western blot analysis of IκBα levels demonstrated that TNF-α treatment was effective in the subsequent degradation of IκBα in all transfected samples, including HTNV N-expressing cells (Fig. 3). Furthermore, IκBα levels were similar in all unstimulated and transfected samples (Fig. 3). These results suggested that the observed decrease in NF-κB activation was not due to upstream signaling molecules' being targeted but rather the inability of NF-κB itself to enter the nucleus.
HTNV N inhibits nuclear translocation of NF-κB p65.
To examine the possibility that HTNV N alters the ability of NF-κB to enter the nucleus, we used a nuclear translocation assay to examine the transport of p65. IκB serves as an inhibitor of NF-κB and is required for maintaining p65 in the cytoplasm. When p65 is overexpressed, endogenous levels of IκB become saturated, causing nuclear translocation of p65 (37). Furthermore, p65 translocates to the nucleus in the absence of TNF-α or other stimuli. To determine if HTNV N could inhibit the translocation of p65, we coexpressed a GFP-tagged p65 construct with our cDNA plasmids. In cells that were expressing GFP-p65 and empty vector, GFP-p65 could be detected in the nucleus, as indicated by the cyan color in the merged image of DAPI and GFP-p65 staining (Fig. 4, first row). Nuclear translocation of GFP-p65 was also identified in cells that were expressing HTNV glycoproteins, indicating that the glycoproteins had no effect on p65 translocation (Fig. 4, third row). When we examined HTNV N-expressing cells, we found mainly cytoplasmic GFP-p65 present in cells (Fig. 4, second row). Interestingly, we also noted that GFP-p65 was accumulating in areas of the cytoplasm (Fig. 4, second row, second column) and that this phenomenon was not seen in the empty vector control or cells expressing HTNV glycoproteins. When we looked at the staining of HTNV N, we also observed areas of accumulating N (Fig. 4, second row, third column), and this accumulation appeared to be in the same location as that of GFP-p65 (Fig. 4, second row, fourth column).
HTNV N protein binds to nuclear import protein importin α.
The above data suggest that NF-κB is being directly sequestered by HTNV N protein or has lost its ability to translocate to the nucleus. To examine if NF-κB was directly targeted, we performed coimmunoprecipitation experiments to determine if HTNV N protein could interact with p50 or p65. We were unable to detect an interaction between HTNV N protein and NF-κB proteins (data not shown), and this result was surprising since p65 and HTNV N proteins appeared in the same areas of cells (Fig. 4). This outcome led us to ask whether HTNV N could target nuclear transport proteins. It has been reported previously that NF-κB accumulates in the nucleus via interaction with importin α3 and importin α4 and its NLS. We wanted to determine if HTNV N could interact with these or possibly other importin α molecules. Cells were cotransfected with a FLAG-tagged importin α1, importin α2, importin α3, or importin α4 construct and an empty or HTNV S expression plasmid. Immunoprecipitation with an anti-HTNV N protein polyclonal antibody followed by Western blotting with anti-FLAG antibody was used to detect importin α proteins. As expected, no interaction of importin α and N was detected in cells that were transfected with empty vector, suggesting that our N antibody was not cross-reacting with any of the importin α proteins (Fig. 5). However, HTNV N protein was able to interact with importin α1, importin α2, and importin α3 but not importin α4, suggesting specificity for importin α proteins (Fig. 5). Furthermore, the interaction of HTNV N and importin α proteins occurred in the absence and presence of TNF-α, suggesting that signaling activation is not required for this interaction (Fig. 5). Western blotting of whole-cell lysates demonstrated that there were similar levels of importin α and N protein in our samples (Fig. 5).
FIG. 5.

Analysis of HTNV N protein interaction with importin α proteins. A549 cells were cotransfected with 0 or 1.25 μg of a FLAG-importin α1 (Iα1), FLAG-importin α2 (Iα2), FLAG-importin α3 (Iα3), or FLAG-importin α4 (Iα4) construct and 1.25 μg of pWRG7077-Empty or pWRG7077-HTNV-S plasmid for 24 h and left untreated (−TNF-α) or treated with 50 ng/ml TNF-α (+TNF-α) for 15 min. Immunoprecipitations (IP) were performed with anti-HTNV N protein antibody bound to Sepharose beads. The single asterisks indicate the location of HTNV N, whereas double asterisks identify the heavy chain. Immunoprecipitates and whole-cell lysates (WCL) were analyzed by Western blotting for the expression of importin α proteins and HTNV N protein. −, no importin α.
