

Summary
Miniproteins provide a bridge between proteins and small molecules. Here we adapt methods from combinatorial chemistry to optimize CD4M33, a synthetic miniprotein into which we had previously transplanted the HIV-1 gp120 binding surface of the CD4 receptor. Iterative deconvolution of generated libraries produced CD4M47, a derivative of CD4M33 that had been optimized at four positions. Surface-plasmon resonance demonstrated 4-to-6-fold improvement in CD4M47 affinity for gp120 to a level about 3-fold tighter than that of CD4 itself. Assessment of the neutralization properties of CD4M47 against a diverse range of isolates spanning from HIV-1 to SIVcpz showed that CD4M47 retained the extraordinary breadth of the parent CD4M33, but yielded only limited improvements in neutralization potencies. Crystal structures of CD4M47 and a phenylalanine variant ([Phe23]M47) were determined at 2.4 and 2.6 Å resolution, in ternary complexes with HIV-1 gp120 and the 17b antibody. Analysis of these structures revealed a correlation between mimetic affinity for gp120 and overall mimetic-gp120 interactive surface. A correlation was also observed between CD4- and mimetic-induced gp120-structural similarity and CD4- and mimetic-induced gp120 affinity for the CCR5 co-receptor. Despite mimetic substitutions, including a glycine to (D)-proline change, the gp120 conformation induced by CD4M47 was as close or closer to the conformation induced by CD4 as the one induced by the parent CD4M33. Our results demonstrate the ability of combinatorial chemistry to optimize a disulfide-containing miniprotein and of structural biology to decipher the resultant interplay between binding affinity, neutralization breadth, molecular mimicry, and induced affinity for CCR5.
Introduction
Macromolecular interfaces, such as those between virus and receptor or between transcription factor and promoter, are tantalizing candidates for therapeutic intervention, and the design of small molecule compounds that interfere with these interfaces has been a long sought, though mostly unfulfilled, goal 1-4. We and others have transplanted macromolecular binding surfaces into synthetic miniprotein scaffolds 5-10, where they might be optimized by the tools of modern organic chemistry. These efforts provide a means to address the gap between therapeutically attractive macromolecular targets and current inhibitor-development technology.
With HIV-1, viral entry is initiated by the binding of the gp120 envelope glycoprotein to the CD4 receptor on the host cell 11-13. This binding induces conformational changes in gp120 that lead to the formation of the site for a requisite co-receptor, either CCR5 or CXCR4 14-17. Crystallographic analysis shows that the interface between gp120 and CD4 is a little under 2000 Å2 shared about equally between both binding partners 18-20. The interface itself is non-ideal, with interfacial cavities and captured water molecules. On CD4, it centers about a β-hairpin, which comprises roughly half the CD4 interface with gp120. We used the similarity between this critical β-hairpin and a similarly exposed β-hairpin found in scorpion toxins, such as charybdotoxin and scyllatoxin, to transplant the gp120-binding surface of CD4 into a toxin scaffold 21,22. Iterative optimization of the transplanted β-hairpin resulted in a synthetic mimetic of only 27 residues. This miniprotein mimetic, named CD4M33, displayed nanomolar affinity to gp120 23. The crystal structure of CD4M33 in complex with core gp120 and Fab 17b revealed extensive mimicry, between the binding surfaces on CD4M33 and CD4 for gp120 as well as between the CD4M33- and CD4-induced conformations of gp120 24.
The synthetic nature of the CD4M33 enabled us to adapt chemical methods of combinatorial optimization to enhance its binding affinity to HIV-1 gp120. One of the most elegant and popular combinatorial strategies, the “split and mix” method 25, 26, uses solid phase-peptide synthesis chemistry to obtain a high statistical sampling of all possible combinations of building blocks. Four positions in the β-hairpin of CD4M33 were selected for and submitted to combinatorial optimization. All (L) amino acids were incorporated at two positions, and both (L) and (D) configurations were incorporated at the two others. The “combinatorially optimized” peptide, named CD4M47, was then analyzed biochemically, in terms of its binding parameters to gp120, virologically, in terms of neutralization potency and breadth, crystallographically, in a ternary complex with YU2 core gp120 and the antigen-binding fragment (Fab) of the 17b antibody, and mechanistically, in terms of its ability to unmask cryptic epitopes related to the binding of the CCR5 co-receptor.
Results
Combinatorial optimization of CD4M33
The β-hairpin of CD4M33 (residues 18-27) accounts for about 75% of its interface with HIV-1 gp120 (Fig. 1) 24. A “hotspot” of interaction involves the biphenylalanine at residue 23, which inserts into a gp120 pocket at the interface of several gp120 domains. The biphenyl moiety enhances CD4M33 affinity for gp120 about 10-fold over its phenyl derivative ([Phe23]M47) 24. To optimize CD4M33, we examined where variability might be best introduced. We avoided altering a number of residues: the six cysteines corresponding to three disulfides, which are vital to the correct miniprotein folding; and residues in the second β-strand of CD4M33, which hydrogen bond to β15-strand of gp120 in a manner similar to the C″-strand of CD4. Instead, we explored the first β-strand of CD4M33, which engages in peripheral interactions with gp120 (Fig. 1), by introducing L-amino acids into positions Lys18 and Ala20. We also explored the top of the β-hairpin, by introducing L- and D- amino acids into position Gly21 and Ser22 (positions i+1 and i+2 of the turn).
Figure 1.
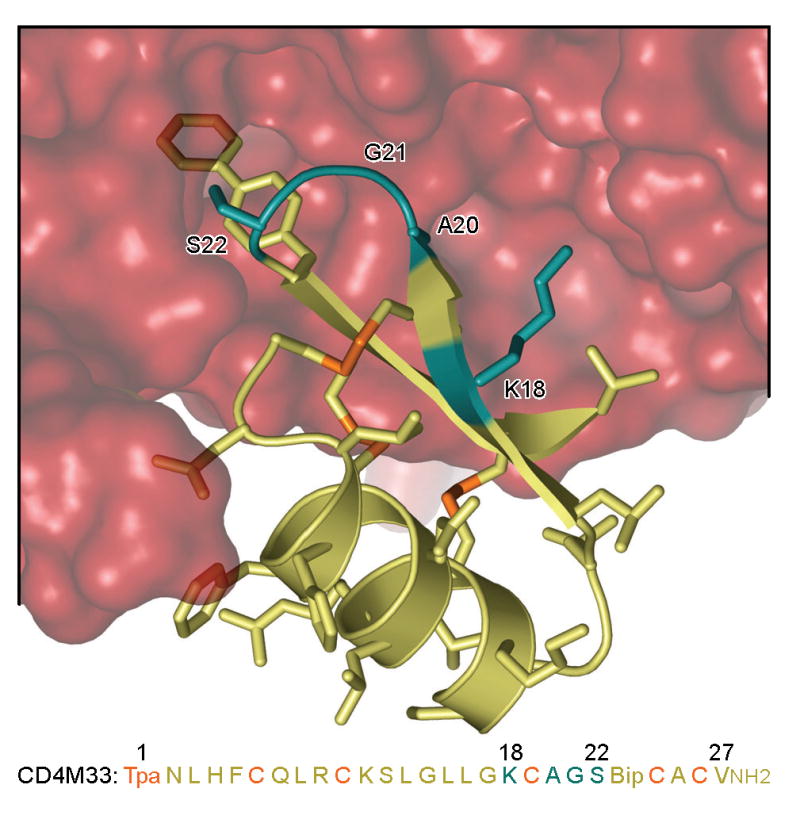
Structure and sequence of CD4M33 bound to gp120. Close-up of the synthetic miniprotein, CD4M33, binding to the HIV-1 gp120 envelope glycoprotein (from pdb accession code 1yyl) 24. Both structure and sequence of CD4M33 are colored yellow, except for residues which contribute to disulfide bonds (in orange) or which were chosen for combinatorial optimization (in cyan). The biphenyl at position 23 can be seen penetrating into the molecular surface of gp120, which is colored red. Sequence abbreviations: Tpa, thioproprionyl; Bip, biphenylalanine; NH2, C-terminal amidation. (Similar views of the CD4M47 and [Phe23]M47 complexes are shown in Fig. 5.)
To enhance solubility and synthesis yield, the biphenylalanine residue at position 23 was replaced by a phenylalanine in the template used for combinatorial synthesis. Cysteines (both L-and D-) were omitted as choices for the four optimized residues to reduce potential misfolding during library synthesis. Iterative deconvolution was used to screen the peptide libraries, so that synthesis was required of only four libraries of, respectively, 19, 19, 34 and 34 sublibraries. Together, the generated variation encompassed 417,316 different peptides.
