

Abstract
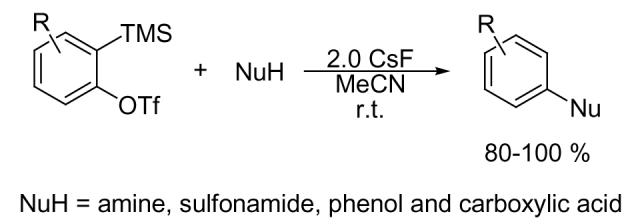
An efficient, transition-metal free procedure for the N-arylation of amines, sulfonamides and carbamates and O-arylation of phenols and carboxylic acids has been achieved by allowing these substrates to react with a variety of o-silylaryl triflates in the presence of CsF. Good to excellent yields of arylated products are obtained under very mild reaction conditions. This chemistry readily tolerates a variety of functional groups.
Introduction
Establishing an efficient, reliable method for the N-arylation of amines, sulfonamides and carbamates, and the O-arylation of phenols and carboxylic acids is currently a very active area of research in organic synthesis. Such aryl subunits are commonly found in a variety of biologically active and natural compounds,1 agrochemicals,2 HIV-1 protease inhibitors,3 and compounds of interest in material science.4 Traditionally, the N-arylation of amines and O-arylation of phenols have been carried out under copper-mediated Ullmann-type conditions involving the coupling of amines and phenols with aryl halides.5 Although these copper-promoted reactions are useful, they usually require harsh reaction conditions and stoichiometric amounts of copper, and the yields are not very reproducible.6 Recently, a number of valuable new methods have been developed for the N-arylation of amines and sulfonamides, and the O-arylation of phenols and carboxylic acids.7 Buchwald8 and Hartwig9 have demonstrated that the Cu- or Pd-catalyzed N-arylation of a variety of amines and O-arylation of phenols by aryl halides is a powerful method for the synthesis of arylamines and diaryl ethers. Evans10 and Chan11 have independently reported the synthesis of diaryl ethers by the copper(II)-promoted cross-coupling of phenols and aryl halides.
Despite these significant recent improvements, there still are limitations in present N- and O-arylation methods. For example, (1) it is still difficult to prepare N-arylated sulfonamides12 and present N-arylation methodology may not accommodate certain organic functionality. Of the functional groups that are incompatible with the Cu- or Pd-catalyzed N-arylation methodology, the most important are probably halides, sulfonates,13 hydroxyl groups14 and probably carbon-carbon triple bonds. (2) Phenols can smoothly be converted to diaryl ethers only if no strong electron-withdrawing group is present.7a,b (3) Most reaction conditions are fairly harsh, usually requiring high temperatures (>110 °C), and strong polar and often toxic solvents.
The direct coupling of aromatic carboxylic acids and aryl halides or the carbonylation of aryl halides using a palladium catalyst to generate the corresponding aryl esters is difficult.15 The direct esterification of phenols by aliphatic or aromatic carboxylic acids is virtually impossible due to the poor nucleophilicity of phenols.16 In the classical methods for esterification, a strong acid, such as sulfuric acid, is needed, which is not suitable for acid sensitive compounds, can be corrosive, and often produces side reactions, such as carbonization, oxidation, etc.17 In this paper, we report an efficient and reliable procedure to N-arylate amines and sulfonamides and O-arylate phenols and carboxylic acids under mild reaction conditions, which can tolerate a wide variety of functional groups, including halide, ester, amide, hydroxyl, nitro, aldehyde, and ketone groups.
Recently, silylaryl triflate 1a18 has been employed to generate benzyne under very mild reaction conditions, which can easily undergo a variety of synthetically useful nucleophilic19 and cycloaddition reactions.20 However, the arylation of amines,21 sulfonamides, phenols and carboxylic acids by arynes has not been widely studied due to the difficulty in generating arynes under convenient reaction conditions. Very recently, we reported that the reaction of a variety of amines, sulfonamides, carbamates, phenols and carboxylic acids with arynes generated from a variety of silylaryl triflates under very mild reaction conditions provides excellent yields of arylated products, while tolerating many functional groups.22 Herein, we wish to report the full details of this very efficient arylation procedure and our studies on the regioselectivity of addition to a number of unsymmetrical substituted silylaryl triflates.
Results and Discussion
Preparation of the Aryne Precursors
The arynes 1a-1e were selected as substrates for our experiments. Aryne precursor 1a was selected as the simplest and most readily available aryne to study the scope of this chemistry. Aryne precursors 1b, 1d and 1e were selected to study the regioselectivity of the arylation procedure. The synthesis of silylaryl triflates 1a,18 1b,23 1c,19c and 1d24 has already been reported, and the precursor 1e25 can easily be prepared by a similar three-step procedure from the corresponding 2-bromo-4,6-dimethylphenol.

