

Abstract
The Hdmx protein restricts p53 activity in vivo and is overexpressed in a significant fraction of human tumors that retain the wild type p53 allele. An understanding of how Hdmx limits p53 activation and blocks apoptosis could therefore lead to development of novel therapeutic agents. We previously showed that Hdmx modulates tumor cell sensitivity to Nutlin-3a, a potent antagonist of the p53/Hdm2 interaction. In this report, we demonstrate that this also applies to MI-219, another Hdm2 antagonist. Thus, the inability to disrupt Hdmx/p53 complexes is a potential barrier to the efficacy of these compounds as single agents. We show that sensitivity to apoptosis in cells with high Hdmx levels is restored by combined treatment with Nutlin and a Bcl-2 family member antagonist to activate Bax. The data are consistent with a model in which Hdmx attenuates p53-dependent activation of the intrinsic apoptotic pathway, and that this occurs upstream of Bax activation. Thus, selectively inhibiting Hdm2 and activating Bax is one effective strategy to induce apoptosis in tumors with high Hdmx levels. Our findings also indicate that preferential induction of apoptosis in tumor versus normal cells occurs using appropriate drug doses.
Keywords: p53, apoptosis, Hdmx, Mdmx, bcl-2
Introduction
The p53 tumor suppressor is inactivated in roughly half of all human cancers due to altered expression of its upstream regulators including Arf and Hdm2.1 Hdm2 is an E3 ubiquitin ligase that antagonizes p53-dependent transactivation and targets it to the proteasome for degradation. Arf is a tumor suppressor that antagonizes Hdm2 function, leading to p53 activation. Correspondingly, loss of arf and/or overexpression of Hdm2 are common events in tumors that retain wild type p53.2, 3 Hdmx, an Hdm2 homolog, is also overexpressed in tumors with wild type p53, usually in the absence of arf or Hdm2 alterations.4 Thus, during tumorigenesis, selection for elevated Hdm2 or Hdmx is an effective mechanism to inhibit p53 tumor suppressor function. Hdmx is an effective inhibitor of p53-dependent transactivation, but has no intrinsic ability to degrade p53.5 However, Hdmx co-operates with Hdm2 to stimulate p53 degradation.6, 7 Importantly, both Hdm2 and Hdmx are essential p53 negative regulators since deleting either of the murine homologs results in early embryonic lethality that is rescued by p53 deletion.8–10
In light of these observations, Hdm2 and Hdmx are attractive targets for development of p53 agonists in tumors that encode wild type p53. In principle this can be achieved in at least three ways. First is the use of antagonists that disrupt Hdm2 and/or Hdmx-p53 interactions. The use of small molecules, including the cis-imidazoline compound Nutlin-3a, demonstrates the success of such an approach.11, 12 However, despite the significant amino acid similarities in the p53 binding sites of Hdm2 and Hdmx, the differences are sufficient to prevent significant disruption of Hdmx/p53 interactions by Nutlin-3a.13–15 Thus, the development of Hdmx-specific antagonists is an attractive strategy for use either as a single agent, or in combination with Hdm2 inhibitors.
A second approach is to use small molecules that promote the destabilization or degradation of Hdm2 or Hdmx, since in vitro studies indicate that their degradation is required for p53 activation.16 However, such molecules are not currently available, and await elucidation of factors that control Hdm2 and Hdmx stability in vivo.
A third strategy is to trigger tumor cell death by directly activating the core apoptotic machinery. This latter approach may be particularly effective when combined with agents that activate p53, since p53 target genes contribute to apoptosis induction (see below). Both ‘extrinsic’ and ‘intrinsic’ pathways can trigger cell death in vitro and in vivo.17 The intrinsic pathway is activated by changes in mitochondrial function that induce cytochrome c release and activate caspases. Pro- and anti-apoptotic Bcl-2 family proteins interact in a complex network, and set the threshold for apoptosis in response to stress.18 Apoptotic signals are transduced through this network and converge on the ‘effector BH3 proteins’, Bax and Bak. Conformational changes in Bax at the mitochondria lead to the release of cytochrome c and caspase activation. This activity of Bax is inhibited by anti-apoptotic Bcl-2 homologs that interact with Bax itself, or with upstream ‘activator BH3 proteins’. Notably the level of Mcl-1, a Bcl-2 family member that antagonizes pro-apoptotic Bax and Bak, is a determinant of cell survival in response to multiple apoptotic stimuli. Mcl-1 levels are increased by survival signals such as cytokines, and decreased by apoptotic stimuli such as UV irradiation.19, 20 Mcl-1 downregulation results from transcriptional repression, increased proteasome-dependent degradation or caspase activation.21, 22
Since anti-apoptotic proteins are frequently upregulated in cancer, they are also attractive therapeutic targets.23 ABT-737, a BH3 mimetic, antagonizes multiple Bcl-2 family members, thereby sensitizing cells to Bak- and Bax-induced apoptosis.24, 25 Importantly, the p53 pathway also converges on the mitochondrion to trigger the intrinsic death pathway by inducing transcriptional targets including the pro-apoptotic proteins Bax, Noxa and PUMA.26 p53 itself may also promote apoptosis independent of its transactivation function by activating Bax or by directly interacting with mitochondria.27, 28 Therefore, drugs such as ABT-737 may enhance p53-dependent apoptosis transduced via the mitochondria.
Here we determine whether another class of Hdm2 antagonist, the spiro-oxindole MI-219 29, can prevent Hdmx binding to p53, and whether this is associated with increased apoptosis compared to Nutlin-3a. We show that MI-219 activates p53 as effectively as Nutlin-3a, but neither drug appreciably affects Hdmx binding. Hdmx overexpression reduced apoptosis by blocking p53 transactivation of targets such as Bax, PUMA and Noxa. Importantly, by engaging the intrinsic apoptotic pathway with ABT-737, we restored Bax activation and sensitivity to Nutlin-induced death in multiple cell lines. These data indicate that priming the intrinsic death pathway can abrogate Hdmx-mediated inhibition of apoptosis. Appropriate dosing revealed that combination treatment selectively induced apoptosis in tumor versus normal cells, suggesting a favorable therapeutic index may be achievable using this drug combination strategy.
