

Abstract
We suggest two extensions of the coalescent effective population size of Sjödin et al. (2005) and make a third, practical point. First, to bolster its relevance to data and allow comparisons between models, the coalescent effective size should be recast as a kind of mutation effective size. Second, the requirement that the coalescent effective population size must depend linearly on the actual population size should be lifted. Third, even if the coalescent effective population size does not exist in the mathematical sense, it may be difficult to reject Kingman's coalescent using genetic data.
MODERN population genetics is data driven and yet relies on modeling to capture the long-term interaction of forces shaping genetic variation. Data are interpreted by comparing observed patterns of variation to the predictions of mathematical models. Minimally, these models incorporate mutation and random genetic drift, but often include other factors, such as population structure and natural selection. The standard neutral coalescent process (Kingman 1982; Hudson 1983; Tajima 1983), also known as Kingman's coalescent, is the accepted null model for the initial interpretation of data. For this reason, Sjödin et al. (2005) argued that Kingman's coalescent is a more relevant idealized model for discussions of effective population size than the traditional Wright–Fisher model (Fisher 1930; Wright 1931).
The idea of effective population size is to map a given population onto a simpler well-known model of a population. The effective size of a population is often defined loosely as the corresponding size of a Wright–Fisher population that would have the same “rate of genetic drift.” Several different definitions of effective population size have been proposed on the basis of single measures of the rate of genetic drift or single measures of polymorphism, such as heterozygosity (Crow and Kimura 1970; Ewens 1982, 1989). As Sjödin et al. (2005) point out, an effective size based on convergence to Kingman's coalescent is preferable because its existence implies that all aspects of genetic variation should conform to the predictions of Kingman's coalescent, meaning that any statistical test applied to data should reject the model only at the nominal level.
A coalescent effective size is also preferable because Kingman's coalescent has been shown to hold for a surprisingly wide variety of population models (Kingman 1982; Möhle 1998; Nordborg and Krone 2002), including the Wright–Fisher model and many others. In short, the complicated details of many populations disappear in the limit as the population size N tends to infinity, with time rescaled appropriately, so that the ancestry of a sample is determined by a very simple process. Each pair of lineages ancestral to the sample coalesces independently with rate 1 and each single lineage experiences mutations independently with rate θ/2. Note that defining an effective population size Ne in this context means we are interested only in its value or behavior asymptotically as the population size N tends to infinity.
We include mutation in “Kingman's coalescent” and argue that this is crucial because, without mutation, Kingman's coalescent (or any other model) cannot make predictions about genetic variation. The mutation parameter is defined as θ = 2Neμ for haploids and θ = 4Neμ for diploids, where μ is the mutation probability during meiosis at a locus under study. In cases where the complicated details of a population collapse to Kingman's coalescent as N → ∞, we advocate calling this Ne in θ the coalescent effective population size. This can be seen as a type of mutation effective size (Ewens 1989), which differs from previous definitions (Maruyama and Kimura 1980; Whitlock and Barton 1997; Charlesworth 2001; Pannell 2003) in that it applies to the parameter of the entire ancestral process, with its manifold predictions about data, rather than just to single measures of variation such as the heterozygosity of the population.
Sjödin et al. (2005) dealt with mutation implicitly. Following Möhle (2001) and Nordborg and Krone (2002), their definition focused instead on the way in which time is rescaled to achieve a coalescence rate equal to 1 for each pair of lineages. If AN(k) denotes the number of lineages ancestral to a sample in generation k in the past for a given population, and A(t) denotes the number of lineages ancestral to the sample at rescaled time t in the past under Kingman's coalescent, then if AN([Nt/c]) → A(t) as N → ∞, the coalescent effective size is N/c. Importantly, Sjödin et al. (2005) restricted their definition to cases in which c is a constant factor. In addition, because they considered populations with nonoverlapping generations, Sjödin et al. (2005) did not define a “generation” explicitly, as needed if the coalescent effective size is to apply to populations more generally (Felsenstein 1971; Hill 1979).