N-Arylation of Aromatic Amines
Our initial studies focused on achieving optimal reaction conditions for the N-arylated product using aniline and silylaryl triflate 1a as the model system. We first allowed 2-(trimethylsilyl)phenyl triflate (1a) to react with 2.0 equiv of CsF and 1.2 equiv of aniline in MeCN at room temperature for 20 h. Diphenylamine was obtained in an 81% yield and only a trace of triphenylamine was isolated. After trying several reactions, we found that THF was also a good solvent for the N-arylation of aniline and CsF can be replaced by n-Bu4NF (TBAF). Although the reaction time can be dramatically decreased to 30 mins when TBAF is allowed to react with the silylaryl triflate 1a to generate benzyne, the yield is a little lower. The optimal reaction conditions thus far developed employ 0.25 mmol of aniline, 1.1 equiv of silylaryl triflate 1a and 2.0 equiv of CsF in MeCN at room temperature for 10 h. Diphenylamine was obtained in a 92% yield (Scheme 1) (Table 1, entry 1).
Scheme 1.
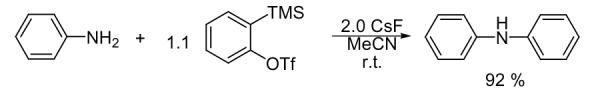
Table 1.
Facile N-Arylation of Aromatic Aminesa
| entry | amine | silylaryl triflate (equiv) |
CsF (equiv) |
time (h) |
product | % isolated yield |
|---|---|---|---|---|---|---|
| 1 |  |
1a (1.1) | 2.0 | 10 |  |
92 |
| 2 | 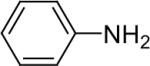 |
1b (1.1) | 2.0 | 10 | 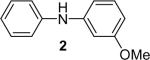 |
94 |
| 3 | 1a (1.1) | 2.0 | 10 |  |
85 | |
| 4 | 1a (1.1) | 2.0 | 10 |  |
90 | |
| 5 | 1a (1.1) | 2.0 | 10 | 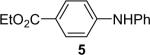 |
94 | |
| 6 | 1a (1.1) | 2.0 | 10 | 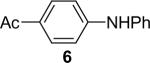 |
94 | |
| 7 | 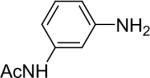 |
1a (1.1) | 2.0 | 10 | 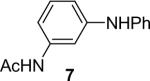 |
95 |
| 8 | 1a (1.1) | 2.0 | 10 | 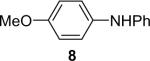 |
89b | |
| 9 | 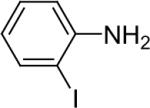 |
1a (1.1) | 2.0 | 10 | 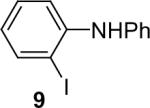 |
91 |
| 10 | 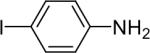 |
1a (1.1) | 2.0 | 10 | 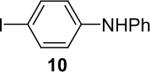 |
92 |
| 11 | 1a (1.1) | 2.0 | 10 | 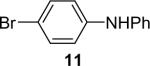 |
91 | |
| 12 | 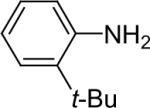 |
1a (1.1) | 2.0 | 10 | 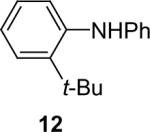 |
77 |
| 13 | 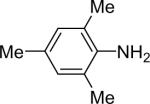 |
1a (1.1) | 2.0 | 10 | 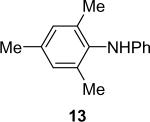 |
90 |
| 14 |  |
1a (1.1) | 2.0 | 5 | 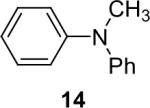 |
96 |
| 15 | 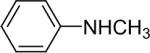 |
1c (1.1) | 2.0 | 10 | 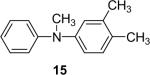 |
97 |
| 16 | 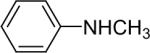 |
1d (1.1) | 2.0 | 5 | 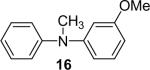 |
94 (1:1) |
| 17 | 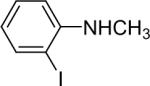 |
1a (1.1) | 2.0 | 5 | 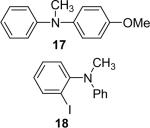 |
97 |
| 18 |  |
1a (1.1) | 2.0 | 5 |  |
97 |
| 19 | 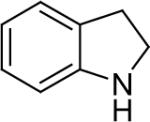 |
1a (1.1) | 2.0 | 5 | 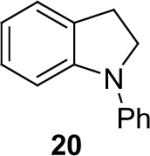 |
97 |
| 20 | 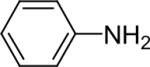 |
1a (2.4) | 4.0 | 72 | 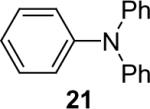 |
98 |
| 21 | 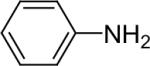 |
1b (2.4) | 4.0 | 72 | 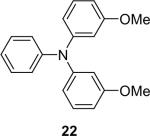 |
94 |
| 22 | 1c (2.4) | 4.0 | 72 | 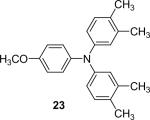 |
93 | |
| 23 | 1a (2.4) | 4.0 | 72 | 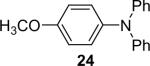 |
100 | |
| 24 | 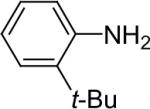 |
1a (2.4) | 4.0 | 72 | 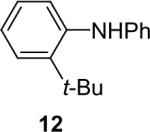 |
81c |
| 25 | 1a (2.4) | 4.0 | 72 | 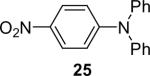 |
55d | |
| 26 | 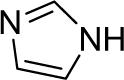 |
1a (1.2) | 2.0 | 24 | 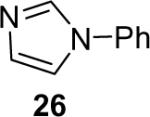 |
76 |
| 27 | 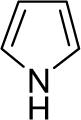 |
1a (3.6) | 6.0 | 72 | 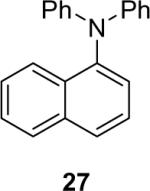 |
81 |