Materials and Methods
Cell culture and drug treatments
SJSA, MCF7 and derivatives were grown in DMEM/10% FBS plus CIP and Fungizone (GIBCO). BL cells (Professor Martin Allday, Imperial College, London) were grown in RPMI/10% heat inactivated FBS. MEFs were grown in DMEM/15% FBS/100 μM beta-mercaptoethanol at 3% oxygen. WS1 cells were from ATCC and grown as described previously.16 Nutlin-3a was supplied by Lyubomir Vassilev (Roche, Nutley, New Jersey). MI-219 and MI-426 were supplied by Dajun Yang and Nathalie Bruey-Sedano (Ascenta Therapeutics, San Diego, CA). ABT-737 was supplied by Stephen Fesik (Abbott Laboratories, Illinois). MG132 was from Calbiochem (San Diego, CA). zVAD-fmk was from MBL International (Woburn, MA). Reagents were dissolved in DMSO to give 10mM stock solutions.
Plasmids and viral infections
Hdmx cDNA was originally from Dr Aart Jochemsen (Leiden University, Netherlands). Hdmx tagged at the N terminus with a 2xHA epitope and the G57A Hdmx mutant were made by PCR and mutagenesis (sequences on request). Untagged and HA tagged Hdmx constructs were subcloned into the pKEY-GFP lentiviral backbone (Dr Kristine Yoder) for infection of SJSA cells. p53-DD was subcloned into pCLNCX retrovirus and resistant SJSA cells were selected in 800ug/ml G418 (GIBCO). Viral infections were as previously described.15
Quantitative PCR
RNA and quantitative PCR were performed as previously described.15, 30 Sequences of primers are in Supplementary Table 1.
Cell death assays
Adherent and floating cells were harvested and subjected to trypan blue counts for initial evaluation of cell death. Following 2 × PBS washes, cells were fixed in 70% ethanol and stained with propidium iodide solution (final 1 μg/ml, SIGMA) containing RNAseA (final 200 μg/ml) for a minimum of 2h prior to FACS analysis (Becton-Dickinson) of the subG1 population. For Hdmx siRNA/ABT-737 experiments, MCF7 were seeded at 25–30% confluence at the time of siRNA transfection, and ABT-737 was added 24h later. Cells were harvested 24–48h post-addition of ABT-737. For colony formation assays, cells were plated at 2000/10cm plate and colonies were allowed to form (~5 days) before addition of Nutlin-3a and/or ABT-737, which were continuously present. After ~7d, cells were fixed in 4% formaldehyde and stained with crystal violet prior to colony counting. For AnnexinV (Pharmingen) assays, samples were normalized for cell number and washed once in PBS- prior to resuspension in 300ul binding buffer (10mM HEPES pH7.4, 150mM NaCl, 5mM KCl, 1mM MgCl2, 1.8mM CaCl2) containing AnnexinV diluted 1:100. After incubation for 10 min on ice, cells were analyzed for AnnexinV binding by FACS.
Immunofluorescence
Cells were fixed in 4% formaldehyde 10 min RT and permeabilized in 0.2% CHAPS/PBS for 5 min RT. Following a wash in PBS/0.02% Tween20, cells were blocked in PBS/0.02% Tween20/10% normal goat serum for 20 min. A mix of anti-Bax (monoclonal 6A7, 1:200, Exalpha) and anti-p21 (monoclonal IgG2a, 1:400, Transduction Labs) was added overnight at 4 degrees. Following 3 washes in PBS/0.02% Tween20, isotype-specific secondaries (1:1500, Molecular Probes) with 1ug/ml Hoechst were added for 1h RT. Cells were washed in PBS/0.02% Tween20 followed by distilled water, dehydrated in 100% ethanol and mounted in Vectashield (Vector Labs).
Western blotting and immunoprecipitations
Blots were performed as described previously.16 Antibodies and dilutions were: Hdm2 (SMP14, IF2, 4B2, all from Oncogene, 1:500 each), Hdmx (BL1258 (1:5000), Bethyl Labs), p53 (mouse 1:2000, DO-1, Oncogene), actin (rabbit 1:10,000, SIGMA), tubulin (mouse 1:500,000, SIGMA), p21 (rabbit 1:1500, Santa Cruz C-19), full length caspase 9 (rabbit, 1:1000, Santa Cruz), full length and cleaved caspase-3 (rabbit 1:1000, Cell Signaling Technology), PUMA (rabbit 1:500, ProSci #3043), Mcl-1 (rabbit 1:2000, S-19, Santa Cruz), Bcl-XL (1:1000, BD Pharmingen #551259). Immunoprecipitations were performed using agarose-conjugated FL-393 for p53 (Santa Cruz), or HA.11 for Hdmx (BabCo) per the manufacturer’s recommendations. 500 μg of total lysate was used for each IP as described previously.15
Results
The spiro-oxindole MI-219 exhibits equivalent potency as Nutlin-3a as a p53 activator, but does not prevent Hdmx binding
Hdmx level is a determinant of sensitivity to the Hdm2 antagonist Nutlin-3a, since Hdmx can still bind to p53 in the presence of the drug.13–15 We therefore evaluated whether the chemically distinct spiro-oxindole Hdm2 antagonist MI-219 29 exhibits broader activity and antagonizes Hdmx/p53 interaction. Figure 1A shows that, although there were differences in the magnitude of p53 stabilization, MI-219 and Nutlin-3a exhibited similar potencies with regard to p53 activation (evaluated by p21 mRNA induction) at 24h post-treatment. This effect is likely general as it occurs in both murine embryonic fibroblasts and human MCF7 breast carcinoma cells. As previously reported, Hdmx degradation is attenuated following treatment of MCF7 cells with Nutlin-3a15, and MI-219 also failed to induce Hdmx degradation in these cells. Both compounds were effective antagonists of the Hdm2/p53 interaction but, strikingly, neither compound disrupted Hdmx/p53 complexes (Figure 1B). Note that the amount of Hdm2 associated with p53 was significantly reduced after Nutlin-3a or MI-219 treatment compared to untreated cells (compare the amount of co-immunoprecipitated Hdm2 in lanes 5–7 with the Hdm2 input in lanes 1–3). Nutlin-3a and MI-219 also resulted in higher p53 and Hdm2 levels than proteasome inhibition (PI), yet less Hdm2 was co-precipitated with p53 in the presence of the Hdm2 antagonists (compare lanes 6–8). This also indicates that p53/Hdm2 complexes are stabilized by PI, but destabilized or prevented from forming by Hdm2 antagonists. Conversely, we observed that the Hdmx/p53 interaction was not reduced by either Nutlin-3a or MI-219. This is concordant with in vitro Biacore studies showing that both Nutlin-3a and MI-219 bind with significantly higher affinities to Hdm2 than to Hdmx (31 and Dajun Yang, personal communication). Consistent with their similar activation of p53 transcription, Nutlin-3a and MI-219 induced a similar level of apoptosis in SJSA osteosarcoma cells (Figure 1C).