HTNV inhibits TNF-α-induced nuclear localization of NF-κB p65 without affecting IκBα degradation.
The results of the HTNV S segment transfection experiments clearly demonstrated the inhibition of NF-κB. To confirm that infection with HTNV also inhibited TNF-α-induced activation of NF-κB by preventing nuclear localization, we examined virus-infected cells by immunofluorescent antibody staining with antibodies to p65 and HTNV N protein. We did not detect nuclear p65 in unstimulated cells that were mock infected or infected with HTNV (Fig. 6A, left panels). TNF-α-stimulated, mock-infected cells had primarily nuclear localization of p65 (Fig. 6A, right panels, first column). Interestingly, as infection progressed to day 5, the staining pattern of p65 became predominantly cytoplasmic in TNF-α-treated cells, and this pattern was similar to that in unstimulated cells (Fig. 6A, right panels, second column).
FIG. 6.

Examination of TNF-α-induced NF-κB p65 nuclear translocation and degradation of IκBα in HTNV-infected cells. A549 cells were mock infected (M) or infected with HTNV (V) at an MOI of 5. On day 5, mock- and HTNV-infected cells were left untreated (−) or treated (+) with 50 ng/ml of TNF-α for 15 min. (A) After fixation, cells were stained with antibodies against NF-κB p65 (green) and HTNV N protein (red) and stained with DAPI to highlight nuclei (blue). (B) Cell lysates were separated into cytoplasmic and nuclear fractions. (C) Total cell lysates were prepared for immunoblotting, and proteins were transferred onto PVDF membranes. Blots were probed with antibodies against IκBα, p50, p65, HTNV N protein, GAPDH, or histone H2B.
To further establish that p65 is being retained in the cytoplasm, we performed fractionation experiments. Mock-infected or HTNV-infected lysates were separated into cytoplasmic and nuclear fractions following no stimulation or stimulation with TNF-α. Western blot analysis of cytoplasmic p65 indicates that similar levels of p65 were present in mock- and HTNV-infected lysates of unstimulated cells (Fig. 6B). However, when cells were treated with TNF-α, more p65 was detected in cytoplasmic fractions from HTNV-infected cell lysates than in those from mock-infected cell lysates (Fig. 6B). No p65 was detected in nuclear fractions of untreated mock- and HTNV-infected cell lysates (Fig. 6B). After the treatment of cells with TNF-α, we observed less p65 present in nuclear lysates of HTNV-infected cells than in those of mock-infected cells (Fig. 6B). Similar levels of GAPDH and histone H2B proteins were observed in cytoplasmic and nuclear fractions, respectively, from infected and mock-infected cells, indicating that differences were not due to loading (Fig. 6B).
To confirm that there was no decrease in the total levels of p50 or p65 protein and that no upstream signaling molecules were being targeted, we performed Western blot analysis. Mock-infected or HTNV-infected lysates harvested after no stimulation or stimulation with TNF-α had no apparent differences in the overall levels of p50 or p65, indicating that a reduction in amounts of p50 or p65 was not the cause of decreased nuclear localization in the presence of virus (Fig. 6C). HTNV was also unable to inhibit the degradation of IκBα after being treated with TNF-α, suggesting that IκBα itself and signaling molecules upstream were not being targeted by HTNV (Fig. 6C).