| 28 | 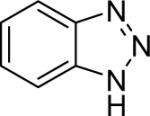 |
1a (1.2) | 2.0 | 24 | 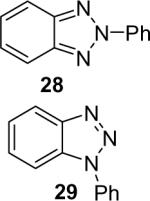 |
92 (1 : 3.5) |
All reactions were run under the following reaction conditions, unless otherwise specified: 0.25 mmol of amine and the indicated amount of silylaryl triflate and CsF were stirred in 4 mL of MeCN at room temperature.
A 4% yield of the diarylated product was obtained.
Only monoarylated product was obtained.
A 40% yield of monoarylated product was obtained.
We next studied the scope of this methodology by allowing a wide variety of aromatic amines to react with the silylaryl triflates 1a, 1b, and 1c.26 The results are summarized in Table 1. Aniline itself and aniline with electron-donating and -withdrawing groups (Table 1, entries 1 and 3-8), such as nitro, cyano, ester, ketone, amide and methoxy groups, all react well with silylaryl triflate 1a and CsF to afford excellent yields of the desired phenyl-substituted products. It is noteworthy that haloanilines also react with silylaryl triflate 1a to generate the desired halo-substituted products in excellent yields (Table 1, entries 9-11). Halides, which are unlikely to survive under the Pd-mediated amination reaction conditions,8,9 are readily accommodated by our reaction conditions. When sterically-hindered amines are used as the starting materials (Table 1, entries 12 and 13), good yields of the N-arylated products are still obtained.
Secondary amines usually react faster than primary amines with the silylaryl triflates plus CsF (Table 1, entries 14 and 16-19), except when silylaryl triflate 1c is employed (Table1, entry 15), and excellent yields are generally obtained. It appears that generation of the aryne is significantly slower using the silylaryl triflate 1c.27
Diarylation products are also readily obtained in excellent yields when an excess of the silylaryl triflate is employed with primary aromatic amines (Table 1, entries 20-23 and 25). However, the sterically-hindered aniline o-t-butylaniline afforded only the monoarylated product, even when an excess of the silylaryl triflate 1a was employed (Table 1, entry 24). While imidazole affords a high yield of the N-arylated product (entry 26), 1-(diphenylamino)naphthalene is obtained in an 81% yield, when pyrrole is allowed to react with 3.3 equiv of silylaryl triflate 1a (Table 1, entry 27).28 This unexpected product can be explained by the mechanism shown in Scheme 2. First pyrrole reacts with benzyne to generate the N-arylated compound A, which then undergoes [4+2] cycloaddition to afford compound B, which can apparently react further with one more equivalent of benzyne to afford the 1-(diphenylamino)naphthalene in an 81% yield. The N-arylation of benzotriazole proceeded in high yield, but afforded a 1:3.5 mixture of N-2 and N-1 arylation products respectively. This result is similar to results obtained using the Pd-catalyzed amination chemistry (Table 1, entry 28).29
Scheme 2.

The regiochemistry of the substitution reaction using unsymmetrical arynes has proven quite interesting. The methoxy-substituted silylaryl triflate 1b reacts cleanly with aniline to generate a single regioisomer of either the monoarylation or diarylation products in good yields (Table 1, entries 2 and 21). When methoxy-substituted silylaryl triflate 1d is employed with N-methylaniline, two isomers were obtained in almost equal amounts (Table 1, entry 16). The very different regioselectivity observed using these two arynes can be readily explained by steric and electronic effects. The use of silylaryl triflate 1b favors nucleophilic attack at the position meta to the methoxy group of the aryne (Scheme 3, path B).30 When using silylaryl triflate 1d, the steric and electronic effects are very similar in the meta and para positions to the methoxy group of the aryne.