A combinatorial deconvolution strategy was iteratively used to study the four screened positions at residues 18, 20, 21 and 22. The strategy involved a series of four libraries comprising mixtures in which the identity of an amino acid at a given position was known, and combinations of amino acids were incorporated at the other positions. The overall procedure is illustrated in Fig. 2.
Figure 2.

Iterative deconvolution and sublibrary synthesis used in combinatorial optimization of CD4M33.
The first peptide library involved 19 defined (L)-amino acids at position 18, with 19×34×34 variants at positions 20, 21 and 22. Reverse-phase HPLC showed a time shift in retention of the main synthetic peak suggesting proper oxidization of the disulfides. Analyses by MALDI-TOF confirmed the presence of a mass pattern at the desired size. CD spectra were indicative of globally correct folding similar to the CD spectrum of CD4M33 (supplementary Fig. S1).
Peptide concentrations were normalized by quantitative amino-acid composition, and each sublibrary of the first library was tested at three different concentrations (10-6, 10-7 and 10-8 M) for the inhibition of gp120 binding to CD4 by fluorescence anisotropy competition assay 27 (Fig. 3) and by ELISA (supplementary Fig. S2). In the initial optimization, inhibition at 10-8 M was low for both methods. On the other hand, both methods showed that with the sublibraries for position 18 (B1A to B1Y), Arg had the highest activity at 10-6 and 10-7 M, followed by Ala in fluorescence anisotropy and Lys and His in ELISA (Fig. 3 and supplementary Fig. S2), and position 18 was fixed as an arginine.
Figure 3.

Combinatorial optimization of CD4M33: screening of peptide libraries by means of a competitive fluorescence polarization assay. Inhibition of the binding of CD4M33-Fl (1 nM) to gp120HXBc2 (12.5 nM) by libraries B1, B2, B3 and B4 was measured by fluorescence anisotropy. The percentage of inhibition of CD4M33-Fl binding is displayed as a mean of duplicate measurements. Each sublibrary was tested at three concentrations: 10-6 (black), 10-7 (grey) and 10-8 M (stripes). An arrow indicates the selected residue at each round of optimization. (X represents an equimolar mixture of (L)-amino acids at position 20 and of (L/D)-amino acids at positions 21 and 22).
The remaining positions were sequentially identified through an iterative procedure of (re)synthesis and screening (Fig. 2). Thus, a second peptide library was generated that comprised 19 sublibraries of 1156 compounds each (B2A to B2Y), with arginine fixed at position 18 and a defined L-amino acid at position 20. After validation of conformity and assessment of gp120 binding by fluorescence anisotropy (Fig. 3) and ELISA (supplementary Fig. S2), position 20 showed a small (10%) preference for Ala (over Arg and Ser), and position 20 was fixed as an alanine.
A third combinatorial library was synthesized and tested (Fig. 3 and supplementary Figs. S1 and S2). This library included 34 sublibraries of 34 different miniproteins each (B3A to B3dY) with a fixed amino acid of either L- or D-configuration at position 21. A pronounced difference was observed between sublibraries containing L-amino acids and those containing D-amino acids. A clear preference was observed for residues of D-configuration and to a lesser extent for glycine (which is achiral). The selection at this position for a whole family of amino acids suggested a structural effect of the mutation, which was confirmed by the small difference in activity between the different sublibraries with a D-amino acid. The type II′ β-turn seemed to be the most favourable turn for the activity of our miniproteins. Based on these considerations, position 21 was fixed as a (D)-proline.
The fourth and last library amounted to a parallel synthesis of 34 different peptides with a defined (L) or (D)-amino acid at position 22 (B4A to B4dY). The preferred amino acid here was not as clear as with previous positions. Several amino acids showed roughly equivalent inhibition at the 10-7 M. At lower concentrations, however, a preference was observed for threonine, which was chosen for position 22. The final selected sequence, shown in Fig. 3, contained Arg, Ala, (D)-Pro and Thr at positions 18, 20, 21 and 22, respectively.
Surface-plasmon resonance characterization of CD4-mimetic binding to HIV-1 gp120
The optimized compound and its homologue with a biphenyl instead of the phenyl moiety at position 23 were individually synthesized and named [Phe23]M47 and CD4M47, respectively. Surface-plasmon resonance was utilized to determine the binding parameters between HIV-1 gp120 and the various CD4-mimetic miniproteins as well as 2-domain CD4 (sCD4) (Table 1).
Table 1.
Binding parameters of sCD4 and CD4-mimetic miniproteins to gp120
| Ligand | gp120 | On-rate (M--1s-1) | Off-rate (s-1) | Kd (nM) |
|---|---|---|---|---|
| sCD4 | HXBc2 | 6.07×104 ± 4.1×102 | 3.42×10-4 ± 4.7×10-5 | 5.64 ± 0.77 |
| CD4M33 | HXBc2 | 3.41×105 ± 3.2×103 | 3.66×10-3 ± 3.4×10-5 | 10.8 ± 0.14 |
| [Phe23]M33 | HXBc2 | 1.73×105 ± 9.6×102 | 3.27×10-3 ± 1.6×10-5 | 18.9 ± 0.14 |
| CD4M47 | HXBc2 | 3.33×105 ± 1.3×103 | 9.01×10-4 ± 2.1×10-5 | 2.71 ± 0.65 |
| [Phe23]M47 | HXBc2 | 1.52×105 ± 5.7×102 | 9.47×10-4 ± 1.6×10-5 | 6.24 ± 0.10 |
| sCD4 | YU2 | 5.13×104 ± 1.3×102 | 3.94×10-4 ± 1.7×10-5 | 7.68 ± 0.33 |
| CD4M33 | YU2 | 2.18×105 ± 7.5×102 | 2.72×10-3 ± 1.3×10-5 | 12.5 ± 0.075 |
| [Phe23]M33 | YU2 | 1.35×105 ± 5.6×102 | 4.75×10-3 ± 1.3×10-5 | 35.1 ± 0.18 |
| CD4M47 | YU2 | 3.49×105 ± 1.1×103 | 7.41×10-4 ± 1.6×10-5 | 2.12 ± 0.047 |
| [Phe23]M47 | YU2 | 1.10×105 ± 1.9×102 | 4.27×10-4 ± 1.1×10-5 | 3.87 ± 0.10 |
The mimetics or sCD4 were immobilized onto CM5 sensor chips using standard amine coupling, and the kinetic parameters to both HXBc2 and YU2 gp120 were measured with a Biacore 3000. Parameters for HXBc2 and YU2 were similar, though not exactly identical; changes in rates are discussed in the context of HXBc2, with those for YU2 shown in parenthesis when different. The on-rate of CD4M47 was mostly unchanged from the parent CD4M33, about 6-fold (7-fold) faster than the on-rate of sCD4. Optimization appeared to have its greatest effect on the CD4M47 off-rate, which was reduced almost 4-fold from that of CD4M33, to within 3-fold (2-fold) the rate of sCD4. Taken together, these changes in on- and off-rates resulted in an overall Kd for CD4M47 of 2.7 nM (2.1 nM), about 2-fold (4-fold) tighter than that of sCD4.
Kinetic parameters were also measured for the Phe23 variants of the CD4 mimetics. Both [Phe23]M33 and [Phe23]M47 had on-rates about half as fast as their biphenyl counterparts. Off-rates between phenyl- and biphenyl- containing ligands, meanwhile, were similar to within 1.1-fold (1.7-fold). The results suggest that the biphenyl moiety enhances the speed of complex formation. The biphenyl effect on off-rate, meanwhile, is variable, and dependent on strain-specific features.
CD4-mimetic neutralization of virus
To explore how enhancement in affinity translated into virus neutralization, we analyzed the ability of the CD4-mimetic miniproteins to neutralize a diverse panel of envelope clones. We used two assays, a broadly sensitive HIV-entry assay based on JC53BL-13 indicator cells, which is able to give quantified infection within a single cycle of replication 28 and an HIV-1 envelope-mediated fusion assay employing as effectors persistently infected PM1 cells that present the native envelope on their surface 23.
CD4 and miniprotein neutralization potencies were evaluated with a diverse range of viral envelopes, ranging from HIV-1 IIIB (which is extremely similar to the HXBc2 envelope used in the optimization procedure) to SIVcpz (which showed a genetic distance of almost 0.5 changes per residue from HXBc2). The neutralization results, expressed as IC50s, are summarized in Table 2.