By pinning the concept of Ne to Kingman's coalescent, we follow Sjödin et al. (2005) in saying that the coalescent effective population size does not exist if AN([Nt/c]) converges to some other kind of ancestral process, such as a coalescent with multiple mergers (Pitman 1999; Sagitov 1999) or simultaneous multiple mergers (Schweinsberg 2000; Möhle and Sagitov 2001; Sagitov 2003). Thus, Ne here is different, and in this sense more restrictive, than the earlier definition by Möhle (2001), which allowed convergence to any of these continuous-time ancestral processes and also applied when the effective size could not be expressed as N/c with a constant c. However, the restriction to Kingman's coalescent seems desirable because multiple mergers can dramatically alter the most basic predictions of the model—for example, see Eldon and Wakeley (2006) and Sargsyan and Wakeley (2008)—so the utility of mapping populations onto a general set of coalescent models is not clear.
The Ne in the rescaled mutation parameter θ of Kingman's coalescent is a composite of two quantities that are crucial to genetic ancestry in any population: (1) the probability that a pair of ancestral lineages are descended from a common ancestor and (2) the probability that a single ancestral lineage is newly born (i.e., is the descendant of a birth or reproduction event). Both of these probabilities are computed for a single time step back into the ancestry of the sample. Importantly this initial unit of time will depend on the details of the population. When generations are nonoverlapping, as in the Wright–Fisher model, time is measured in units of generations. When generations are overlapping, as in the model of Moran (1958), time may be measured in other units, at least initially. Ultimately, we would like to measure time in comparable units in every model, namely in generations, and this is the purpose of the probability (2) above.
To illustrate, let cN and bN denote the probabilities (1) and (2) above, with subscripts to indicate possible dependence on the population size. In the haploid Wright–Fisher model, cN = 1/N and bN = 1, the latter because in each unit of time every individual in the population is replaced by a newborn. Compare this to the discrete-time Moran model, where in each time step a single offspring is produced and replaces a single adult who dies, including possibly the parent. For the Moran model we have cN = 2/N2, because one of the lineages we are following must be the offspring and the other must be the parent and there are two ways for this to occur, and bN = 1/N, because in this case the single lineage we are following must be the offspring itself.
These same probabilities apply in every time step, so in both cases the waiting time back to the event is geometrically distributed. A generation is defined as the average time back to the birth of a single lineage, or 1/bN. For the Wright–Fisher model, 1 time step constitutes 1 generation. For the Moran model, it takes N time steps to make 1 generation.
The convergence of ancestral processes as N → ∞ is described in detail in Möhle and Sagitov (2001), and we emphasize that our Ne exists only when multiple mergers become negligible and the limiting ancestral process is Kingman's coalescent. Convergence is achieved by measuring time in units of 1/cN time steps, which is the average time back to a coalescent event for a pair of lineages. In the Wright–Fisher model, 1/cN = N, and in the Moran model, 1/cN = N2/2. Note that this means that the Moran model does not have a coalescent effective population size according to the definition of Sjödin et al. (2005) because they require that 1/cN is a linear function of N. The Ne we proposed above avoids this potential problem.
Here, after Eldon and Wakeley (2006) and Sargsyan and Wakeley (2008), we focus not only on the way time must be rescaled by 1/cN time steps to obtain a coalescence rate of 1 for each pair of lineages, but also on the additional role that the opportunity for mutation plays in establishing a mutation rate of θ/2 for each single lineage in Kingman's coalescent. This additional scaling in θ is especially important when generations are overlapping. Convergence to Kingman's coalescent, with mutation rate θ/2 = 2Neμ for haploids or θ/2 = 4Neμ for diploids, occurs with a coalescent effective population size defined as
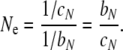 |
The intermediate step in this equation illustrates that Ne is the average time to a coalescent event measured in units of the average time back to a birth event (i.e., a generation). We have Ne = N for the Wright–Fisher model and Ne = N/2 for the Moran model, as expected. For any model of nonoverlapping generations, bN = 1, and our Ne becomes identical to the time-scale-only definitions of Möhle (2001) and Sjödin et al. (2005).
This new definition of Ne and the two points we make below are motivated by recent work (Sargsyan and Wakeley 2008) on a population model inspired by the biology of sessile marine organisms that reproduce by broadcast spawning. Individuals of these species, for example mussels, periodically release huge numbers of gametes into the water, which then may unite with gametes from other individuals to form larvae. Larvae spend varying amounts of time in the water column before settling in hopes of beginning adult life. Many gametes fail to unite and only a small fraction of larvae become successful adults. In addition, disturbance can be an important factor in opening up patches of habitat for colonization by larvae (Dayton 1971; Paine and Levin 1981).