Scheme 3.
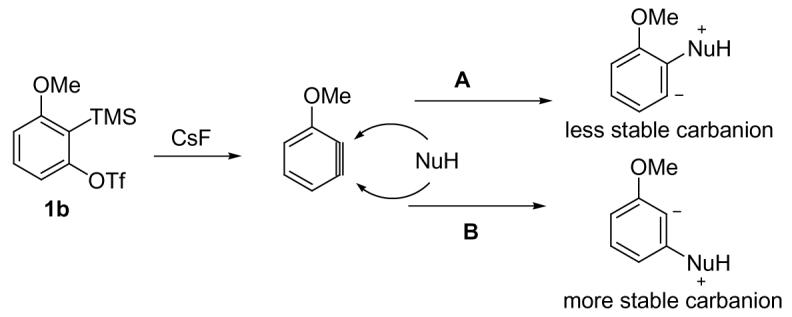
In summary, from Table 1, one can see that almost all aromatic amines react well with silylaryl triflates and CsF under very mild reaction conditions to afford good to excellent yields of the desired products. Furthermore, one can selectively prepare secondary or tertiary arylamines by simply changing the ratio of the reactants (compare entries 1 and 21, and 8 and 23).
N-Arylation of Alkyl Amines
Alkylamines also react well with the silylaryl triflates and CsF to afford mono or diarylated alkylamines in good to excellent yields under very mild reaction conditions. The results are summarized in Table 2. The mono N-arylation of primary alkylamines is accomplished in fairly good yield when allowing an excess of the primary alkylamine to react with the silylaryl triflate and CsF, although about 10% of the diarylated product is also usually obtained (Table 2, entries 1-4). When an excess of the silylaryl triflate and CsF is employed with primary amines or secondary amines, tertiary amines can be obtained in excellent yields (Table 2, entries 5-13). Even sterically hindered t-BuNH2 affords an excellent yield of the diarylated product. The silylaryl triflate 1a can also selectively react with an amino group in the presence of an alcohol group to afford the corresponding N-arylation product in an 83% yield, although 8% of the O-arylation product N-(2-phenoxyethyl)-N-phenylaniline was isolated as a side product (entry 7). An amino ester has also proven to be a good substrate for N-arylation (Table 2, entry 14). Alcohols, and carbon-carbon double and triple bonds are readily accommodated by this process. It should be pointed out that a variety of other functional groups, including halide and ester groups, should be readily tolerated under our reaction conditions.
Table 2.
Facile N-Arylation of Alkylaminesa
| entry | amine | silylaryl triflate (equiv) |
CsF (equiv) |
time (h) |
product | % isolated yield |
|---|---|---|---|---|---|---|
| 1 |  |
1a (0.9) | 2.0 | 5 |  |
70b |
| 2 | 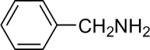 |
1b (0.9) | 2.0 | 5 | 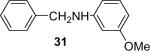 |
71b |
| 3 | HC≡CCH2NH2 | 1a (0.9) | 2.0 | 5 | HC≡CCH2NHPh 32 |
62c |
| 4 | 1a (0.9) | 2.0 | 5 | 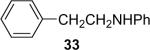 |
66b | |
| 5 | 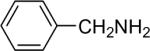 |
1a (2.4) | 4.0 | 72 | 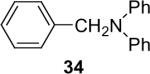 |
97 |
| 6 | 1a (2.4) | 4.0 | 72 | 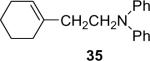 |
99 | |
| 7 | HOCH2CH2NH2 | 1a (2.4) | 4.0 | 72 | 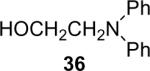 |
83d |
| 8 | HC≡CCH2NH2 | 1a (2.4) | 4.0 | 72 | 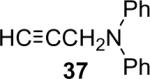 |
78 |
| 9 | t-Bu—NH2 | 1a (2.4) | 4.0 | 72 | 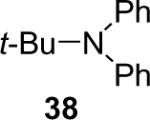 |
96 |
| 10 | 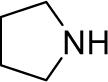 |
1a (1.2) | 2.0 | 5 | 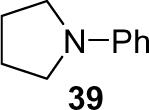 |
95 |
| 11 | 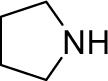 |
1b (1.2) | 2.0 | 5 | 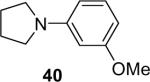 |
97 |
| 12 |  |
1a (1.2) | 2.0 | 5 | 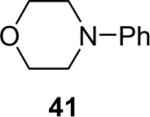 |
81 |
| 13 | (Bn)2NH | 1a (1.2) | 2.0 | 5 | 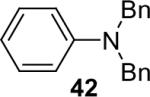 |
99 |
| 14 | 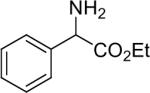 |
1a (1.2) | 2.0 | 5 | 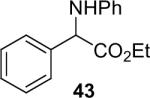 |
65 |
All reactions were run under the following reaction conditions, unless otherwise specified: 0.25 mmol of the amine and the indicated amount of silylaryl triflate and CsF were stirred in 4 mL of MeCN at room temperature.