Table 2.
Comparison of antiviral activities between sCD4, CD4M33, [Phe23]M33, CD4M47 and [Phe23]M47
| Virus | Species | Genetic Distance to HXBc2 | sCD4 (IC50, nM) | CD4M33 (IC50, nM) | [Phe23]M33 (IC50, nM) | CD4M47 (IC50, nM) | [Phe23]M47 (IC50, nM) |
|---|---|---|---|---|---|---|---|
| IIIB* | HIV-1 group M | 0.03060 | 6 | 12 | 260 | 8 | 7 |
| NL4.3 env | HIV-1 group M | 0.05769 | 2 | 5 | 13 | 3 | 4 |
| BaL* | HIV-1 group M | 0.13532 | 41 | 120 | 200 | 27 | 33 |
| YU-2 env | HIV-1 group M | 0.16766 | 8 | 210 | 1000 | 64 | 150 |
| MN-25 | HIV-1 group M | 0.17951 | 36 | 350 | 3600 | 150 | 320 |
| SUMA d736-73 | HIV-1 group M | 0.18120 | 56 | 480 | 8500 | 370 | 950 |
| MN-8 | HIV-1 group M | 0.18147 | 0.5 | 34 | 85 | 11 | 15 |
| SUMA d736-68 | HIV-1 group M | 0.18273 | 2 | 4 | 28 | 3 | 7 |
| WEAU 16-8M | HIV-1 group M | 0.20241 | 1 | 85 | 130 | 64 | 47 |
| GAB-1 fl | SIVcpz (P.t.t) | 0.43711 | 4 | 340 | 350 | 150 | 69 |
| GAB-2 62 fl | SIVcpz (P.t.t) | 0.47819 | 16 | 400 | 1000 | 230 | 290 |
Assays were performed with PM1 cells; all other assays performed with JC53BL-13 cells (see text).
CD4M47 was active over the entire range of diverse isolates, though its potency (IC50 of neutralization) varied substantially from 3 to 370 nM. Only minor enhancements in neutralization potency were observed between CD4M33 and CD4M47 (1.3-4.3-fold over the entire range), less than the observed increase in relative affinity.
Overall, results from neutralization potency agreed with the rank order of measured affinities (Kd), with one notable exception, that of sCD4. Thus, the mimetics showed a rank order of gp120 affinity ([Phe23]M33 > CD4M33 > [Phe23]M47 > CD4M47) that matched the rank order of mimetic neutralization potency. sCD4, however, had an intermediate affinity, but the most potent neutralization. Two possibilities might account for this. First, the larger size of sCD4 (7-times the mass of the miniproteins) might boost its neutralization potency, and second, the contribution of on- and off-rate to neutralization potency is unclear, and the low off-rate of sCD4, slower than any of the mimetics, may enhance its neutralization potency relative to its Kd.
Although CD4M47 displayed only minor improvements in neutralization breadth versus CD4M33, this improvement was seen over the entire range of isolates. Thus, despite increased divergence from the CD4 sequence (Gly to (D)-Pro and Ser to Thr), CD4M47 retained the remarkable breadth of the parent CD4M33 (Fig. 4).
Figure 4.

Free energy differences of CD4 and CD4-mimetics as a function of gp120 genetic distance. We previously showed that the difference in free energy (ΔΔG) for two ligands, n=1 and n=2 with 50% inhibitory concentrations of IC50(1) and IC50(2) can be approximated as ΔΔG ≈ −RT ln [IC50(1)/IC50(2)], which we define as ΔΔGN 24. Here we graph ΔΔGN (vertical axis) as a function of genetic distance (horizontal axis). The slope relates the change in relative potency as a function of evolutionary distance, and the y intercept gives a normalized relative potency against isolates like HXBc2. CD4M47 has an intercept potency similar to that of CD4, whereas [Phe23]M47 is reduced by about 0.5 kcal/mol (left and middle graph). The breadth of CD4M47, however, is not as good as that of [Phe23]M47. When compared to CD4M33, however, CD4M47 is about 0.5 kcal/mol more potent, with virtually indistinguishable breadth (right graph).
Structures of mimetic miniproteins in complex with HIV-1 gp120 core
Both CD4M47 and [Phe23]M47 were crystallized in ternary complexes with core gp120 from the primary HIV-1 isolate YU2 and the antigen binding fragment (Fab) of the 17b antibody. The structures were isomorphous with the previously determined CD4M33 crystal structure 24 and were refined to nominal resolutions of 2.4 and 2.6 Å for the CD4M47 and the [Phe23]M47-containing complexes, respectively (Table 3). Crystal grew as thin plates, and while lower resolution shells were complete, higher resolution shells were less so. Nonetheless, collected data were sufficient to obtain a detailed atomic-level picture of the mimetic-gp120 interactions. Moreover, the crystal lattice contained two independent ternary complexes in the crystal asymmetric unit, allowing the effects of lattice packing and refinement uncertainty to be gauged.
Table 3.
X-ray crystallographic data and refinement statistics
| Data collection | ||
|---|---|---|
| Complex | CD4M47 with core YU2 gp120 and Fab 17b | [Phe23]M47 with core YU2 gp120 and Fab 17b |
| Space group | P21 | P21 |
| Molecules per ASU | 2 | 2 |
| Wavelength, Å | 1.00 | 1.00 |
| Unit cell dimensions | a=51.4 Å | a=51.4 Å |
| b=158.2 Å | b=157.8 Å | |
| c=109.9 Å | c=109.8 Å | |
| β=93.7° | β=93.7° | |
| Resolution, Å | 50-2.20 | 50-2.30 |
| Completeness, %* | 83.1 (67.3, 47.8, 32.6) | 71.8 (40.1, 30.7, 19.9) |
| No. of reflections † | 247392 (74201) | 216213 (67879) |
| Redundancy* | 3.3 (2.5, 2.1, 1.6) | 3.2 (1.9, 1.6, 1.3) |
| I/σ* | 15.8 (2.2, 2.1, 1.9) | 15.3 (3.2, 2.6, 1.9) |
| Rsym*,‡ | 8.7 (34.6, 34.2, 33.0) | 9.6 (25.7, 25.3, 28.2) |
| Refinement statistics (|F|>0 σ) | ||
|
| ||
| Resolution, ŧ | 20.00-2.20 | 20.00-2.40 |
| No. of reflections | 69673 | 56951 |
| Rcrys, %‖ | 21.3 | 21.5 |
| Rfree, %‖,¶ | 27.2 | 28.1 |
| No. of protein atoms | 11758 | 11715 |
| No. of water atoms | 549 | 485 |
| No. of other atoms | 224 | 232 |
| Rmsd bond length, Å | 0.0071 | 0.0065 |
| Rmsd bond angles, ° | 1.14 | 1.31 |
| Luzzatti error, Å | 0.31 | 0.33 |
| Average B-factor, Å2 | ||
| Protein overall | 39.7 | 36.3 |
| Water | 35.9 | 32.4 |
| Others | 55.0 | 57.9 |
| Ramachandran plot | ||
| Most favored, % | 87.1 | 87.3 |
| Additionally allowed, % | 11.9 | 11.6 |
| Generously allowed, % | 0.6 | 0.5 |
| Disallowed, %# | 0.4 | 0.5 |
Values in parentheses are for the outermost three resolution shell: 2.48-2.37, 2.37-2.28, 2.28-2.20 Å for the CD4M47 complex and 2.59-2.48, 2.48-2.38, 2.38-2.30 Å for the [Phe23]M47 complex.
Numbers in parentheses are for the unique reflections.
Rsym=Σ|I-<I>|/Σ<I>, where I is the observed intensity, and <I> is the average intensity of multiple observations of symmetry related reflections.
The resolution at which the number of measured reflections would represent 100% complete data is 2.4 Å and 2.6 Å for the CD4M47 and [Phe23]M47 complexes, respectively.
R=Σhkl‖Fobs|-|Fcalc‖/Σhkl|Fobs|
Rfree is calculated from 10% of the reflections excluded from refinement.
Disalllowed residues included Gln 258 of gp120, Ser 30 and Ala 50 of the 17b light chain and Gln 65 of the 17b heavy chain. Real-space correlations of these residues ranged from 0.899 to 0.958.