This combination of life-history characteristics is not captured in the standard population genetic models or Wright–Fisher and Moran models. For example, there may be a “sweepstakes effect,” in which a relatively small number of individuals may have very large numbers of offspring, possibly even replacing a substantial fraction of the population in a single reproduction event (Beckenbach 1994; Hedgecock 1994). Events of this sort can never happen in the Moran model, with its single, paired birth–death events. In the Wright–Fisher model, even though all adults die and are replaced by offspring every generation, the chance that a substantial fraction of the population are the offspring of a few individuals is vanishingly small because every individual is equally likely to be the parent of every offspring.
The model in Sargsyan and Wakeley (2008) contains both the Wright–Fisher model and the Moran model as special cases and also includes the possibility of sweepstakes-like reproduction. Consider a discrete-time model of a finite population of constant size N, in which the default mode of reproduction is given by the Moran model. However, with probability ɛN a disturbance event occurs that removes XN individuals from the population. These are replaced by XN new individuals that are the offspring of YN adults whose larvae happen to the present at that time. The XN and YN individuals are chosen at random without replacement from the population, and each of the YN adults is equally likely to be the parent of each of the XN offspring. Subscripts denote that the dynamics in the limit N → ∞ will depend on the relative magnitudes of these parameters.
After defining Ne above, the second point we wish to make is that the coalescent effective population size should not be limited to cases in which Ne depends linearly on N. The model described above can converge to several different kinds of processes in the limit N → ∞, including a discrete-time Markov process, Kingman's coalescent, or a coalescent process with multiple mergers or simultaneous multiple mergers. For a detailed analysis, see Sargsyan and Wakeley (2008). Here we consider one of the special cases of the model in which the coalescent effective size exists, as we have defined it, but is not a linear function of the population size N.
Table 1 gives the parameters of the model, including some of those used to classify the limiting ancestral processes (Sargsyan and Wakeley 2008). Here we consider the case in which the probability of a disturbance event (ɛN) and the fraction of the population that is replaced in a disturbance event (XN/N) converge to finite, nonzero constants in the limit: 0 < ɛ < ∞ and 0 < φ < ∞. At the same time, we assume that the number of potential parents at each disturbance event is large, that is, YN → ∞ as N → ∞. However, we assume that YN grows more slowly than N, in particular YN/N → 0 as N → ∞. Two simple examples are 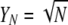 and YN = log(N), but it is not necessary to adopt any particular function form.
and YN = log(N), but it is not necessary to adopt any particular function form.
TABLE 1.
Parameters of the model of Sargsyan and Wakeley (2008)
| Discrete model parameters | |
|---|---|
| N | Population size, the no. of (haploid) individuals |
| ɛN | Probability of a disturbance event per time unit |
| XN | No. of individuals that die in a disturbance event |
| YN | No. of potential parents of the offspring that will replace the (XN) individuals that died in a disturbance event |
| Limiting model parameters
| |
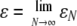 |
|
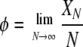 |
Fraction of population removed in each disturbance event |
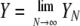 |
|
The details of why the ancestral process is Kingman's coalescent in this case are in Sargsyan and Wakeley (2008), but heuristically it follows from the fact that the number of potential parents (YN) is large. Note that, although it may in fact be reasonable to suppose, we do not necessarily imply that there is a biological dependence between YN and N. We simply offer the limiting model as a potentially useful approximation to the behavior of a very large population in which disturbance events occur with measurable frequency (ɛ) and intensity (φ)—perhaps as described for the mussel Mytilus californianus by Paine and Levin (1981)—and in which there are a large number of potential parents at each disturbance event; but for whatever reasons 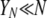 .
.
For this version of the model, using Equation 1 in Sargsyan and Wakeley (2008) gives
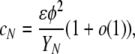 |
where o(1) denotes terms that go to zero as N → ∞. Ignoring the o(1) term, this formula is easily understandable as a simple product: the probability of a disturbance event times the probability that both ancestral lineages are newborns times the probability that they have the same parent. Similarly, using Equation 10 in Sargsyan and Wakeley (2008) gives
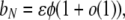 |
which, ignoring the o(1), is the product of the probability of a disturbance event and the probability that the ancestral lineage is among the newborns (and so may be a mutant). Thus, we have
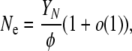 |
which is a less-than-linear function of N due to our assumption about YN. We argue that the coalescent effective size should extend to cases like this since genetic variation in a sample from such a population should agree in every way with the predictions of Kingman's coalescent.