Figure 1.

(A) MCF7 (upper panel) or MEFs (lower panel) were treated with Nutlin-3a, MI-219 or MI-426 (inactive control) all at 10uM for the indicated times and lysates evaluated for the indicated proteins. Quantitative qPCR of p21 mRNA abundance was used as a metric of p53 activation. (B) MCF7 were treated with Nutlin-3a, MI-219 or proteasome inhibitor MG132 (all 10uM) for 5h prior to immunoprecipitation with anti-p53 antibody FL393. Long and short refer to exposure times for Hdm2 and Hdmx. (C) SJSA were treated with Nutlin-3a or MI-219 for 36h and subjected to FACS analysis following propidium iodide staining.
Hdmx binding to p53 mediates protection against Nutlin-induced apoptosis
Since Nutlin-3a and MI-219 do not effectively antagonize p53/Hdmx interaction, we reasoned that Hdmx overexpression would reduce apoptosis induced by these agents. We tested this hypothesis by comparing apoptosis induction in SJSA cells, which express high levels of Hdm2, very little Hdmx and are highly sensitive to Nutlin-induced cell death32, to an SJSA derivative we engineered to stably overexpress Hdmx (SJSA-X). Western analysis and viability assays showed that both Nutlin and MI-219 were equally effective in reducing SJSA viability. Furthermore, Hdmx blocked caspase-3 activation, and increased survival following treatment with Nutlin-3a and MI-219 (Figure 2A and B). The protection required Hdmx binding to p53, since the Hdmx p53 binding mutant, G57A, failed to protect against Nutlin-induced apoptosis despite being expressed at similar levels as WT Hdmx (Figure 2C). Apoptosis can be triggered by p53-dependent transcription of pro-apoptotic genes and/or by microRNAs that downregulate survival proteins.26, 33 Therefore, Hdmx suppression of Nutlin-induced death may be due to reduced p53 target gene expression. To investigate this, we compared the expression of p53 gene targets in SJSA and SJSA-X cells. Induction of p21 mRNA and protein was significantly decreased in SJSA-X cells (Figures 2D and 3A). At 48h, the apparent increase in p21 in SJSA-X cells relative to SJSA is likely due to significant apoptosis and consequent p21 degradation in SJSA cells. The induction of most pro-apoptotic genes, including those encoding the BH3 proteins PUMA and Noxa, was also significantly reduced by Hdmx overexpression (Figure 3A and data not shown). Taken together, these data show that Hdmx suppresses p53 transactivation of multiple pro-apoptotic target genes, thereby reducing the apoptotic response.
Figure 2.

(A) SJSA or SJSA-X were treated with 10uM Nutlin-3a for the indicated times and lysates were analyzed for the indicated proteins. (B) Quantification of cell viability by trypan blue staining in SJSA versus SJSA-X following treatment with 2.5 or 10uM Nutlin-3a or MI-219 (48h). (C) Upper panel: Cell viability in parental SJSA, or in cells overexpressing WT Hdmx (WT) or the p53 binding mutant G57A. Lower panel: SJSA expressing HA tagged WT or G57A were treated with Nutlin for 6h and Hdmx was immunoprecipitated using anti-HA antibody. (D) Parental SJSA (P) or SJSA-X (X) were treated with 10uM Nutlin-3a for the indicated times and subjected to western analysis.
Figure 3.

(A) qPCR analysis of p53 gene targets 24h post Nutlin-3a treatment. Data are average of 2 independent experiments. (B) Western analysis showing (left panel) downregulation of Mcl-1 in parental SJSA, but not in SJSA-Hdmx cells following Nutlin-3a treatment and (C) rescue of Nutlin-3a induced Mcl-1 degradation by preincubation with zVAD to block caspase activation. SJSA (C) and BL cells (D) were treated with Nutlin-3a in the presence or absence of zVAD for 24h prior to blotting for the indicated proteins.
In addition to examining pro-apoptotic Bcl-2 family members, we also analyzed levels of the anti-apoptotic proteins Bcl-XL and Mcl-1 (Figures 2D and 3B). Bcl-XL levels were unchanged until 48h in SJSA cells, by which time almost all the cells had undergone apoptosis. In contrast, significant decreases in Mcl-1 protein levels occurred as early as 8h following Nutlin-3a treatment in SJSA cells, but did not change in apoptosis-resistant SJSA-X cells (Figure 3B).
Downregulation of Mcl-1 can be a cause or consequence of apoptosis, depending on the stimulus, cell type and time of analysis.21 To determine whether Mcl-1 degradation occurred prior to or following apoptosis, we pretreated SJSA cells with the caspase inhibitor zVAD before treatment with Nutlin-3a. zVAD blocked both caspase-3 activation and downregulation of Mcl-1, suggesting that Mcl-1 downregulation is caspase-dependent, (Figure 3C). Additionally, this indicates that Mcl-1 cleavage is a consequence, rather than a cause of, apoptosis induced by Nutlin-3a. Together, these data indicate that Nutlin-induced death involves induction of pro-apoptotic p53 target genes, and downregulation of anti-apoptotic Bcl-2 family members. By preventing p53 activation, Hdmx suppresses both events, thereby blocking cell death.