The structure of the newly determined CD4M47 complex is shown in Fig. 5 (along with the structure of the [Phe23]M47 variant). Superpositions with CD4M33 are shown in Figs. 5D and 5E and with CD4 in Figs 5F and 5G. The mimetic structures, before and after combinatorial optimization, were extremely similar, with root-mean-square (rms) deviations for the entire backbone of 0.27-0.67 Å, and of 0.19-0.26 Å for residues 18-22. Residue 18 was not closely constrained by gp120, with considerable distance between this portion of the mimetic and gp120. The added bulk of Arg 18 in CD4M47 versus the parent CD4M33 Lys allowed for additional contact surface area with gp120, although these contacts were somewhat tenuous. Residues 20 and 21 were more constrained by gp120; optimization added a total of four carbon atoms to these two residues, increasing the fit between mimetic and gp120 (Fig. 5E).
Figure 5.

Crystal structures of CD4M47 and [Phe23]M47 in complex with YU2 core gp120 and Fab 17b. (A) Overall view of gp120 (red), CD4M47 (teal), and Fab 17b (gray). The orientation shown is 90° about a vertical axis from the orientations in C-H, and the viral membrane would be positioned towards the top of the page. Molecules are displayed as Cα cartoons with the biphenyl moiety at position 23 of CD4M47 highlighted all atoms stick representation. (B) Sequences of CD4, CD4M33, CD4M47, [Phe47]M47. Sequence abbreviations: Tpa, thioproprionyl; Bip, biphenylalanine; NH2, C-terminal amidation. (C) Interaction hot-spot between CD4M47 (teal) and gp120 (red). The three residues at positions 18, 21, and 22 that have been altered by combinatorial optimization are labeled. A simulated “omit” map was calculated, removing the entire mimetic, and the Fo-Fc electron density from this map is shown, contoured at 3σ. (D) Same as (C) but for [Phe23]M47. (E) Close-up of the critical mimetic β-turn, at residues 20-23, showing CD4M47 (teal) and CD4M33 (brown). (F) Close-up of the critical mimetic β-turn, at residues 20-23, showing [Phe23]M47 (teal) and the C′C″ loop of CD4 (yellow). (G) Molecular surface of gp120 shown in reference to the molecular surfaces of CD4M47 (teal) and CD4M33 (yellow). Stick representations (Cα and all atoms for residues 18, 21 and 22) are shown. (H) Molecular surface of gp120 shown in reference to [Phe23]M47 (teal) and CD4 (yellow). Stick representations of the mimetic and the C′C″ loop of CD4 are shown as in F (only the surface is shown for most of CD4 for clarity). As can be seen, the optimization increases the bulk of CD4M47 and its fit to gp120 relative to CD4M33 and CD4. (Coordinates for CD4M33, [Phe23]M33 and CD4 containing complexes with YU2 gp120 can be found at pdb accession codes 1YYL, 1YYM and 1RZK 19, 24).
One measure of the degree of fit between two molecules is their surface complementarity 29. The surface complementarity between CD4 and core gp120 ranged from 0.674 for HXBc2 core gp120 and 0.716 for the YU2 core (with the value for an optimal fit being 1.000). With the CD4-mimetic miniproteins, surface complementarities ranged from 0.769 for both lattice molecules of CD4M33 to 0.801 for molecule 1 of [Phe23]M33 (supplementary Table S1). Within this range, differences from lattice variation had a considerable effect, with the two molecules of CD4M47 showing surface complementarities of 0.769 and 0.793. Perhaps because of the highly restricted range, no correlation was observed between surface complementary and overall affinity.
A second way to analyze structural interfaces is to quantify the molecular contact surface. For mimetic residues 18, 20 and 21, in each case, optimization resulted in an increase in the contact surface. For residue 18, the increase was roughly 20Å2, and for residues 21 and 22, the increase was roughly 10Å2 (Table 4). Thus, the total increase in contact surface from the optimized residues was ∼40Å2, or about 10% in the total mimetic contact surface.
Table 4.
Contact surface area of gp120 by CD4 and or CD4-mimetics*
| CD4 or CD4-mimetic (asymmetric unit) Å2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD4:HXB2 | CD4:YU2 | CD4M33(1) | CD4M33(2) | [Phe23]M33(1) | [Phe23]M33(2) | CD4M47(1) | CD4M47(2) | [Phe23]M47(1) | [Phe23]M47(2) | |
| YU2 residue | ||||||||||
|
|
||||||||||
| 105 H | 1.2 | |||||||||
| 123 T | 4.1 | |||||||||
| 124 P | 32.3 | 47.2 | 9.7 | 8.8 | 5.7 | 8.6 | 10.3 | 8.2 | 2.4 | 8.8 |
| 125 L | 3.3 | 1.7 | ||||||||
| 126 C | 45.9 | 45.5 | ||||||||
| 127 V | 9.5 | 2.8 | ||||||||
| 128 G | 2.8 | 15.0 | ||||||||
| 194 G | 12.6 | |||||||||
| 196 C | 6.9 | 20.1 | ||||||||
| 255 V | 18.1 | 16.5 | 16.3 | 17.9 | ||||||
| 256 S | 8.2 | 7.4 | 5.8 | 7.5 | ||||||
| 257 T | 5.7 | 4.1 | 15.1 | 10.8 | 6.5 | 6.5 | 12.4 | 11.2 | 6.9 | 6.4 |
| 278 T | 0.8 | |||||||||
| 279 N | 18.3 | 15.6 | ||||||||
| 280 N | 42.3 | 42.6 | 2.5 | 1.9 | 1.1 | 0.9 | 12.6 | 10.5 | 4.7 | 13.8 |
| 281 A | 39.4 | 39.6 | 12.6 | 12.3 | 14.7 | 2.2 | 21.1 | 23.7 | 13.8 | 18.8 |
| 282 K | 9.2 | 14.4 | ||||||||
| 283 T | 16.1 | 16.4 | 0.2 | 2.1 | 1.9 | |||||
| 364 S | 1.4 | 2.8 | 1.1 | 1.5 | 1.8 | 1.7 | ||||
| 365 S | 47.1 | 37.1 | 32.3 | 32.9 | 32.9 | 35.0 | 34.5 | 33.3 | 33.3 | 29.5 |
| 366 G | 20.1 | 20.8 | 18.3 | 23.5 | 16.7 | 23.8 | 19.0 | 24.0 | 18.1 | 23.7 |
| 367 G | 21.8 | 21.9 | 17.0 | 26.6 | 17.9 | 25.4 | 19.4 | 24.7 | 17.9 | 27.3 |
| 368 D | 52.3 | 50.4 | 36.8 | 49.6 | 37.8 | 52.8 | 36.8 | 52.0 | 37.0 | 45.4 |
| 369 P | 0.7 | 1.0 | 1.7 | |||||||
| 370 E | 19.1 | 17.6 | 29.2 | 30.4 | 21.2 | 18.7 | 26.9 | 30.8 | 21.7 | 18.9 |
| 371 I | 39.3 | 38.1 | 44.8 | 40.2 | 40.3 | 41.1 | 39.3 | 37.7 | 42.6 | 38.3 |
| 375 S | 15.7 | 14.1 | 13.6 | 13.7 | ||||||
| 376 F | 10.7 | 13.0 | 8.7 | 8.3 | ||||||
| 377 N | 7.4 | 8.8 | 5.5 | 7.3 | ||||||
| 382 F | 19.4 | 23.1 | 16.8 | 17.6 | ||||||
| 384 Y | 9.0 | 7.4 | 5.8 | 3.5 | ||||||
| 424 I | 9.8 | 7.9 | 6.2 | 9.0 | ||||||
| 425 N | 26.8 | 31.6 | 21.1 | 22.4 | 15.5 | 15.4 | 21.3 | 22.0 | 13.4 | 12.7 |
| 426 M | 13.8 | 12.7 | 15.3 | 15.4 | 14.8 | 13.9 | 15.4 | 17.2 | 13.6 | 13.7 |
| 427 W | 30.5 | 25.7 | 42.2 | 39.3 | 25.6 | 27.5 | 40.5 | 39.8 | 25.2 | 28.4 |
| 428 Q | 2.4 | 2.6 | 2.0 | 3.2 | 1.3 | 2.8 | 3.0 | 2.6 | 4.5 | 5.1 |
| 429 E | 13.7 | 12.1 | 11.3 | 11.2 | 9.7 | 11.5 | 11.6 | 12.3 | 11.0 | 10.1 |
| 430 V | 69.0 | 73.6 | 42.2 | 41.0 | 39.8 | 44.8 | 42.1 | 38.5 | 36.7 | 47.5 |
| 431 G | 1.4 | 2.3 | 1.5 | 2.1 | 1.5 | 1.1 | 1.5 | 1.4 | 0.6 | 0.2 |
| 432 K | 1.7 | 2.8 | 1.3 | |||||||
| 455 T | 12.8 | 10.0 | 3.1 | 2.8 | 0.2 | 0.9 | 8.9 | 9.2 | 12.5 | 13.8 |
| 456 R | 7.0 | 3.8 | ||||||||
| 457 D | 26.9 | 27.0 | 4.9 | 2.6 | 2.2 | 6.2 | 4.8 | |||
| 458 G | 5.2 | 9.8 | 2.6 | 1.5 | 1.5 | |||||
| 459 G | 29.2 | 11.8 | ||||||||
| 460 K | 33.5 | 61.4 | ||||||||
| 461 D | 1.7 | 9.6 | ||||||||
| 469 R | 6.6 | 7.0 | 1.8 | |||||||
| 471 G | 0.7 | 0.8 | ||||||||
| 472 G | 22.4 | 17.6 | 5.1 | 6.5 | 6.7 | 6.2 | 6.2 | 6.2 | 5.4 | 7.3 |
| 473 G | 23.5 | 23.2 | 25.5 | 26.1 | 21.4 | 21.1 | 24.9 | 22.3 | 25.6 | 25.9 |
| 474 D | 38.9 | 32.7 | 21.1 | 23.1 | 22.2 | 21.6 | 25.3 | 22.4 | 28.6 | 26.7 |
|
| ||||||||||
| Total | 821.7 | 874.1 | 527.6 | 543.1 | 363.8 | 398.0 | 537.0 | 553.5 | 397.2 | 439.2 |
Mimetic surface areas are show in red, when CD4 does not have any contact surface, and in blue, when the values for CD4M47 and [Phe23]M47 are larger than their CD4M33 or [Phe23]M33 counterparts.