DISCUSSION
This is the first reported study to show that HTNV can antagonize the activation of NF-κB via the TNF-α pathway. In addition, our study revealed a novel function of the HTNV N protein. Hantavirus N protein is known to play a vital role in viral replication and is the most abundant protein detected in the cytoplasm of infected cells (16). Based on our results and those of others, the N protein appears to be emerging as a multifunctional protein with roles in several virus-host cell interactions in addition to viral replication. In earlier studies, Puumala virus N protein was found to interact with the apoptotic protein Daxx (23) and HTNV and Tula virus N proteins have been shown to interact with ubiquitin-conjugating (Ubc) enzyme and small ubiquitin-like modifier 1 (SUMO-1) (18, 25). Despite the identification of these interactions, the significance of the interactions remains uncertain. Here, we determined that NF-κB is sequestered in the cytoplasm and we identified an interaction of HTNV N protein with the nuclear transport proteins importin α1, importin α2, and importin α3. Importin α1 and importin α3 molecules were previously found to interact with NF-κB, although it is not clear if importin α1 is used for transport. Similarly, our assays could not determine if the binding of HTNV N protein to the various importin α proteins was related only to preventing the transport of NF-κB or if other, as-yet-undiscovered host pathways were also being targeted by the HTNV N protein interaction. This remains a possibility since HTNV N could interact with multiple importin α proteins even in the absence of TNF-α.
NF-κB can be rapidly activated by various cytokines, including TNF-α, and activation is followed by swift translocation to the nucleus, where it serves as a transcription factor for a number of genes. In its inactive state, IκB masks the NLSs of the NF-κB subunits, preventing the interaction of NF-κB with the transport molecule importin α. Once IκB is degraded, the NLS becomes unmasked and the importin α-NF-κB complex interacts with importin β. Importin β serves as an adaptor protein to tether the cargo-importin α complex to the cytoplasmic side of the nuclear pore so that it can be shuttled into the nucleus (21). We hypothesize that the HTNV N protein is acting as a decoy to bind and retain importin α molecules in the cytoplasm, thus preventing their interaction with NF-κB and its transport to the nucleus. We were unable to detect a direct interaction between HTNV N and NF-κB p50 and p65; however, this outcome may have been due to conditions in our assay. Alternatively, it remains possible that HTNV N is binding to both NF-κB and importin α, perhaps in a complex, to prevent the nuclear translocation of NF-κB. Further experiments need to be performed to clarify the exact mechanism of NF-κB inhibition.
Importin α proteins interact with the NLS via a domain that contains tandem repeats of 40 amino acids called armadillo motifs (6). All importin α molecules have two possible NLS binding sites contained within the N-terminal and C-terminal portions of the armadillo motifs (4, 8). NLSs allow importin α to distinguish between cargo that requires transport and other cellular proteins (21). NLSs consist of one (monopartite) or two (bipartite) stretches of the basic amino acids arginine and lysine (21). There are some reports of viral proteins, including VP24 of Ebola virus and ORF6 of severe acute respiratory syndrome coronavirus (9, 33), that contain NLSs allowing for the recruitment of importin α molecules and thus the inhibition of IFN pathways. Using the algorithm PSORT II (31), we were able to identify a putative, monopartite NLS within the HTNV N protein, beginning at amino acid position 172. This finding was surprising, as HTNV N protein has never been detected in the nucleus during infection. However, our results showing an interaction between HTNV N protein and importin α1, importin α2, and importin α3 lead us to hypothesize that the NLS within the N protein may be responsible for this interaction. Interestingly, further sequence analysis suggests that a putative NLS is found within the N proteins of other HFRS and HPS viruses, so further studies are needed to determine if the NLS sequences of hantaviruses are indeed the portion of the molecules that interact with the importin α proteins. It remains possible that if hantaviruses are utilizing an NLS to bind importin α molecules, different strains of hantaviruses may target different importin α molecules, thus inhibiting different pathways. Alternatively, binding importin α molecules to inhibit cellular pathways may be a common mechanism of HFRS and HPS viruses. We are especially interested in determining what these differences or similarities between the HFRS and HPS viruses are.