Our third point is more practical than theoretical. Namely, our ability to discern from genetic data whether a coalescent effective population size is an appropriate concept for a given species may be limited. In our model there are cases in which the limiting ancestral process is not Kingman's coalescent, but rather a coalescent process with multiple mergers or simultaneous multiple mergers, and yet many of the predictions of the model are similar to those of Kingman's coalescent (Sargsyan and Wakeley 2008). We use another special case of the model, not too different from the one above, to show that the ability to distinguish these other ancestral processes from the standard coalescent can depend heavily on the sample size.
We consider a situation in which the probability of a disturbance event is small, but is still much larger than the probability of a coalescent event in the background Moran model, specifically N2ɛN → ∞ as N → ∞. In this case, a continuous-time coalescent process with simultaneous multiple mergers is obtained if YN → Y, with 2 ≤ Y < ∞, and XN/N → φ, with φ > 0. The resulting model should approximate the behavior of a very large population in which disturbance events occur infrequently, but still dominate the ancestral process, and where the offspring of a possibly small number of parents replace a substantial fraction of the population. This corresponds to the classic “sweepstakes” model of reproduction (Beckenbach 1994; Hedgecock 1994) and is captured in simulation Algorithm 1 of Sargsyan and Wakeley (2008).
There is no coalescent effective population size in this case because the ancestral process is not Kingman's coalescent but rather a coalescent with simultaneous multiple mergers. One of the ways that data from such a sweepstakes population should differ from data from a coalescent population is in having an excess of low-frequency polymorphisms (Beckenbach 1994; Hedgecock 1994; Sargsyan and Wakeley 2008). The commonly used test statistic D (Tajima 1989) should tend to be negative and may be used to reject Kingman's coalescent in favor of a coalescent with simultaneous multiple mergers. We use Tajima's test to illustrate that some aspects of genetic variation under this model may be similar to those under Kingman's coalescent, but we note that there may be more powerful tests (e.g., based on patterns of linkage disequilibrium).
We generated 100,000 pseudodata sets under the above model, with θ = 10 and φ = 0.5, and for a range of sample sizes and values of Y. We assumed that mutations occurred according to the infinite-sites model without intralocus recombination (Watterson 1975). We compared the values of Tajima's D to the lower 5% cutoff obtained for each sample size under Kingman's coalescent with θ = 10, also using simulations. Figure 1 shows the fraction of simulation replicates for which the value of Tajima's D is below the 5% cutoff, which is denoted q5% in Figure 1. Almost no pseudodata sets had significant positive values of D (results not shown), consistent with our expectation that D would deviate in the negative direction.
Figure 1.—

The power to reject the standard neutral coalescent at the 5% level using Tajima's D under the coalescent with simultaneous multiple mergers described in the text. Each point is based on 100,000 simulation replicates with θ = 10, and q5% is the lower 5% quantile computed under Kingman's coalescent with θ = 10, also using simulations.
As Figure 1 shows, with very small samples there is little power to reject Kingman's coalescent regardless of Y. For larger samples, the power increases, but the rate of increase depends strongly on Y. Even with a sample of size 1500 (not shown in the graph), the probability that D < q5% becomes only ∼0.32 for Y = 80. This is understandable because Y is the number of potential parents at each disturbance event. Unless the sample size is greater than Y, the chance of observing a multiple-merger coalescent event may be very small; see Wakeley and Takahashi (2003) for a similar result in a different model. Even though the ancestral process is not Kingman's coalescent, so that the coalescent effective population size does not exist, it may be very difficult to know this on the basis of samples of genetic data. On the other hand, it would not make sense to apply the concept of a coalescent effective population size in this case because a large sample (or perhaps multiple loci) would show patterns of variation distinctly different from those predicted by Kingman's coalescent and with some power would reject that model.
References
- Beckenbach, A. T., 1994. Mitochondrial haplotype frequencies in oyster: neutral alternatives to selection models, pp. 188–198 in Non-Neutral Evolution: Theories and Molecular Data, edited by B. Gelding. Chapman & Hall, New York.