While these results suggest a correlation between affinity for gp120 and contact surface area between mimetic and gp120, they must be tempered by the caveat that other amino acids that might increase surface area were not observed to enhance affinity in the optimization. Thus, while interactive surface area was observed to correlate with enhanced affinity, other factors such as surface complementarity and the specific chemistry of the interactions need to be taken into account.
Structural mimicry induced in gp120 by miniproteins and correlation with induced affinity for CCR5 coreceptor
Another structural measure of the interaction between two molecules is the similarity in their induced conformational change. In the case of the gp120-CD4 interaction (and also the gp120-CD4-mimetic interaction 24), gp120 undergoes substantial structural rearrangement, with much of the secondary structure of the gp120-inner domain rearranging 30-32.
We used distance-sorted distance matrices 24, 33 to quantify conformational comparisons of gp120 around the interaction hotspot (Fig. 6). In the 10Å shell extending from the Cα of residue 23 on the mimetic, the structures of the gp120 molecules complexed to CD4M47 showed normalized linear difference distances of 0.22-0.23Å, similar to that seen with CD4M33 (0.22-0.24Å). The linear difference distances decreased with [Phe23]M47, with normalized linear difference distances of 0.16-0.20Å, a bit larger than with the [Phe23]M33 analogue (0.13Å). Thus, in terms of induced gp120 structure, [Phe23]M33 remained the closest mimic to CD4, with [Phe23]M47 intermediate to CD4M47 and CD4M33.
Figure 6.
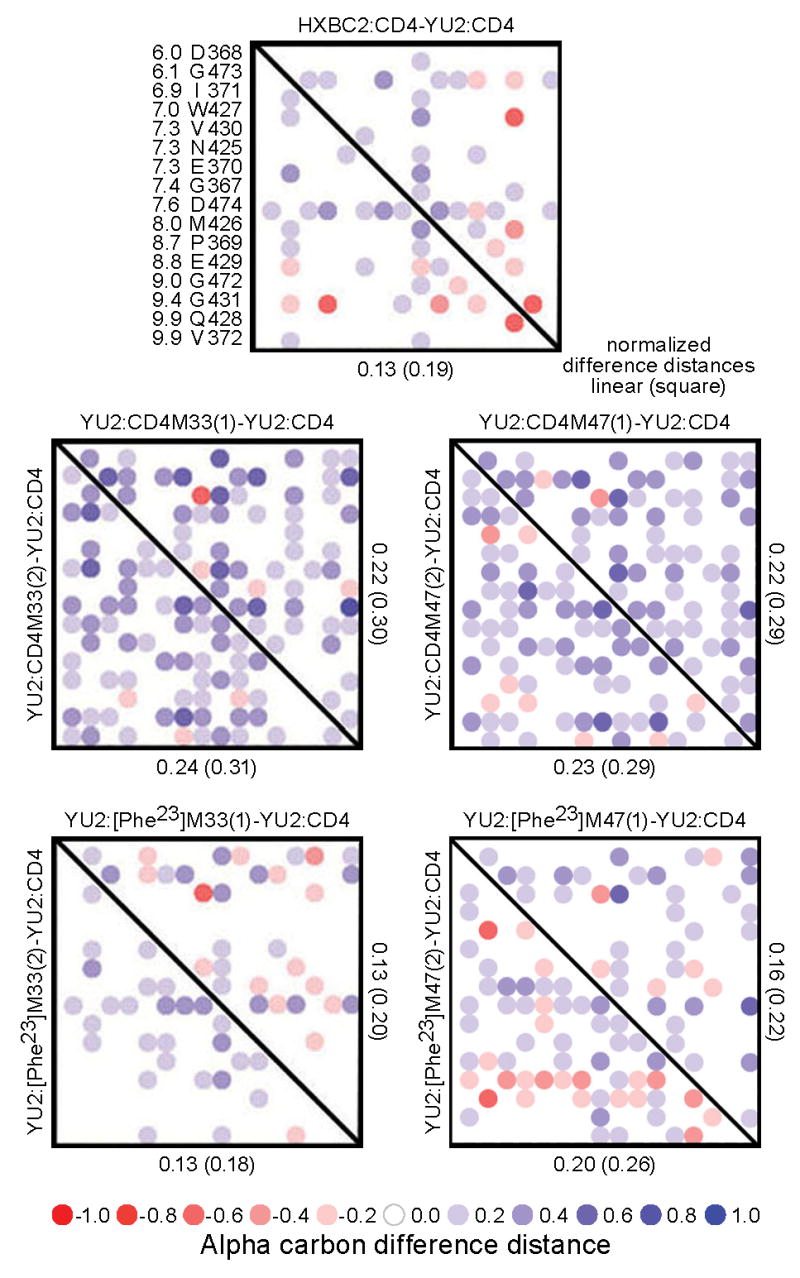
Distance-sorted difference distance matrix analysis of induced gp120 conformation. Difference distance matrices provide an alignment-independent means to assess structural similarity 33, 62, and we previously showed that this type of analysis could be focused to an interaction hotspot though distance sorting and quantified by normalization of the difference distances 24. The gp120-CD4 interaction hotspot is focused on residue 43 of CD4 (the equivalent of residue 23 in the mimetic miniproteins). The YU2 gp120 Cα-distances from residue 43 of CD4 were sorted and are displayed to the left of the top panel along with the corresponding gp120 residue. Difference distance matrices were calculated with these residues for gp120-17b ternary complexes containing 2-domain CD4, CD4M33, [Phe23]M33, CD4M47, and [Phe23]M47. Each i,j matrix element shows the difference between the atom i and atom j distance in the first specified structure and the atom i and atom j distance in the second specified structure. For non-diagonal elements, the linear summation of the absolute value of the difference distances normalized by the number of matrix elements is shown as well as the rms of the difference distances (“square”). The matrices provide both visual and quantitative assessment of the similarity in gp120 conformations in the defined interaction hotspot. As can be seen, both [Phe23]M47 and [Phe23]M33 induce confirmations more closely related to CD4 than the biphenyl containing mimetics, CD4M47 and CD4M33.
Here the two independent copies of the mimetic in the P21 lattice provide a means to verify the relevance of the distance-sorted difference distance metrics. However, the overall linear and square difference distances were small, only a bit larger than coordinate error. Moreover, the structural measurements were actually made in the ternary complex of mimetic, gp120 and 17b, and thus the gp120 conformation was influenced by 17b. We therefore sought to determine whether these small matrix differences were biologically relevant.