There is still much uncertainty regarding the mechanisms of hantavirus pathogenesis. It has been suggested previously that virus-activated immune cells and/or a combination of cytokines mediate this complex process. Although it is likely that all of these factors are involved, a firm understanding of the disease process remains elusive and is complicated by the fact that there is no animal disease model for the severe HFRS-causing viruses. Also, hantavirus replication does not produce cytopathic effects in infected cells in vitro. Furthermore, examination of postmortem human samples demonstrates no sustained damage to endothelial cells from hantavirus replication (17), yet patients suffer from increased permeability and capillary leakage (26). It has also been reported that patients have elevated levels of TNF-α, soluble TNF-α receptor, and interleukin-6 (IL-6) (24). Interestingly, in vitro, hantaviruses do not directly activate proinflammatory cytokines (TNF-α, IL-6, and IL-1) in endothelial cells (19, 40). However, TNF-α has been detected in kidney biopsy samples from HFRS patients during acute infection, suggesting that a local inflammatory response occurred (41). These findings, along with the presence of interstitial mononuclear infiltrates at sites of infection (30), suggest that these mononuclear cells are recruited and activated and release proinflammatory cytokines. This process may account for the presence of TNF-α that is detected in biopsy specimens. This local inflammatory response may be detrimental to viral replication. The results from our study would provide a mechanism whereby HTNV could protect itself from the antiviral effects of TNF-α released by immune cells in infected areas.
Unfortunately, there is no reverse genetic system or animal model to manipulate HTNV to establish virulence factors. This makes it difficult to determine the importance of our findings in relation to pathogenesis. What is clear is that the hantavirus N protein in the cytoplasm is in a prime location to target host cell proteins important in the inflammatory and innate immune response. Furthermore, the small number of proteins encoded by hantaviruses makes it likely that these proteins will have multiple functions in replication and in creating an environment to sustain replication.
Acknowledgments
We acknowledge Megan Shaw for cloning of the FLAG-importin α and pCAGGS-GFP-p65 constructs. All experiments involving infectious hantavirus were performed in a biosafety level 3 laboratory at the U.S. Army Medical Research Institute of Infectious Diseases.
The research described herein was sponsored by the Defense Threat Reduction Agency contract number W81XWH-07-2-0028. This research was performed while Shannon Taylor held a National Research Council research associateship award at the U.S. Army Medical Research Institute of Infectious Diseases.
The opinions, interpretations, conclusions, and recommendations presented herein are ours and are not necessarily endorsed by the U.S. Army or the Department of Defense.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Alff, P. J., I. N. Gavrilovskaya, E. Gorbunova, K. Endriss, Y. Chong, E. Geimonen, N. Sen, N. C. Reich, and E. R. Mackow. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 809676-9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baeuerle, P. A., and D. Baltimore. 1996. NF-kappa B: ten years after. Cell 8713-20. [DOI] [PubMed] [Google Scholar]
- 3.Beg, A. A., S. M. Ruben, R. I. Scheinman, S. Haskill, C. A. Rosen, and A. S. Baldwin, Jr. 1992. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 6:1899-1913. [DOI] [PubMed] [Google Scholar]
- 4.Conti, E., M. Uy, L. Leighton, G. Blobel, and J. Kuriyan. 1998. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell 94193-204. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham, M. D., J. Cleaveland, and S. G. Nadler. 2003. An intracellular targeted NLS peptide inhibitor of karyopherin alpha:NF-kappa B interactions. Biochem. Biophys. Res. Commun. 300403-407. [DOI] [PubMed] [Google Scholar]