Approximately a 10% yield of the diarylated product was obtained.
A 12% yield of the diarylated product was obtained.
An 8% yield of N-(2-phenoxyethyl)-N-phenylaniline was also obtained.
N-Arylation of Sulfonamides and Carbamates
While some effort has been devoted recently to the N-arylation of sulfonamides,31 the scope of this chemistry with respect to either the sulfonamide or the aryl halide is still very limited. As shown in Table 3, entries 1-13, alkane- and arenesulfonamides both efficiently undergo N-arylation under our reaction conditions. Using primary sulfonamides and an excess of silylaryl triflate, one obtains the corresponding diarylation product in high yield in a fairly short reaction time compared with the times required for the N-arylation of amines. Again, a variety of functional groups, including iodine, chloride, and ester groups, are tolerated under our reaction conditions. Both electron-donating and electron-withdrawing groups on the arene moiety of the arenesulfonamide give excellent yields (Table 3, entries 4-8). Interestingly, p-aminobenzenesulfonamide reacts with 3.3 equiv of silylaryl triflate 1a to afford a product with a monoarylated amine and the diarylated sulfonamide (Table 3, entry 9). It is important to point out that we cannot obtain monoarylation products of primary sulfonamides under our reaction conditions, even when one uses an excess of the sulfonamide. One can also start with secondary sulfonamides and produce the corresponding N-arylated tertiary sulfonamides in excellent yields (Table 3, entries 10-13). The methoxy-substituted silylaryl triflate 1b also reacts cleanly with N-methyl-p-toluenesulfonamide to afford a high yield and excellent regioselectivity (Table 3, entry 11).30 Only the product of substitution meta to the methoxy substituent is observed. Saccharin also reacts well with silylaryl triflate 1a to afford the N-arylated product in a 72% yield (Table 3, entry 14).
Table 3.
Facile N-Arylation of Sulfonamides and Carbamatesa
| entry | sulfonamides | silylaryl triflate (equiv) |
CsF (equiv) |
time (h) |
product | % isolated yield |
|---|---|---|---|---|---|---|
| 1 | 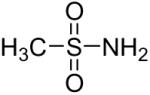 |
1a (2.4) | 4.0 | 24 | 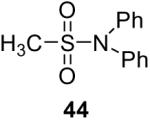 |
80 |
| 2 | 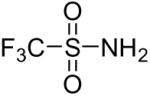 |
1a (2.4) | 4.0 | 24 | 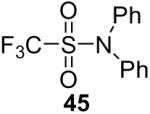 |
78 |
| 3 | 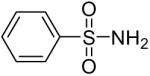 |
1a (2.4) | 4.0 | 24 | 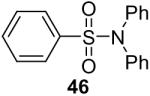 |
99 |
| 4 | 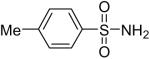 |
1a (2.4) | 4.0 | 24 | 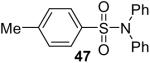 |
100 |
| 5 | 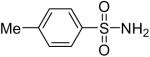 |
1b (2.4) | 4.0 | 24 | 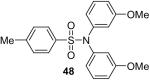 |
92 |
| 6 | 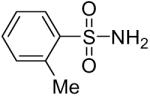 |
1a (2.4) | 4.0 | 24 | 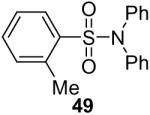 |
99 |
| 7 | 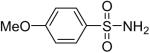 |
1a (2.4) | 4.0 | 24 | 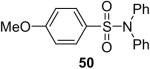 |
100 |
| 8 | 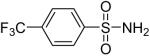 |
1a (2.4) | 4.0 | 24 | 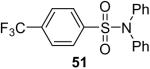 |
91 |
| 9 | 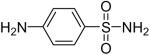 |
1a (3.3) | 2.0 | 24 | 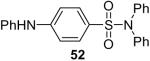 |
75 |
| 10 | 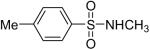 |
1a (1.2) | 2.0 | 24 | 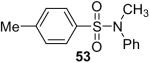 |
87 |
| 11 | 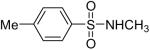 |
1b (1.2) | 2.0 | 12 | 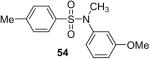 |
91 |
| 12 | 1a (1.2) | 2.0 | 12 | 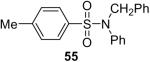 |
94 | |
| 13 | 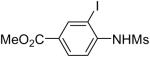 |
1a (1.2) | 2.0 | 12 | 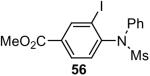 |
98 |
| 14 | 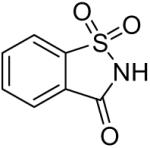 |
1a (1.2) | 2.0 | 12 | 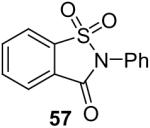 |
72 |
| 15 | 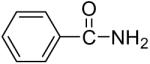 |
1a (1.2) | 2.0 | 24 | 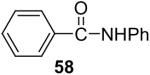 |
0 |
| 16 | 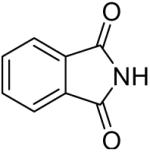 |
1a (1.2) | 2.0 | 12 | 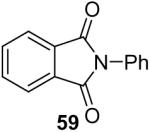 |
60 |
| 17 | 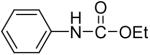 |
1a (1.2) | 2.0 | 5 | 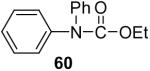 |
96 |
| 18 | 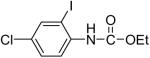 |
1a (1.2) | 2.0 | 5 | 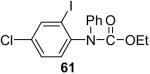 |
93 |
All reactions were run under the following reaction conditions, unless otherwise specified: 0.25 mmol of the sulfonamide or amide and the indicated amount of silylaryl triflate and CsF were stirred in 3 mL of MeCN at room temperature.