Unliganded gp120 binds poorly to the CCR5 coreceptor, with high affinity CCR5-gp120 interaction requiring a CD4-induced conformational change 34. To define the biological effects of the mimetic-induced changes in gp120, we compared the amount of gp120 bound to CCR5+ cell lines in the absence or in the presence of CD4 or of one of the CD4-mimetic miniproteins. The amount of gp120 bound to CCR5+cell lines was evaluated with two CCR5-tropic gp120 (SF162 and YU2, the same as used for co-crystals). Sequential incubations were carried out with the gp120-reactive antibody, D7324, and with a secondary PE-conjugated antibody. As shown in Fig. 7, [Phe23]M33 and [Phe23]M47 were more capable than the biphenylalanine derivatives in inducing a CCR5-binding conformation. Moreover, the induced affinity of gp120 to CCR5 correlated well with induced structural mimicry, as quantified by distance-sorted distance matrices.
Figure 7.

gp120-induced affinity for CCR5 and correlation with structural mimicry. The binding of recombinant gp120 envelope glycoprotein to CCR5+-CHO cells in absence or in presence of saturating concentrations of sCD4 or CD4-mimetic miniprotein was analyzed by FACS. (A) Induced binding of YU2 gp120. (B) Induced binding of SF162 gp120. The horizontal axes display fluorescence and the vertical axes the number of sorted events for representative experiments. (C) Correlation of induced gp120 affinity and difference-sorted difference distance matrix analysis of induced gp120 conformational similarity for YU2 (left panel) and SF162 (right panel). The geometric mean value of the amount of bound gp120 was determined in triplicate and is shown on the vertical axis. The normalized rms of the non-diagonal distance-sorted difference distance matrices elements for gp120 within a 10 Å radius of the CD4-gp120 interactive hotspot (Fig. 6) is plotted on the horizontal axis.
Discussion
Previous applications of combinatorial optimization have generally been with small molecules, with attempts of up to 2000 Da limited to linear peptides, for example, with antigens and peptide ligands of MHC class molecules 35, 36. As none of these prior efforts required disulfide formation for folding, methodological optimizations were required.
The first synthesized sublibrary did not pass quality controls of integrity: HPLC profiles showed large quantities of truncated peptide, a result confirmed by CD spectroscopy. Synthesis with monitored deprotection, moreover, showed that double coupling was required starting from residue 10. The presence of the sequence –GLLG- may have also complicated miniprotein synthesis, as glycine-containing sequences have been reported to favour aggregation 37, 38, thereby hindering the N-terminal accessibility required for chain elongation. To resolve these difficulties, the homogeneity of each sublibrary was monitored by HPLC spectra before and after disulfide bond formation. These spectra served to validate both miniprotein folding and synthesis completion. Results were confirmed by CD- and mass-spectroscopic analysis of each sublibrary.
Incomplete peptides were assumed to have neither inhibitory nor interference properties in terms of the activity of the complete peptide. As the last coupled residue of the mimetic was a thiopropionyl, which was required to form the third disulfide bond, we felt presence of this residue would likely be required for proper folding. However, the first pool of miniproteins comprised more than 20,000 different molecules – and potentially many more incomplete ones – and we could not be certain that all of the incomplete ones were inactive. To compensate for this uncertainty, we used two different biochemical tests to evaluate the biological activity of each sublibrary. The tests were based on different physical properties: one relying on molecular adsorption at a liquid-solid interface (ELISA); and the other on light depolarization and solution equilibrium (fluorescence anisotropy).
For the first peptide library, an arginine was selected at position 18, whereas the parent CD4M33 residue was a lysine. The increased bulk of the arginine appeared to increase contact surface with gp120. For the second peptide library, the residue at position 20 retained its original amino acid, an alanine. However, sublibrary B2A was only slightly more active than B2R. This position at the beginning of the β-hairpin plays a critical structural role: its main-chain atoms hydrogen bond to those of residue 23 to stabilize the β-turn. Potential interactions between residue 20 and gp120 appeared to be counterbalanced by steric clashes, which destabilize the turn, thereby limiting clear selection of new amino acids at this position. On the other hand, the third library presented a striking difference between amino acids of configuration (D) and (L). The pronounced selection of (D)-amino acids may relate to a strong preference in the mimetic for a type II′ β-turns, combined with a strong propensity with these turns for a (D)-amino acids at the i+1 position of the turn 39, 40. Sublibraries of the fourth peptide library were similarly divided in two groups, though in this case, the biologically active amino acids were of (L)-configuration. The negative selection of (D)-residues at the fourth position might correspond to the destabilization of type II′ β-hairpins by consecutive amino acids of the same configuration 41. Moreover, steric hindrance strongly selects for small amino acids, particularly polar ones such as Thr, Ser and Asn, which are capable of hydrogen bonding to the backbone carbonyl oxygen of gp120 Met426.
The combinatorial optimizations carried out here are similar in some respects to phage display methods of combinatorially enhancing protein affinity 42-44. One advantage of combinatorial chemistry is that non-natural amino acids can be incorporated, for example, the (D)-amino acids used to augment the variation in optimization of positions 21 and 22.
Although the non-natural biphenyl moiety at position 23 has a clear advantage in affinity relative to the natural Phe, because of solubility considerations, we optimized [Phe23]M33, not its biphenyl variant. Direct optimization of CD4M33 might have lead to another set of amino acids with increased compatibility for the biphenyl moiety. Nonetheless the current work allows [Phe23]M47 to be used as the basis for further optimizations, especially off the zeta position of the phenyl ring, where it opens into a large interfacial binding pocket that extends into the hydrophobic interior of gp120 19, 20. Extensions into this conserved pocket show potential for increasing mimetic affinity, as has been demonstrated for thio-modifications of a Phe43Cys variant of CD4 45 and more recently with this present scaffold 46. Multivalent gp120 inhibitors, containing several copies of the CD4-mimetic miniprotein, may also exhibit increased neutralization potency 47, 48. Finally, we demonstrate the ability of our mimetics to induce epitopes, in this case, the binding surface for the co-receptor CCR5. This induction raises the possibility that CD4-mimetics of the type described here may synergize in a therapeutic setting with the high titer of antibodies reactive with these epitopes, which are present in most HIV-1 infected individuals 49.
Experimental Procedures
Reagents
Fmoc-Bip was from Advanced Chemtech (Louisville, KT, USA). Alexa-488 succinimidyl ester was from Molecular Probes (Leiden, The Netherlands). X4-tropic HXB2 and R5-tropic YU2 recombinant gp120s were obtained from Dr F. Veas (Retroviral and Molecular Immunology Laboratory, Montpellier, France). Recombinant gp120 from the HIV-1 strains R5-tropic SF162 was provided by Dr Indresh Srivastava (Chiron Vaccine Co., Emeryville, USA). 4-domain soluble CD4 was obtained from Progenics (Progenics, Tarrytown, USA) and 2-domain soluble CD4 from the NIH AIDS Research and Reference Reagent Program. CHO-CCR5+ cells were obtained from Dr Marc Parmentier (University of Brussels, Belgium). All other reagents and solvents used were from Sigma-Aldrich and Fluka (St Quentin Fallavier, France) or SDS (Solvants Documentation Syntheses, Peypin, France).
Peptide Synthesis
All combinatorial libraries and individual peptides were synthesized on an Advanced Chemtech 357 Multisynthesizer by solid phase method using fluorenylmethyloxycarbonyl(Fmoc)-protected amino acids and 2-(1-H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) coupling reagent 50. Each sublibrary was synthesized using 33 μmol Fmoc-PAL-PEG-PS-resin (PerSeptive Biosystems, Warrington, GB). The 10 first residues were introduced by 10 molar equivalent Fmoc-protected amino acids, using 9.1 equivalent HBTU and 22 equivalent N,N-Diisopropylethylamine (DIEA), during 30 minutes. The end of the synthesis was performed using double coupling steps with half the previous quantities. The last residue was incorporated as a 3,3′-dithiodipropionic acid moiety. To introduce variability at the desired positions, the resin was collected before coupling, mixed in N-methylpyrrolidone/Dichloromethane (50/50) under nitrogen flow, and divided equally into n wells (where n is the number of different amino acids we wanted to couple). This process, called “split and mix” method, was repeated for each variable position so that, if we consider a peptide with a variability of n, n′ and n″ amino acids at three different position, we end up with n wells containing n′×n″ peptides each. The resin was finally treated with tributylphosphine for 1.5 h, washed and the sublibraries cleaved by treatment with reagent K′ (81.5% trifluoroacetic acid, 5% water, 5% phenol, 5% thioanisole, 2.5% ethanedithiol, 1% triisopropylsilane) for 2.5 h at room temperature. After one hour precipitation in diethyl oxide, the peptides were washed and lyophilized. Disulfide bridges were formed with the sublibraries dissolved at 0.4 mg/mL in 100 mM TrisHCl buffer (pH 7.8) in the presence of 0.5 mM/5 mM oxidized/reduced glutathione. The sublibraries were purified by reverse-phase HPLC on a C18 Discovery column (10 × 250 mm) (Supelco, Bellefonte, USA) and their composition was verified by quantitative amino acid analysis and MALDI-TOF mass spectrometry. Finally, the peptide libraries were analyzed by circular dichroism (CD) on a Jobin Yvon CD6 dichrograph.