- 6.Fagerlund, R., L. Kinnunen, M. Kohler, I. Julkunen, and K. Melen. 2005. NF-κB is transported into the nucleus by importin α3 and importin α4. J. Biol. Chem. 28015942-15951. [DOI] [PubMed] [Google Scholar]
- 7.Fagerlund, R., K. Melen, L. Kinnunen, and I. Julkunen. 2002. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin alpha 5. J. Biol. Chem. 27730072-30078. [DOI] [PubMed] [Google Scholar]
- 8.Fontes, M. R., T. Teh, and B. Kobe. 2000. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-alpha. J. Mol. Biol. 2971183-1194. [DOI] [PubMed] [Google Scholar]
- 9.Frieman, M., B. Yount, M. Heise, S. A. Kopecky-Bromberg, P. Palese, and R. S. Baric. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 819812-9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganchi, P. A., S. C. Sun, W. C. Greene, and D. W. Ballard. 1992. I kappa B/MAD-3 masks the nuclear localization signal of NF-kappa B p65 and requires the transactivation domain to inhibit NF-kappa B p65 DNA binding. Mol. Biol. Cell 31339-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorlich, D., and U. Kutay. 1999. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15607-660. [DOI] [PubMed] [Google Scholar]
- 12.Hayden, M. S., and S. Ghosh. 2004. Signaling to NF-kappaB. Genes Dev. 182195-2224. [DOI] [PubMed] [Google Scholar]
- 13.Henkel, T., U. Zabel, K. van Zee, J. M. Muller, E. Fanning, and P. A. Baeuerle. 1992. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell 681121-1133. [DOI] [PubMed] [Google Scholar]
- 14.Herbein, G., and W. A. O'Brien. 2000. Tumor necrosis factor (TNF)-alpha and TNF receptors in viral pathogenesis. Proc. Soc. Exp. Biol. Med. 223:241-257. [DOI] [PubMed] [Google Scholar]
- 15.Hooper, J. W., D. M. Custer, E. Thompson, and C. S. Schmaljohn. 2001. DNA vaccination with the Hantaan virus M gene protects hamsters against three of four HFRS hantaviruses and elicits a high-titer neutralizing antibody response in rhesus monkeys. J. Virol. 758469-8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson, C. B., and C. S. Schmaljohn. 2001. Replication of hantaviruses. Curr. Top. Microbiol. Immunol. 25615-32. [DOI] [PubMed] [Google Scholar]
- 17.Kanerva, M., J. Mustonen, and A. Vaheri. 1998. Pathogenesis of puumala and other hantavirus infections. Rev. Med. Virol. 867-86. [DOI] [PubMed] [Google Scholar]
- 18.Kaukinen, P., A. Vaheri, and A. Plyusnin. 2003. Non-covalent interaction between nucleocapsid protein of Tula hantavirus and small ubiquitin-related modifier-1, SUMO-1. Virus Res. 9237-45. [DOI] [PubMed] [Google Scholar]
- 19.Khaiboullina, S. F., D. M. Netski, P. Krumpe, and S. C. St. Jeor. 2000. Effects of tumor necrosis factor alpha on Sin Nombre virus infection in vitro. J. Virol. 7411966-11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krakauer, T., J. W. Leduc, and H. Krakauer. 1995. Serum levels of tumor necrosis factor-alpha, interleukin-1, and interleukin-6 in hemorrhagic fever with renal syndrome. Viral Immunol. 875-79. [DOI] [PubMed] [Google Scholar]
- 21.Lange, A., R. E. Mills, C. J. Lange, M. Stewart, S. E. Devine, and A. H. Corbett. 2007. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J. Biol. Chem. 2825101-5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, H. W., P. W. Lee, and K. M. Johnson. 1978. Isolation of the etiologic agent of Korean hemorrhagic fever. J. Infect. Dis. 137298-308. [DOI] [PubMed] [Google Scholar]
- 23.Li, X. D., T. P. Makela, D. Guo, R. Soliymani, V. Koistinen, O. Vapalahti, A. Vaheri, and H. Lankinen. 2002. Hantavirus nucleocapsid protein interacts with the Fas-mediated apoptosis enhancer Daxx. J. Gen. Virol. 83759-766. [DOI] [PubMed] [Google Scholar]
- 24.Linderholm, M., C. Ahlm, B. Settergren, A. Waage, and A. Tarnvik. 1996. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 17338-43. [DOI] [PubMed] [Google Scholar]
- 25.Maeda, A., B. H. Lee, K. Yoshimatsu, M. Saijo, I. Kurane, J. Arikawa, and S. Morikawa. 2003. The intracellular association of the nucleocapsid protein (NP) of Hantaan virus (HTNV) with small ubiquitin-like modifier-1 (SUMO-1) conjugating enzyme 9 (Ubc9). Virology 305288-297. [DOI] [PubMed] [Google Scholar]