We examined additional cell lines to determine whether there was a general ability of Hdmx to inhibit apoptosis. The Nutlin-sensitive Burkitt lymphoma line BL2 15, also exhibited Mcl-1 downregulation concomitant with Nutlin-mediated p53 activation and Hdmx degradation (Figure 3D). While Mcl-1 degradation is caspase-dependent, Hdmx degradation is not (Figure 3D), and is mainly due to p53-mediated activation of Hdm2. By contrast, in Nutlin-resistant BL40 cells, neither Hdmx nor Mcl-1 was downregulated, and caspase-3 was not activated (lanes 4–6). Absence of caspase activation in BL40 cells was not due to lack of caspase-3 expression (Supplementary Figure 1). These data indicate that the ability of Nutlin-3a to induce Hdmx degradation precedes caspase activation, and is one of the triggers of the apoptotic response.
Bcl2 family inhibition overcomes Hdmx-dependent protection
Changes in the ratio and specific interactions between pro- and anti-apoptotic proteins are integrated at the mitochondria, which are critical mediators of the intrinsic apoptosis pathway. Our observations suggest that the ability of Hdmx to antagonize p53 transactivation affects the levels of pro- and anti-apoptotic Bcl-2 family members, which determines cell fate following Nutlin-3a treatment. Therefore, agents that activate the intrinsic death pathway might synergize with p53 agonists to trigger apoptosis in tumor cells expressing high Hdmx levels.
We compared components of the intrinsic apoptotic pathway between SJSA and SJSA-X cells. To determine which molecular events Hdmx might inhibit, we initially focused on Bax activation, since it is a pro-apoptotic molecule implicated in p53-dependent apoptosis.34, 35 Since Bax insertion into the mitochondria triggers release of cytochrome c and caspase activation36, we quantified Bax activation by the appearance of immunofluorescent foci following incubation with the conformation-specific antibody 6A7.37 Untreated cells exhibited diffuse cytosolic 6A7 immunoreactivity, without significant focal localization at mitochondria. By contrast, parental SJSA cells treated with Nutlin-3a exhibited obvious punctate cytoplasmic fluorescence by 48h treatment, consistent with marked apoptosis at this time (Figure 4A). Notably, maximal Bax activation occurs in the presence of the caspase inhibitor zVAD. This is most likely because cells are lost rapidly once Bax is inserted into the mitochondria and apoptosis is initiated.38, 39 Importantly, Hdmx overexpression in SJSA-X cells significantly reduced p53 activation of endogenous target genes such as p21. Bax activation was significantly reduced, and the number of viable cells was increased (Figures 2 and 4B). These data indicate that Hdmx attenuates p53 transactivation and consequent Bax activation, mitochondrial disruption and induction of apoptosis.
Figure 4.
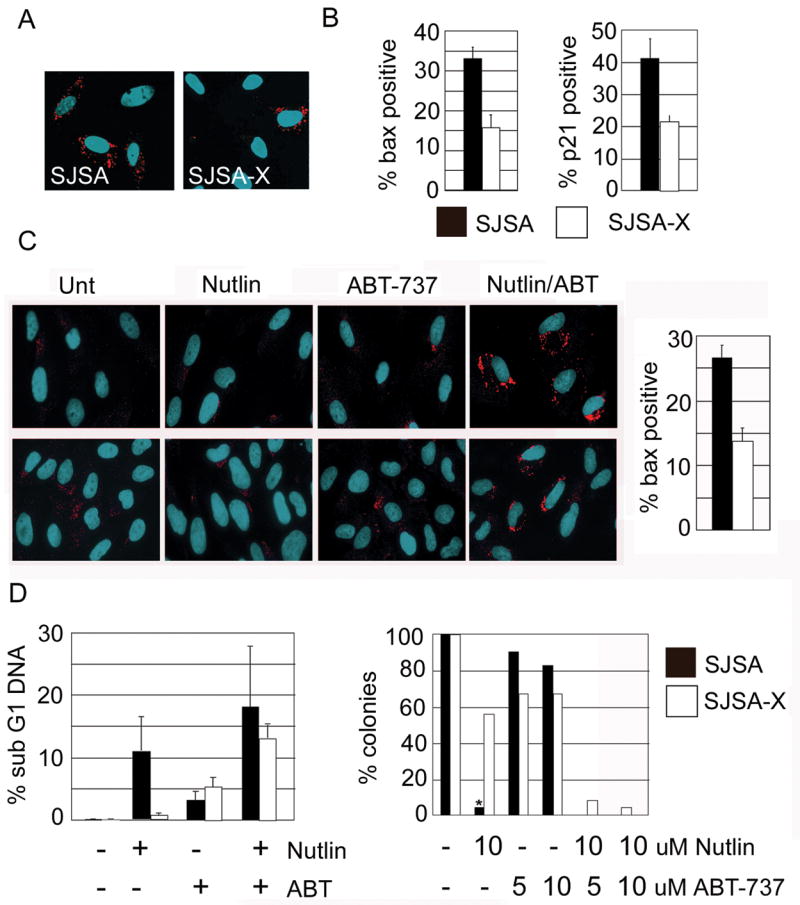
SJSA and SJSA-X were pretreated with 100uM zVAD for 1h prior to 48h treatment with 10uM Nutlin-3a. Cells were immunostained for active Bax using 6A7 antibody (A) and quantified for active Bax and p21 expression (B). (C) SJSA (upper panels) and SJSA-X (lower panels) were pre-treated with 100uM zVAD for 1h prior to addition of 20uM ABT-737, 10uM Nutlin-3a or both. 24h later, cells were processed for activated Bax by immunofluorescence. (D) Quantification of apoptosis (left) and colony formation (right) induced by Nutlin-3a or ABT-737 treatment alone, or the combination. SubG1 is likely an underestimate of the actual amount of death in the combined treatment, since many cells were already in late stage apoptosis under these conditions. For colony formation, SJSA were seeded onto 10cm plates and colonies were allowed to form. (*): no colonies were observed following Nutlin treatment of parental SJSA. Drugs were added and left in the medium for 7 days.