We have also investigated the use of carboxamides to generate N-arylamides from the silylaryl triflate 1a and CsF at room temperature. Unfortunately, only a trace of the corresponding N-arylamide could be obtained from benzamide (Table 3, entry 15). Simple amides are apparently neither acidic enough to form the corresponding anion or nucleophilic enough to directly attack the aryne. However, one can obtain a 60% yield of N-arylation product from phthalimide (Table 3, entry 16). However, N-arylcarbamates react well with the silylaryl triflate 1a, affording the N-arylation products in excellent yields in 5 h (Table 3, entries 17 and 18). Note that once again iodine and chloride groups are readily accommodated by this process.
O-Arylation of Phenols
After our success with the N-arylation of amines, sulfonamides and carbamates under very mild reaction conditions, it was natural for us to investigate the O-arylation of phenols and carboxylic acids using silylaryl triflates and CsF. We first examined the reaction of phenol with 1.2 equiv of 2-(trimethylsilyl)phenyl triflate (1a) and 2 equiv of CsF in MeCN at room temperature, our standard reaction conditions for the N-arylation of amines and sulfonamides. We obtained only a 66% yield of the desired diphenyl ether in 20 h. After optimizing this reaction system using phenol and 1.5 equiv of silylaryl triflate 1a plus 3.0 equiv of CsF in MeCN for 1 d at room temperature, we were able to obtain diphenyl ether in a 92% isolated yield (Table 4, entry 1).
Table 4.
Facile O-Arylation of Phenolsa
| entry | phenol | silylaryl triflate (equiv) |
CsF (equiv) |
time (h) |
product | % isolated yield |
|---|---|---|---|---|---|---|
| 1 | 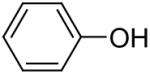 |
1a (1.5) | 3.0 | 24 | 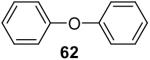 |
92 |
| 2 | 1a (1.5) | 3.0 | 24 | 98 | ||
| 3 | 1a (1.5) | 3.0 | 24 | 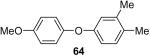 |
93 | |
| 4 | 1a (1.5) | 3.0 | 24 | 85 | ||
| 5 | 1a (1.5) | 3.0 | 24 | 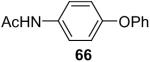 |
91 | |
| 6 | 1a (1.5) | 3.0 | 24 | 96 | ||
| 7 | 1a (1.5) | 3.0 | 24 | 96 | ||
| 8 | 1a (1.5) | 3.0 | 24 | 91 | ||
| 9 | 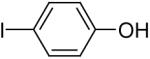 |
1a (1.5) | 3.0 | 24 | 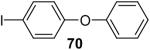 |
94 |
| 10 | 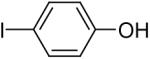 |
1a (1.5) | 3.0 | 24 | 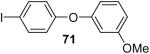 |
95 |
| 11 | 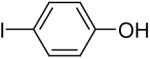 |
1e (1.5) | 3.0 | 24 | 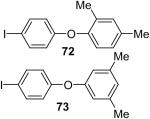 |
92 (1 : 1.5) |
| 12 |  |
1a (1.5) | 3.0 | 24 | 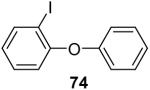 |
90 |
| 13 | 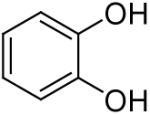 |
1a (3.0) | 6.0 | 60 | 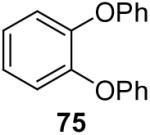 |
72 |
| 14 | 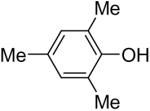 |
1a (1.5) | 3.0 | 24 | 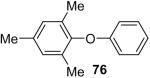 |
68 |
| 15 | 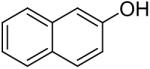 |
1a (1.5) | 3.0 | 24 | 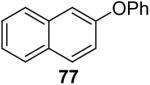 |
84 |
| 16 | 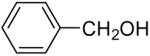 |
1a (1.5) | 3.0 | 24 | 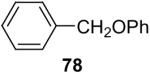 |
25b |
| 17 | 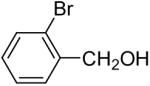 |
1a (1.5) | 3.0 | 24 | 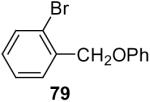 |
36 |
| 18 | 1a (1.5) | 3.0 | 24 | 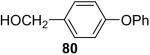 |
75 | |
| 19 | 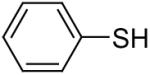 |
1a (1.5) | 3.0 | 24 | 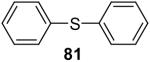 |