- Charlesworth, B., 2001. Effect of life history and mode of inheritance on neutral genetic variation. Genet. Res. 77 153–166. [DOI] [PubMed] [Google Scholar]
- Crow, J. F., and M. Kimura, 1970. Introduction to Population Genetics Theory. Harper & Row, New York.
- Dayton, P. K., 1971. Competition, disturbance, and community organization: the provision and subsequent utilization of space in a rocky intertidal community. Ecol. Monogr. 41 351–389. [Google Scholar]
- Eldon, B., and J. Wakeley, 2006. Coalescent processes when the distribution of offspring number among individuals is highly skewed. Genetics 172 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1982. On the concept of effective size. Theor. Popul. Biol. 21 373–378. [Google Scholar]
- Ewens, W. J., 1989. The effective population size in the presence of catastrophes, pp. 9–25 in Mathematical Evolutionary Theory, edited by M. W. Feldman. Princeton University Press, Princeton, NJ.
- Felsenstein, J., 1971. Inbreeding and variance effective numbers in populations with overlapping generations. Genetics 68 581–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon, Oxford.
- Hedgecock, D., 1994. Does variance in reproductive success limit effective population sizes of marine organisms?, pp. 122–134 in Genetics and Evolution of Aquatic Organisms, edited by A. R. Beaumont. Chapman & Hall, London.
- Hill, W. G., 1979. A note on effective population size with overlapping generations. Genetics 92 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Testing the constant-rate neutral allele model with protein sequence data. Evolution 37 203–217. [DOI] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. On the genealogy of large populations. J. Appl. Probab. 19A 27–43. [Google Scholar]
- Maruyama, T., and M. Kimura, 1980. Genetic variability and effective population size when local extinction and recolonization of subpopulations are frequent. Proc. Natl. Acad. Sci. USA 77 6710–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhle, M., 1998. Robustness results for the coalescent. J. Appl. Probab. 35 438–447. [Google Scholar]
- Möhle, M., 2001. Forward and backward diffusion approximations for haploid exchangeable population models. Stoch. Proc. Appl. 95 133–149. [Google Scholar]
- Möhle, M., and S. Sagitov, 2001. A classification of coalescent processes for haploid exchangeable population models. Ann. Appl. Probab. 29 1547–1562. [Google Scholar]
- Moran, P. A. P., 1958. Random processes in genetics. Proc. Camb. Philos. Soc. 54 60–71. [Google Scholar]
- Nordborg, M., and S. M. Krone, 2002. Separation of time scales and convergence to the coalescent in structured populations, pp. 194–232 in Modern Developments in Theoretical Population Genetics: The Legacy of Gustave Malécot, edited by M. Slatkin and M. Veuille. Oxford University Press, Oxford.
- Paine, R. T., and S. A. Levin, 1981. Intertidal landscapes: disturbance and the dynamics of pattern. Ecol. Monogr. 51 145–178. [Google Scholar]
- Pannell, J. R., 2003. Coalescence in a metapopulation with recurrent local extinction and recolonization. Evolution 57 949–961. [DOI] [PubMed] [Google Scholar]
- Pitman, J., 1999. Coalescents with multiple collisions. Ann. Probab. 27 1870–1902. [Google Scholar]
- Sagitov, S., 1999. The general coalescent with asynchronous merges of ancestral lines. J. Appl. Probab. 36 1116–1125. [Google Scholar]
- Sagitov, S., 2003. Convergence to the coalescent with simultaneous mergers. J. Appl. Probab. 40 839–854. [Google Scholar]
- Sargsyan, O., and J. Wakeley, 2008. A coalescent process with simultaneous multiple mergers for approximating the genealogy of many marine organisms. Theor. Popul. Biol. 74 104–114. [DOI] [PubMed] [Google Scholar]
- Schweinsberg, J., 2000. Coalescents with simultaneous multiple collisions. Electron. J. Probab. 5 1–50. [Google Scholar]
- Sjödin, P., I. Kaj, S. Krone, M. Lascoux and M. Nordborg, 2005. On the meaning and existence of an effective population size. Genetics 169 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and T. Takahashi, 2003. Gene genealogies when the sample size exceeds the effective size of the population. Mol. Biol. Evol. 20 208–213. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., and N. H. Barton, 1997. The effective size of a subdivided population. Genetics 146 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]