If Bax activation is a limiting event in apoptosis in cells overexpressing Hdmx, then small molecule activation of Bax in combination with Nutlin-3a should efficiently kill SJSA-X cells. We investigated this by treating cells with Nutlin-3a alone or in combination with ABT-737, a BH3 mimetic that antagonizes pro-survival Bcl-2 family members and promotes the release of Bax and Bak.40 In this liberated, latent form, Bax is susceptible to activation by other pro-apoptotic BH3 proteins. In SJSA cells, 24h treatment with Nutlin-3a induced low levels of Bax activation and apoptosis (Figure 4C and data not shown). Similarly, Bax was not activated when cells were treated solely with ABT-737. This is consistent with previous data indicating that antagonism of Bcl-2 family members alone is insufficient to activate Bax.25 Strikingly, combined Nutlin-3a and ABT-737 treatment for 24h robustly activated Bax compared to either treatment alone (Figure 4C). We did not observe enhanced suppression of colony outgrowth after combination treatment in parental SJSA cells compared to Nutlin alone (Figure 4D). This is probably because sustained Nutlin exposure alone potently induces apoptosis in SJSA cells, which express very low levels of Hdmx. Importantly however, we found that the Nutlin-3a/ABT-737 combination sensitized SJSA-X cells to apoptosis (Figure 4D). Despite the sensitization of SJSA-X cells, the frequency of Bax-positive cells was lower than in parental cells (Figure 4C). This presumably relates to heterogenous Hdmx expression levels in this non-clonal population, leading to more robust Hdmx-dependent suppression of p53 activation in some cells. However as shown in longer term colony formation assays (Figure 4D), all SJSA-X cells eventually died, indicating that the Hdmx expression levels were not sufficient to block the effects of combined Nutlin-3a/ABT-737 treatment. This effect was not observed in SJSA cells in which p53 function was disabled by overexpression of a dominant-negative p53 mutant (p53-DD, Supplementary Figure 2). Thus, although ABT-737 can induce p53-independent cell death40, its major effect in these cells is to enhance p53-dependent apoptosis. These data indicate that suppression of p53-dependent apoptosis by Hdmx occurs upstream of Bax activation. Furthermore, they suggest that a combination of p53 activation and pro-apoptotic BH3 agonists is a potent death stimulus in cells that overexpress Hdmx.
We next determined whether ABT-737 could be used to sensitize MCF7 cells to Nutlin-induced apoptosis, since they are resistant to Nutlin-induced cell death due to elevated levels of Hdmx.15 Consistent with our previous observations, we observed little death following treatment with Nutlin-3a alone, and few cells displayed Bax immunoreactivity (Figure 5A and 5B). This was despite induction of the pro-apoptotic p53 target, PUMA (Figure 5C). ABT-737 alone induced a modest level of apoptosis, in agreement with a recent study of this cell line (Figure 5A and 25). However, there was a dramatic increase in both Bax activation and cell killing when MCF7 cells were treated with Nutlin-3a and ABT-737 simultaneously (Figure 5A and B). These data confirm our observation in SJSA cells, and clearly demonstrate that ABT-737 enhances the cytotoxicity of Hdm2 antagonists in cancer cells expressing high endogenous Hdmx levels.
Figure 5.

(A) MCF7 were treated with the indicated drug combinations and subjected to FACS analysis of sub-G1 DNA content. (B) MCF7 were treated with Nutlin-3a or ABT-737, or with the combination for 24h, and stained for p21 (green) and active Bax (red). Comparison of lower panels reveals the extent of Bax activation in the combination treatment when caspase activity is blocked. (C) Western analysis of MCF7 treated with the indicated dose of Nutlin-3a or ABT-737 for 24h. Note that Puma is induced following Nutlin-3a treatment in these cells, but that Mcl-1 is not degraded, even in cells treated with both Nutlin-3a and ABT-737. (D) Nutlin-3a sensitive (NutS) or Nutlin-3a resistant (NutR) Burkitt lymphoma lines were treated as indicated for 24h with 5uM Nutlin-3a, or 10uM ABT-737 or a combination of the two. Death was evaluated by AnnexinV staining and the summary of three independent experiments is shown.
Mcl-1, an anti-apoptotic molecule, is not efficiently antagonized by ABT-737, and the levels of Mcl-1 therefore limit efficacy of the drug.41, 42 Since we observed Mcl-1 downregulation in SJSA cells following Nutlin-3a treatment, the enhanced toxicity following combined Nutlin-3a and ABT-737 treatment in MCF7 cells could be due to decreased Mcl-1 levels. However, we found that Mcl-1 is not downregulated in MCF7 cells following Nutlin-3a treatment alone, or in combination with ABT-737 (Figure 5C). An alternative explanation is that ABT-737 could enhance p53-dependent transactivation. However, we observed no increase in p21 or Hdm2 levels following treatment with ABT-737 alone, and no enhancement of Nutlin-induced p53 transcription by ABT-737 (Figure 5B, C and data not shown). We infer that Nutlin-3a-mediated induction of pro-apoptotic BH3 proteins such as PUMA, which can antagonize Mcl-118 enhances activation of Bax liberated by ABT-737 in MCF7 cells. In effect, Nutlin-3a increases the ratio of pro- to anti-apoptotic proteins, circumventing the need for downregulating Mcl-1. We note that UV irradiation induced Mcl-1 degradation in MCF7 cells, and that this is only partly rescued by the caspase inhibitor, zVAD (data not shown). This indicates that Mcl-1 in MCF7 cells is not resistant to degradation per se. Rather, our data indicate that MCF7 cells lack additional factor(s) required for Nutlin-induced downregulation of Mcl-1.