Optimization of the combinatorial protocols and synthesis of individual peptides were performed on an ABI-433 automated peptide synthesizer (Applied Biosystem, Applera Corporation). The fluorescent peptides were labelled by the specific incorporation of Alexa-488 or fluorescein at Lys11, Nε-protected during synthesis with 1-(4,4′ dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl (ivDde) 51.
Amino acid compositions were made under standard conditions: samples were vacuum-dried, sealed in glass tubes and hydrolyzed under vapor phase of 6N HCl with a crystal of phenol through 17h at 110°C. Amino acids were derivatized with ninhydrin and quantified on an aminoTac JLC-500/V amino acids analyzer (JEOL, Japan).
Competitive Binding Assays
Competition binding assays in ELISA were performed in 96-wells plates (Maxisorb, Nunc). Briefly, 50 ng per well of antibody D7324 (Aalto Bio Reagents, Dublin) were coated overnight at 4 °C. Wells were then saturated with PBS 3% BSA buffer, washed 3 times, and 5 ng per well of gp120HXBc2 were added, followed by addition of 250 pg of sCD4 (Progenics, Tarrytown, NY) and different concentrations of soluble competitors. After one night at 4 °C, we successively added anti-CD4 mAb L120 (Centralised Facility for AIDS Reagents, NIBSC, UK), goat anti-mouse peroxidase-conjugated antibody (Jackson ImmunoResearch, West Grove, PA), and the 3,3′,5,5′-tetramethylbenzidine substrate (Sigma). After acidification, optical density was measured at 450 nm and expressed as the mean of duplicate 52.
Binding assays by fluorescence anisotropy were made in a LJL Analyst (LJL Biosystems, Sunnyvale, CA) microplate reader using 384-wells plates. All peptides were diluted in 10 mM sodium phosphate buffer, pH 7.0, with 135 mM NaCl and 0.05% Tween 20.
As previously described 27, competitive binding assays for screening were performed in a final volume of 21 μL by using 1 nM CD4M33-Fl, 12.5 nM gp120HxB2 and different concentrations (10-6, 10-7 and 10-8 M) of each peptide sublibrary. Fluorescence anisotropy was determined after 40 min equilibration time at room temperature, using 485 nm excitation and 530 nm emission filters and an additional 505 nm dichroic filter. All experiments were performed in duplicate.
FACS analysis of the mimetic-induced envelope conformational
Adherent CHO-K1 cells expressing CCR5 53, 54 were used to analyze envelope conformational change by FACS. Briefly, 0.06 μg of gp120 (from either strain SF162 or strain YU2) were incubated overnight at 4°C in the absence or the presence of 500 nM CD4 or 500 nM CD4-mimetic miniprotein, and then added to 2 × 105 cells in HAM F12 supplemented with 10% SVF, 1% penicillin/streptomycin. After 1 h incubation at room temperature, cells were washed 3 times with phosphate buffer saline (PBS) containing 5% BSA. Bound gp120 was stained with antibody D7324, washed, and then labeled with goat anti-sheep secondary antibody conjugated to phycoerythrin (PE) (R&D Systems). After a final wash, cells were analyzed for envelope binding by FACS.
Crystallization and structure determination
Crystals of CD4M47 (or [Phe23]M47) were grown as a ternary complex between YU2 core gp120 and the antigen-binding fragment (Fab) of the 17b antibody. Protein production, crystal growth, and data collection were carried out using procedures described previously for CD4M33 24. Briefly, YU2 core gp120 were produced in Drosophila S2 cells and deglycosylated with Endoglycosidases H and D. The deglycosylated protein was combined with CD4M47 (or [Phe23]M47) and purified by Superdex S200 gel filtration chromatography. The complex peak was combined with Fab 17b, concentrated, and again subjected to Superdex S200 chromatography to obtain the purified ternary complex.
As previously described for CD4M33 24, crystals were grown from mixtures of high molecular weight PEG and isopropanol, cross-linked by vapour diffusion with glutaraldehyde 55, briefly placed in a cryo-solution of 20% ethylene glycol, 10% 2R-3R butanediol, mounted in a cryo-loop, and flashcooled in a nitrogen cryostat set to 100° K). X-ray diffraction data were collected at 1.0 Å wavelength, beamline ID-22, Advanced Photon Source, and processed with HKL2000 56 (Table 1). Refinement was carried out with CNS 57, using O 58 for interactive model building. The spacegroup, P21, and lattice were isomorphous with the previously described CD4M33 ternary complex crystals (pdb accession code 1YYL) 24. With the CD4M33 ternary complex as a starting model, after rigid-body refinement, the CD4M47 and [Phe23]M47 structures were built in accordance with mimetic sequences and inspection of difference electron-density maps. Refinement used torsion-angle simulated annealing with slow cooling, iterative manual fitting, automated water placement, and positional and individual isotropic B-value refinement. Noncrystallographic symmetry restraints were used in the initial stages of refinement and later removed. An Rfree test set consisting of 10% of the data was used as a monitor throughout the refinement. Crystallographic statistics are listed in Table 1.
Structure Analysis
Surface complementarities were calculated with SC (CCP4) 59, distance-sorted distance matrices with DDMP 24, 33, and intermolecular contact surfaces with MS 60. Structure figures were made with PyMol 61.
Surface-plasmon resonance
Kinetic parameters of sCD4 and CD4-mimetics were determined with a Biacore 3000 surface-plasmon resonance spectrometer (GE Healthcare). CD4M33, [Phe23]M33, CD4M47, [Phe23]M47, and sCD4 were immobilized onto CM5 sensor chips using standard amine coupling to surface densities of 485, 239, 238, 286, and 141 response units (RU), respectively. Full length HXBc2 and YU2 gp120 were injected sequentially over the coupled sensor chips with increasing concentrations of 2.7 nM, 5.4 nM, 10.8 nM, 21.7 nM, 43.5 nM, and 87 nM at a flow rate of 30 μl/min for 300 s at 25 °C. The dissociation rates of the complexes were monitored for 600 s at a flow rate of 30 μl/min. The buffer used for preparation of the gp120 samples and buffer blanks contained 10 mM HEPES, pH7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P-20, and 0.1% carboxymethyl dextran. The same buffer was used as running buffer during the binding studies. A 1:1 Langmuir binding model with drifting baseline (BiaEvaluation 4.1) was used to fit data globally and to extract kinetic parameters of interaction with gp120.
Viral neutralization assays
Viral assays of the neutralization potency of the CD4-mimetic miniproteins and of soluble CD4 against a panel of diverse viral isolates were carried out as described previously 23; 28.
Supplementary Material
Acknowledgments
We dedicate this work to the memory of Dr Claudio Vita and Dr André Ménez. We thank Lawrence Shapiro for comments on the manuscript, Francesca Sironi for assistance with neutralization assays, Indresh Srivastava (Chiron Co., Emeryville, CA) for gp120, and Jonathan Stuckey for assistance with figures. Support was provided by the European Microbicides Project (EMPRO), the Intramural AIDS Targeted Antiviral Program of the NIH, and a grant from the Bill and Melinda Gates Foundation Grand Challenges in Global Health Initiative. We acknowledge Becton Dickinson and the Centre for AIDS Reagents supported by EU programme EVA/MRC (contract QLKZ-CT-1999-00609) and the UK Medical Research Council. Use of SER-CAT at the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract W-31-109-Eng-38.