70 |
| 20 | 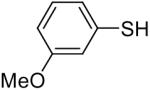 |
1a (1.5) | 3.0 | 24 | 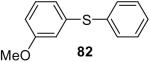 |
66 |
All reactions were run under the following reaction conditions, unless otherwise specified: 0.25 mmol of the phenol and the indicated amount of silylaryl triflate and CsF were stirred in 4 mL of MeCN at room temperature.
When 1.1 equiv of the strong base DBU were added, the yield did not improve.
Using these optimal reaction conditions, this method has been applied to the O-arylation of a wide variety of phenols. The results are summarized in Table 4. Phenol itself and a phenol with an electron-donating methoxy group react well with silylaryl triflate 1a to give the corresponding diaryl ethers in high yields (Table 4, entries 1 and 2). If 4,5-dimethyl-2-(trimethylsilyl)phenyl trifluoromethanesulfonate (1c) is allowed to react with p-methoxyphenol, we can obtain the corresponding diaryl ether in a 93% yield (Table 4, entry 3). p-tert-Butylphenol also works well, affording the corresponding O-arylated product in an 85% yield (Table 4, entry 4). One still can obtain a 91% yield of the O-arylated product from phenol bearing an acetamide group (Table 4, entry 5). This result is consistent with our earlier observation that simple amides do not readily undergo N-arylation. Phenols bearing an electron-withdrawing substituent generally behave poorly or are completely inert towards diaryl ether formation when using aryl halides and a Pd catalyst to effect O-arylation.8e,f However, under our reaction conditions, phenols with an electron-withdrawing group, such as a nitro or aldehyde group, work quite well to generate the corresponding diaryl ethers in high yields (Table 4, entries 6-8). When ortho- or para-substituted iodophenol were employed, one can still obtain the corresponding O-arylated phenols in over 90% yields (Table 4, entries 9-12). The analogous reaction with silylaryl triflate 1c affords two isomers, compounds 72 and 73, in a 1:1.5 ratio (Table 4, entry 11). This is easily explained by steric effects. Clearly, nucleophiles prefer reacting at the position meta to the methyl group, rather than ortho to the methyl group.19 Interestingly, if we use 1,2-benzenediol, we obtain a 72% yield of the double O-arylation product (Table 4, entry 13). The sterically hindered phenol 2,4,6-trimethylphenol also reacts smoothly with silylaryl triflate 1a to afford a 68% yield of the desired product. With β-naphthol, one can obtain the O-arylated product in an 84% yield (Table 4, entry 15).
In general, alcohols do not react with these arynes to give good yields of aryl ethers. For example, benzyl alcohol afforded only a 25% yield of the corresponding aryl ether, even when a stronger base, such as DBU, was added (Table 4, entry 16). o-Bromobenzyl alcohol gave a slightly better yield of 36% (Table 4, entry 17). Consistent with these results, 4-hydroxybenzyl alcohol reacts with silylaryl triflate 1a to afford 4-phenoxybenzyl alcohol in a 75% yield (Table 4, entry 18). Arene thiols also react well with silylaryl triflate 1a to afford the S-arylated product in a fairly good yield (Table 4, entries 19 and 20). This chemistry provides a very convenient method to prepare a wide variety of diaryl ethers and sulfides that tolerates a number of functional groups and produces very high yields under very mild reaction conditions.