- 26.Maes, P., J. Clement, P. H. Groeneveld, P. Colson, T. W. Huizinga, and M. Van Ranst. 2006. Tumor necrosis factor-alpha genetic predisposing factors can influence clinical severity in nephropathia epidemica. Viral Immunol. 19558-564. [DOI] [PubMed] [Google Scholar]
- 27.McBride, K. M., G. Banninger, C. McDonald, and N. C. Reich. 2002. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 211754-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melen, K., R. Fagerlund, J. Franke, M. Kohler, L. Kinnunen, and I. Julkunen. 2003. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J. Biol. Chem. 27828193-28200. [DOI] [PubMed] [Google Scholar]
- 29.Mori, M., A. L. Rothman, I. Kurane, J. M. Montoya, K. B. Nolte, J. E. Norman, D. C. Waite, F. T. Koster, and F. A. Ennis. 1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179295-302. [DOI] [PubMed] [Google Scholar]
- 30.Muranyi, W., U. Bahr, M. Zeier, and F. J. van der Woude. 2005. Hantavirus infection. J. Am. Soc Nephrol. 163669-3679. [DOI] [PubMed] [Google Scholar]
- 31.Nakai, K., and P. Horton. 1999. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 2434-36. [DOI] [PubMed] [Google Scholar]
- 32.Prescott, J., C. Ye, G. Sen, and B. Hjelle. 2005. Induction of innate immune response genes by Sin Nombre hantavirus does not require viral replication. J. Virol. 7915007-15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reid, S. P., L. W. Leung, A. L. Hartman, O. Martinez, M. L. Shaw, C. Carbonnelle, V. E. Volchkov, S. T. Nichol, and C. F. Basler. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 805156-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmaljohn, C., and B. Hjelle. 1997. Hantaviruses: a global disease problem. Emerg Infect. Dis. 395-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmaljohn, C., L. Vanderzanden, M. Bray, D. Custer, B. Meyer, D. Li, C. Rossi, D. Fuller, J. Fuller, J. Haynes, and J. Huggins. 1997. Naked DNA vaccines expressing the prM and E genes of Russian spring summer encephalitis virus and Central European encephalitis virus protect mice from homologous and heterologous challenge. J. Virol. 719563-9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmaljohn, C. S., G. B. Jennings, J. Hay, and J. M. Dalrymple. 1986. Coding strategy of the S genome segment of Hantaan virus. Virology 155633-643. [DOI] [PubMed] [Google Scholar]
- 37.Schmid, J. A., A. Birbach, R. Hofer-Warbinek, M. Pengg, U. Burner, P. G. Furtmuller, B. R. Binder, and R. de Martin. 2000. Dynamics of NF kappa B and Iκ Bα studied with green fluorescent protein (GFP) fusion proteins. Investigation of GFP-p65 binding to DNA by fluorescence resonance energy transfer. J. Biol. Chem. 27517035-17042. [DOI] [PubMed] [Google Scholar]
- 38.Shaw, M. L., W. B. Cardenas, D. Zamarin, P. Palese, and C. F. Basler. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and Toll-like receptor 3-triggered signaling pathways. J. Virol. 796078-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spiropoulou, C. F., C. G. Albarino, T. G. Ksiazek, and P. E. Rollin. 2007. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 812769-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundstrom, J. B., L. K. McMullan, C. F. Spiropoulou, W. C. Hooper, A. A. Ansari, C. J. Peters, and P. E. Rollin. 2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 756070-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Temonen, M., J. Mustonen, H. Helin, A. Pasternack, A. Vaheri, and H. Holthofer. 1996. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin. Immunol. Immunopathol. 7847-55. [DOI] [PubMed] [Google Scholar]
- 42.Thanos, D., and T. Maniatis. 1995. NF-kappa B: a lesson in family values. Cell 80529-532. [DOI] [PubMed] [Google Scholar]
- 43.Verma, I. M., J. K. Stevenson, E. M. Schwarz, D. Van Antwerp, and S. Miyamoto. 1995. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 92723-2735. [DOI] [PubMed] [Google Scholar]
- 44.Wajant, H., K. Pfizenmaier, and P. Scheurich. 2003. Tumor necrosis factor signaling. Cell Death Differ. 1045-65. [DOI] [PubMed] [Google Scholar]
- 45.Wang, P., P. Palese, and R. E. O'Neill. 1997. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza A virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 71:1850-1856. [DOI] [PMC free article] [PubMed] [Google Scholar]