Footnotes
Accession Numbers: Coordinates and structure factors have been deposited with the Protein Data Bank under accession numbers 2I5Y (CD4M47 complex) and 2I60 ([Phe23]M47 complex).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 2.Berg T. Modulation of protein-protein interactions with small organic molecules. Angew Chem Int Ed Engl. 2003;42:2462–2481. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]
- 3.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 4.Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem Int Ed Engl. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 5.Martin F, Toniatti C, Salvati AL, Venturini S, Ciliberto G, Cortese R, Sollazzo M. The affinity-selection of a minibody polypeptide inhibitor of human interleukin-6. Embo J. 1994;13:5303–5309. doi: 10.1002/j.1460-2075.1994.tb06864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Biotechnol. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 7.Chin JW, Schepartz A. Design and Evolution of a Miniature Bcl-2 Binding Protein. Angew Chem Int Ed Engl. 2001;40:3806–3809. doi: 10.1002/1521-3773(20011015)40:20<3806::AID-ANIE3806>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 8.Martin L, Barthe P, Combes O, Roumestand C, Vita C. Engineering Novel Bioactive Mini-Proteins on Natural Scaffolds. Tetrahedron. 2000;56:9451–9460. [Google Scholar]
- 9.Mathonet P, Fastrez J. Engineering of non-natural receptors. Curr Opin Struct Biol. 2004;14:505–511. doi: 10.1016/j.sbi.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Stricher F, Martin L, Vita C. Protein Design (Biology, M i M, Ed) Vol. 340. Humana Press; Totowa, New Jersey: 2006. Design of mini-proteins by the Transfer of Active Sites. [Google Scholar]
- 11.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–4. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 12.Dimitrov DS. Cell biology of virus entry. Cell. 2000;101:697–702. doi: 10.1016/s0092-8674(00)80882-x. [DOI] [PubMed] [Google Scholar]
- 13.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 14.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 15.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 17.Lusso P. HIV and the chemokine system: 10 years later. Embo J. 2006;25:447–456. doi: 10.1038/sj.emboj.7600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8:1329–1339. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 20.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drakopoulou E, Vizzavona J, Vita C. Engineering a CD4 mimetic inhibiting the binding of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein gp120 to human lymphocyte CD4 by the transfer of a CD4 functional site to a small natural scaffold. Lett Pep Sci. 1998;5:241–245. [Google Scholar]
- 22.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Menez A, Roumestand C, Yang YS, Ylisastigui L, Benjouad A, Gluckman JC. Rational engineering of a mini-protein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc Natl Acad Sci U S A. 1999;96:13091–13096. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Menez A, Lusso P, Veas F, Vita C. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- 24.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure. 2005;13:755–768. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Dooley CT, Chung NN, Wilkes BC, Schiller PW, Bidlack JM, Pasternak GW, Houghten RA. An all D-amino acid opioid peptide with central analgesic activity from a combinatorial library. Science. 1994;266:2019–2022. doi: 10.1126/science.7801131. [DOI] [PubMed] [Google Scholar]
- 26.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 27.Stricher F, Martin L, Barthe P, Pogenberg V, Mechulam A, Menez A, Roumestand C, Veas F, Royer C, Vita C. A high-throughput fluorescence polarization assay specific to the CD4 binding site of HIV-1 glycoproteins based on a fluorescein-labelled CD4 mimic. Biochem J. 2005;390:29–39. doi: 10.1042/BJ20041953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 30.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Determining the structure of an unliganded and fully glycosylated SIV gp120 envelope glycoprotein. Structure. 2005;13:197–211. doi: 10.1016/j.str.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 32.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. Energetics of the HIV gp120-CD4 binding reaction. Proc Natl Acad Sci U S A. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleming P. Differences Distance Matrix Plot (DDMP) 2004 ( http://www.roselab.jhu.edu/ddmp/
- 34.Wu L, Paxton WA, Kassam N, Ruffing N, Rottman JB, Sullivan N, Choe H, Sodroski J, Newman W, Koup RA, Mackay CR. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 36.Udaka K, Wiesmuller KH, Kienle S, Jung G, Walden P. Tolerance to amino acid variations in peptides binding to the major histocompatibility complex class I protein H-2Kb. J Biol Chem. 1995;270:24130–24134. doi: 10.1074/jbc.270.41.24130. [DOI] [PubMed] [Google Scholar]
- 37.Hyde C, Johnson T, Owen D, Quibell M, Sheppard RC. Some ‘difficult sequences’ made easy. A study of interchain association in solid-phase peptide synthesis. Int J Pept Protein Res. 1994;43:431–440. [PubMed] [Google Scholar]
- 38.Larsen BD, Holm A. Incomplete Fmoc deprotection in solid-phase synthesis of peptides. Int J Pept Protein Res. 1994;43:1–9. doi: 10.1111/j.1399-3011.1994.tb00368.x. [DOI] [PubMed] [Google Scholar]
- 39.Haque TS, Gellman SH. Insights on b-Hairpin Stability in Aqueous Solution from Peptides with Enforced Type I′ and Type II′ β-turns. J Am Chem Soc. 1997;119:2303–2304. [Google Scholar]
- 40.Matter H, Kessler H. Structures, Dynamics, and Biological-Activities of 15 Cyclic Hexapeptide Analogs of the Alpha-Amylase Inhibitor Tendamistat (Hoe-467) in Solution. J Am Chem Soc. 1995;117:3347–3359. [Google Scholar]
- 41.Haque TS, Little JC, Gellman SH. Stereochemical Requirements for b-Hairpin Formation: Model studies with Four-Residue Peptides and Depsipeptides. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]
- 42.Benhar I. Biotechnological applications of phage and cell display. Biotechnol Adv. 2001;19:1–33. doi: 10.1016/s0734-9750(00)00054-9. [DOI] [PubMed] [Google Scholar]
- 43.Devlin JJ, Panganiban LC, Devlin PE. Random peptide libraries: a source of specific protein binding molecules. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 44.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 45.Xie H, Ng D, Savinov SN, Dey B, Kwong PD, Wyatt R, Smith AB, 3rd, Hendrickson WA. Structure-activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J Med Chem. 2007;50:4898–4908. doi: 10.1021/jm070564e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Herrewege Y, Morellato L, Descours A, Aerts L, Michiels J, Heyndrickx L, Martin L, Vanham G. CD4 mimetic miniproteins: potent anti-HIV compounds with promising activity as microbicides. J Antimicrob Chemother. 2008;61:818–826. doi: 10.1093/jac/dkn042. [DOI] [PubMed] [Google Scholar]
- 47.Li H, Guan Y, Szczepanska A, Moreno-Vargas AJ, Carmona AT, Robina I, Lewis GK, Wang LX. Synthesis and anti-HIV activity of trivalent CD4-mimetic miniproteins. Bioorg Med Chem. 2007;15:4220–4228. doi: 10.1016/j.bmc.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Song H, Heredia A, Le N, Redfield R, Lewis GK, Wang LX. Synthetic bivalent CD4-mimetic miniproteins show enhanced anti-HIV activity over the monovalent miniprotein. Bioconjug Chem. 2004;15:783–789. doi: 10.1021/bc049960r. [DOI] [PubMed] [Google Scholar]
- 49.Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med. 2005;201:1407–1419. doi: 10.1084/jem.20042510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carpino LA. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J Am Chem Soc. 1993;115:4397–4398. [Google Scholar]
- 51.Chhabra SR, Hothi B, Evans DJ, White PD, Bycroft BW, Chan WC. An appraisal of new variants of Dde amine protecting group for solid phase peptide synthesis. Tetrahedron Lett. 1998;39:1603–1606. [Google Scholar]
- 52.Moore JP. Simple methods for monitoring HIV-1 and HIV-2 gp120 binding to soluble CD4 by enzyme-linked immunosorbent assay: HIV-2 has a 25-fold lower affinity than HIV-1 for soluble CD4. Aids. 1990;4:297–305. doi: 10.1097/00002030-199004000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Blanpain C, Lee B, Tackoen M, Puffer B, Boom A, Libert F, Sharron M, Wittamer V, Vassart G, Doms RW, Parmentier M. Multiple nonfunctional alleles of CCR5 are frequent in various human populations. Blood. 2000;96:1638–1645. [PubMed] [Google Scholar]
- 54.Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996;35:3362–3367. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- 55.Lusty CJ. A gentle vapor-diffusion technique for cross-linking of protein crystals for cryocrystallography. J Appl Crystallogr. 1999;32:106–112. [Google Scholar]
- 56.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Academic Press; New York: 1997. [DOI] [PubMed] [Google Scholar]
- 57.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 58.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 59.CCP4, C. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 60.Connolly ML. The molecular surface package. J Mol Graph. 1993;11:139–141. doi: 10.1016/0263-7855(93)87010-3. [DOI] [PubMed] [Google Scholar]
- 61.DeLano WL. The PyMOL Molecular Graphics System. Delano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 62.Nishikawa K, Ooi T, Ysogai Y, Saito N. Representation and computation of the conformations. J Phys Soc Jpn. 1972;32:1331–1337. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.