We next sought to determine the generality of the enhanced toxicity generated by Nutlin-3a and ABT-737 co-treatment. We observed potentiation of apoptosis in Burkitt lymphoma cell lines that are sensitive to Nutlin-3a, as well as those that are resistant to Nutlin3a-induced apoptosis. Figure 5D shows that BL2 cells, despite their sensitivity to apoptosis induced by Nutlin-3a as a single agent, underwent increased cell death in the presence of ABT-737. As previously shown15, Nutlin-3a alone did not induce Hdmx degradation or apoptosis in BL40 cells. However, combination treatment significantly increased BL40 cell death. The death in these cells requires p53, since the Nutlin-3a/ABT-737 combination did not induce apoptosis in BL41 cells, which express transactivation-deficient mutant p53. Taken together, our data demonstrate that the attenuation of p53-dependent apoptosis by Hdmx can be abrogated by BH3 activation.
Combined treatment with Nutlin-3a and ABT-737 selectively induces apoptosis in cancer versus normal cells
Ideally, novel anticancer therapeutics should exhibit low toxicity toward normal cells. To date, the effect of combined Nutlin-3a and ABT-737 treatment in normal cells has not been described. In order to evaluate this in our system, we quantified the effect of combination treatment in WS1 normal human fibroblasts compared to MCF7 cells. Figure 6 illustrates that each drug, when used as a single agent, exhibited little toxicity to either cell type. When used alone, Nutlin-3a treatment of both WS1 and MCF7 induced little apoptosis (Figure 6A). Instead, there was an accumulation of WS1 cells at G2/M, and a slight increase in MCF7 cells in G1 (see Supplementary Table 2). The cell cycle profile of WS1 cells following combined treatment resembled that of Nutlin treatment alone, suggesting that the arrest induced by Nutlin is dominant in these cells (Figure 6A and Supplementary Table 2). Importantly, combination treatment induced significantly more apoptosis in MCF7 cells, but not WS1 cells, compared to single agent treatment (Figure 6B). The selective induction of apoptosis in MCF7 versus WS1 cells was lost at higher concentrations of Nutlin and ABT-737 (data not shown). These data suggest that preferential targeting of tumor versus normal cells using these drugs may be achieved with an appropriate dosing strategy.
Figure 6.

WS1 and MCF7 cells were treated with the indicated doses (uM) of Nutlin or ABT-737 for 48h prior to flow cytometric analysis of DNA content by propidium iodide staining. (A) Representative FACS profiles from both cell types. (B) Quantification of apoptosis (sub G1 DNA content) following the indicated treatments (mean and standard deviation of 3 experiments is shown).
Discussion
Activation of apoptosis in tumor cells by non-genotoxic agents is an attractive anticancer strategy, as it should limit the induction of secondary cancers resulting from genomic damage. Both Nutlin-3a and ABT-737 have proven efficacy in tumor regression studies in animal models, and can induce apoptosis as single agents in some tumor cell lines. However, while p53 is robustly activated in many cell lines treated with Nutlin-3a, a significant fraction of them do not undergo apoptosis.43 Similarly, ABT-737 is extremely potent in specific subgroups of tumors, yet is less effective as a single agent in others.24 Together, these observations indicate the existence of additional factors that govern apoptotic sensitivity to such agents. While some of these factors are beginning to be elucidated, such as Hdmx in the case of Nutlin-3a and Mcl-1 in the case of ABT-737, there are likely others to be discovered. The intersection of p53 signaling with intrinsic death pathways suggests that combined treatment with these agents may be an effective strategy. Indeed, this has been demonstrated recently in acute myeloid leukemia.44 Here, we generalize the efficacy of this combination to Burkitt lymphoma and to epithelial-derived solid tumors, which constitute the majority of human cancers. These studies reveal several novel findings. First, Hdmx antagonizes Nutlin-induced bax activation by binding to p53 and preventing p53-dependent transactivation. Second, the use of BH3 mimetics can abrogate this Hdmx-dependent suppression of apoptosis. Finally, a selective induction of apoptosis in cancer versus normal cells indicates this combination has therapeutic potential.
Nutlin-3a and MI-219 both bind and antagonize Hdm2, yet neither effectively antagonizes Hdmx in most cell types tested. An exception may occur when Nutlin-3a is administered in sites where local delivery and high drug concentrations can be achieved. For example, in one recent study, Nutlin3a was used to treat mouse retinoblastoma, where overexpressed Mdmx mediates p53 inhibition.31 Nonetheless, new agents that target Hdmx more effectively than Nutlin-3a or MI-219 should be powerful additions to the existing repertoire of targeted therapeutic agents. Hdmx-specific antagonists would also be valuable tools for future molecular mechanistic studies of p53 regulation.
Our data show that Hdmx must be competent for binding the p53 transactivation domain to suppress Nutlin-induced apoptosis, indicating that p53 is the key target for Hdmx’s anti-apoptotic effect. Additionally, Hdmx attenuated the induction of most pro-apoptotic genes after Nutlin-3a treatment. These data are consistent with a model in which Hdmx predominantly prevents cell death by reducing p53-dependent transactivation of pro-apoptotic genes. Hdmx could limit p53 transactivation by binding to it in the nucleus, or by sequestering it in the cytoplasm. We observe that in SJSA-X cells, Hdmx can translocate to the nucleus following Nutlin-3a treatment (Supplementary Figure 3), consistent with a nuclear function of Hdmx in p53 inhibition. However, other models suggest p53 has a cytoplasmic role in induction of apoptosis27, 28, and our data do not exclude the possibility that Hdmx blocks cytoplasmic p53 functions.
Downregulation of Mcl-1, cleavage of caspase-9 and activation of Bax following Nutlin-3a treatment implicates the intrinsic (mitochondrial) apoptotic pathway as an important mechanism by which Nutlin-3a induces p53-dependent cell death. Mcl-1 downregulation occurs primarily due to caspase activation, and therefore is a consequence, rather than a cause of Nutlin-induced cell death. However, Mcl-1 cleavage should amplify the apoptotic signal, as reported for other systems.45 The p53 gene targets required for Bax activation, caspase cleavage and apoptosis in response to Nutlin-3a remain to be determined. Screens of multiple tumor types will be required to elucidate the critical effectors and antagonists of Nutlin-induced apoptosis. This type of molecular profiling has already proven useful in predicting sensitivity to BH3 agonists including ABT-737.46 We propose an analogous classification system might be applied to the response to Nutlin-3a. For example, lack of the effector proteins Bax and Bak lead to a “Class B” apoptotic block in the terminology of Deng et al (46). The lack of apoptosis in BL40 and MCF7 in response to Nutlin-3a is not due to a Class B block, since both cell lines express Bax, and both undergo some apoptosis in response to ABT-737, which requires these effector proteins (Figure 5 and data not shown). Alternatively, upstream activators of Bak/Bax may be absent or inadequately induced by p53 following Nutlin-3a treatment of these cell lines (a “Class A” block). p53 target genes that fall into the category of upstream activators of Bax/Bak are Noxa and PUMA.18 Noxa levels were higher in SJSA than in SJSA-X cells after Nutlin-3a treatment, which correlates with their sensitivity to apoptosis. However, we did not find a strict correlation between Noxa and sensitivity to Nutlin-3a in other cell types (data not shown). By contrast, PUMA induction was sustained in the Nutlin-sensitive BL2 cells, but transient in Nutlin-resistant BL40 cells, and might therefore contribute to a Class A block in these Burkitt lymphoma cells. A third type of block (Class C) is defined as protection against death via expression of anti-apoptotic Bcl-2 family members. Although our survey of this family was not exhaustive, we can exclude Bcl-2, since it was barely detectable in Nutlin-resistant BL40 cells, and high in Nutlin-sensitive BL2 cells (Supplementary Figure 4). Given the complexity of factors that promote or antagonize apoptosis, analyses of individual BH3 proteins is not likely to reveal the factors preventing death induction by p53 agonists in a cell line or patient biopsy. Rather, global approaches to predict “death signatures” are needed to define the optimal steps in the apoptotic pathway that synergize with p53 activation.
Other mechanisms of resistance to Nutlin-3a that do not directly involve BH3 proteins have been proposed. For example, some cell lines that arrest in G1 due to p21 activation following Nutlin-3a treatment are resistant to apoptosis.44 However, we note that the majority of cell types treated with Nutlin-3a initially undergo a p53-dependent cell cycle arrest, but not all are protected against apoptosis.43 We suggest that the apoptotic signals induced by Nutlin-3a might be dominant over any cell cycle effects in some cell types. Nevertheless, in agreement with studies in other cell types44, the Nutlin-3a/ABT-737 combination does accelerate the death of SJSA cells. This indicates that p53-dependent apoptosis is enhanced once sufficient Bax is freed from anti-apoptotic Bcl-2 family members. The increased death could be explained in two ways. First, degradation of Mcl-1 triggered by Nutlin-mediated activation of p53 target genes may improve the efficacy of ABT-737, since the latter compound does not antagonize Mcl-1.25 Alternatively, the liberation of Bax from Bcl-2 and related anti-apoptotic proteins by ABT-737 may lower the threshold at which p53-induced pro-apoptotic proteins trigger apoptosis. We consider the second possibility more likely, since Mcl-1 levels do not decrease in SJSA-X cells, but these cells die following combined Nutlin and ABT-737 treatment. Additionally, increased apoptosis in MCF7 cells following combined Nutlin-3a/ABT-737 treatment is not accompanied by Mcl-1 downregulation.
Together these data indicate that Nutlin-induced apoptosis likely depends on the balance between pro- and anti-apoptotic proteins, which varies between tumor types and subgroups (this study, 42, 46). Since Hdmx knockdown can sensitize cells to Nutlin-3a15, targeting Hdmx directly may sensitize cells to activation of the intrinsic apoptotic pathway. In support of this, we observed a modest sensitization to ABT-737 following Hdmx knockdown in MCF7 cells (Supplementary Figure 5). The p53 target genes PUMA and Bax were not detectably increased after Hdmx knockdown, possibly explaining the modest apoptotic increase (Supplementary Figure 5, and data not shown). Together these data indicate that Hdmx knockdown alone does not alter the ratio of pro- to anti-apoptotic genes sufficiently to induce apoptosis. Rather, additional p53 activation (such as that provided by Nutlin-3a) is required to initiate cell death.
In vivo studies in mice show that Nutlin-3a as a single agent is well tolerated during a 20-day dosing schedule, with only moderate depletion of lymphocytes (32 and L. Vassilev, personal communication). Similarly, ABT-737 is tolerated in mice at doses that effectively reduce tumor burden, with some depletion of the platelet population.24, 40 Here, we show for the first time that combined treatment with Nutlin3a and ABT-737 can selectively induce apoptosis in tumor but not normal cells. While our in vitro culture conditions do not recapitulate the tissue architecture and cell-cell contacts in intact animals, they nevertheless encourage future studies examining this drug combination in vivo. Such studies should be designed to determine the minimal doses and drug exposure times that lead to preferential toxicity in tumor versus normal tissues. Importantly, despite its sub-optimal antagonism of Hdmx, Nutlin-3a in combination with ABT-737 is effective even in tumor cells with high Hdmx levels. This treatment may therefore find application in the growing list of tumors that overexpress Hdmx, including retinoblastoma 31, breast carcinoma 47 and leukemia. 48
Supplementary Material
Supplementary Figure 1. BL cells were treated with Nutlin-3a for 24h and subjected to western analysis for caspase-3 expression. MCF7 were included as a negative control.
Supplementary Figure 2. SJSA cells expressing empty retroviral vector (neo), or dominant negative p53 (DD) were treated with Nutlin-3a or ABT-737 alone or in combination at the indicated dose. Apoptosis was evaluated by AnnexinV staining (left) and brightfield imaging (right).
Supplementary Figure 3. Hdmx nuclear localization after treatment of SJSA-X with Nutlin-3a. Cells were treated with Nutlin-3a for the indicated times and processed for immunofluorescence using the polyclonal anti-Hdmx antibody pAb1857 at 1:5000 dilution.
Supplementary Figure 4. BL cells were treated with 5 uM Nutlin-3a for the indicated times prior to western analysis. NCS = radiomimetic drug neocarzinostatin
Supplementary Figure 5. MCF7 were transfected with 100pmol of control or Hdmx siRNA. 24h later, cells were either pretreated with zVAD or left untreated for 1h prior to the addition of ABT-737 for 48h. (A) Western analysis of the indicated proteins. (B) Graph represents the average of two experiments and demonstrates that Hdmx knockdown increases the frequency of Bax activation following treatment with ABT-737. (C) FACS profile of AnnexinV staining (x-axis) indicates that Hdmx knockdown sensitizes MCF7 to apoptosis induced by ABT-737. Figure in brackets indicates % apoptotic cells.
Acknowledgments
We thank all members of the Wahl lab for discussion and suggestions related to this manuscript. We thank Dajun Yang and Nathalie Bruey-Sedano for provision of MI-219, MI-426 and open discussion of results, and Stephen Fesik for ABT-737. We gratefully acknowledge the work of Pam Bock, Anne-Marie Mueller and the Salk OTM, Lori Gressick and Jane Hoff-Smith at Abbott for work regarding MTAs, and Lyubomir Vassilev at Roche for fruitful discussions and provision of Nutlin-3a.
MW is funded by a grant from NIH awarded to GMW (CA61449). JME is funded by grants from NIH (CA117907), March of Dimes (5-FY05-1217) and DOD-CDMRP (CM05054).
Abbreviations and acronyms
- WT
wild type
- zVAD
benzylocarbonyl-Val-Ala-Asp-fluromethyl-ketone
References
- 1.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 2.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindstrom MS, Klangby U, Wiman KG. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene. 2001;20:2171–7. doi: 10.1038/sj.onc.1204303. [DOI] [PubMed] [Google Scholar]
- 4.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 5.Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ, Jochemsen AG. Hdmx stabilizes Mdm2 and p53. J Biol Chem. 2000;275:28039–44. doi: 10.1074/jbc.M003496200. [DOI] [PubMed] [Google Scholar]
- 6.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–4. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- 7.Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008 doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- 8.Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 10.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 11.Dudkina AS, Lindsley CW. Small molecule protein-protein inhibitors for the p53-MDM2 interaction. Curr Top Med Chem. 2007;7:952–60. doi: 10.2174/156802607780906762. [DOI] [PubMed] [Google Scholar]
- 12.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–5. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 14.Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 2006;66:3169–76. doi: 10.1158/0008-5472.CAN-05-3832. [DOI] [PubMed] [Google Scholar]
- 15.Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. Hdmx modulates the outcome of p53 activation in human tumor cells. J Biol Chem. 2006;281:33036–44. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- 16.Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. Embo J. 2004;23:1547–56. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 18.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, Tabarias H, Degli-Esposti MA, Dewson G, Willis SN, Motoyama N, Huang DC, Nutt SL, Tarlinton DM, Strasser A. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol. 2007;8:856–63. doi: 10.1038/ni1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–71. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–38. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 24.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 25.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 27.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 28.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–12. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 29.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–5. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 30.Gomes NP, Bjerke G, Llorente B, Szostek SA, Emerson BM, Espinosa JM. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;20:601–12. doi: 10.1101/gad.1398206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 32.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 33.Raver-Shapira N, Oren M. Tiny actors, great roles: microRNAs in p53’s service. Cell Cycle. 2007;6:2656–61. doi: 10.4161/cc.6.21.4915. [DOI] [PubMed] [Google Scholar]
- 34.Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–9. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ, Lowe SW. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci U S A. 1997;94:2345–9. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–9. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 37.Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates subcellular location and cell death. Embo J. 1999;18:2330–41. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 40.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, Fesik SW, Shen Y. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 42.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 43.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5:2778–86. doi: 10.4161/cc.5.23.3520. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Guerrero AD, Huang L, Shabier Z, Pan M, Tan TH, Wang J. Caspase-9-induced Mitochondrial Disruption through Cleavage of Anti-apoptotic BCL-2 Family Members. J Biol Chem. 2007;282:33888–95. doi: 10.1074/jbc.M702969200. [DOI] [PubMed] [Google Scholar]
- 46.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, Marine JC. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–43. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han X, Garcia-Manero G, McDonnell TJ, Lozano G, Medeiros LJ, Xiao L, Rosner G, Nguyen M, Fernandez M, Valentin-Vega YA, Barboza J, Jones DM, Rassidakis GZ, Kantarjian HM, Bueso-Ramos CE. HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol. 2007;20:54–62. doi: 10.1038/modpathol.3800727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. BL cells were treated with Nutlin-3a for 24h and subjected to western analysis for caspase-3 expression. MCF7 were included as a negative control.
Supplementary Figure 2. SJSA cells expressing empty retroviral vector (neo), or dominant negative p53 (DD) were treated with Nutlin-3a or ABT-737 alone or in combination at the indicated dose. Apoptosis was evaluated by AnnexinV staining (left) and brightfield imaging (right).
Supplementary Figure 3. Hdmx nuclear localization after treatment of SJSA-X with Nutlin-3a. Cells were treated with Nutlin-3a for the indicated times and processed for immunofluorescence using the polyclonal anti-Hdmx antibody pAb1857 at 1:5000 dilution.
Supplementary Figure 4. BL cells were treated with 5 uM Nutlin-3a for the indicated times prior to western analysis. NCS = radiomimetic drug neocarzinostatin
Supplementary Figure 5. MCF7 were transfected with 100pmol of control or Hdmx siRNA. 24h later, cells were either pretreated with zVAD or left untreated for 1h prior to the addition of ABT-737 for 48h. (A) Western analysis of the indicated proteins. (B) Graph represents the average of two experiments and demonstrates that Hdmx knockdown increases the frequency of Bax activation following treatment with ABT-737. (C) FACS profile of AnnexinV staining (x-axis) indicates that Hdmx knockdown sensitizes MCF7 to apoptosis induced by ABT-737. Figure in brackets indicates % apoptotic cells.