O-Arylation of Carboxylic Acids
The O-arylation of carboxylic acids has also been investigated as a method to generate aryl esters. One needs to use 2.0 equiv of silylaryl triflate plus 4.0 equiv of CsF in the reaction with carboxylic acids in order to obtain good yields of the corresponding O-arylation products. The results are summarized in Table 5. As shown in Table 5, benzoic acid and a number of functionally-substituted benzoic acids react smoothly with silylaryl triflates 1a, 1b and 1c to generate the corresponding aryl esters in high yields. Benzoic acids with an electron-donating group, like a methoxy group (Table 5, entries 4-6), generally afford better yields than benzoic acids with an electron-withdrawing group, like a nitro group (Table 5, entry 2). Halogen-substituted carboxylic acids also work well with silylaryl triflates 1a, 1b and 1c to afford good yields of aryl esters, even when a bulky iodo group is present in the ortho position (Table 5, entries 7-10). Unfortunately, aliphatic carboxylic acids do not react well with silylaryl triflates and CsF under our reaction conditions. For example, phenylacetic acid reacts with 2.0 equiv of silylaryl triflate 1a and 4.0 equiv of CsF to afford only a 45% yield of the corresponding aryl ester (Table 5, entry 11). The reason for the low yields of aryl esters from aliphatic carboxylic acids is not clear at this time. When benzenesulfonic acid was employed to react with silylaryl triflate 1a, only a 33% yield of phenyl benzenesulfonate was obtained (Table 5, entry 12).
Table 5.
Facile O-Arylation of Carboxylic Acidsa
| entry | carboxylic acid | silylaryl triflate (equiv) |
CsF (equiv) |
time (h) |
product | % isolated yield |
|---|---|---|---|---|---|---|
| 1 | 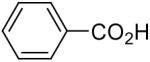 |
1a (2.0) | 4.0 | 24 | 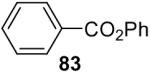 |
81 |
| 2 | 1a (2.0) | 4.0 | 24 | 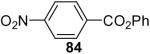 |
66 | |
| 4 | 1a (2.0) | 4.0 | 24 | 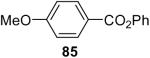 |
96 | |
| 5 | 1b (2.0) | 4.0 | 24 | 98 | ||
| 6 | 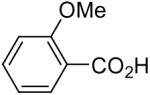 |
1a (2.0) | 4.0 | 24 | 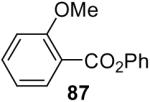 |
84 |
| 7 | 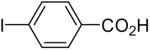 |
1c (2.0) | 4.0 | 24 | 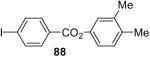 |
81 |
| 8 | 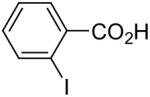 |
1a (2.0) | 4.0 | 24 | 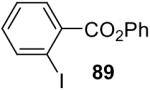 |
75 |
| 9 | 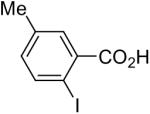 |
1a (2.0) | 4.0 | 24 | 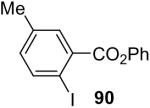 |
89 |
| 10 | 1b (2.0) | 4.0 | 24 | 89 | ||
| 11 | 1a (2.0) | 4.0 | 24 | 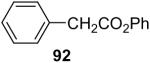 |
45 | |
| 12 | 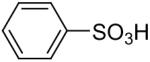 |
1a (2.0) | 4.0 | 24 | 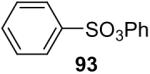 |
33 |
All reactions were run under the following reaction conditions, unless otherwise specified: 0.25 mmol of the carboxylic acid and the indicated amount of silylaryl triflate and CsF were stirred in 4 mL of MeCN at room temperature.
Conclusions
We have developed an efficient, mild, transition metal-free method for the N-arylation of amines, sulfonamides, and carbamates, and O-arylation of phenols and aromatic carboxylic acids. The regioselectivity of the methoxy-substituted silylaryl triflate 1b is excellent. With other silylaryl triflates, such as 1d and 1e, two isomers are obtained. Mono-arylated and diarylated amines can easily be obtained from primary amines by simply controlling the ratio of the reactants. A variety of functional groups are compatible with the reaction conditions, including nitro, hydroxyl, aldehyde, ketone, ester and amide groups, as well as double bonds and triple bonds. We have also shown that halides, which are not compatible with Pd-mediated arylation procedures, readily tolerate our reaction conditions. This chemistry nicely complements classical methods for the N-arylation of amines and sulfonamides, and the O-arylation of phenols and carboxylic acids.
Experimental Section
General Procedure for the Mono N-Arylation of Aromatic Amines
To a solution of MeCN (4 mL), aromatic amine (0.25 mmol) and silylaryl triflate (0.275 mmol) was added CsF (0.4 mmol). The reaction mixture was allowed to stir at room temperature and monitored by TLC to establish completion of the reaction. The resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product.
Diphenylamine (Table 1, entry 1)
Purification by flash chromatography (30:1 hexane/EtOAc) afforded the indicated compound (38 mg) in a 92% yield as a white solid: mp 49-50 °C (lit.32 mp 50-52 °C). The 1H and 13C NMR spectra match the literature data.33
General Procedure for the Mono N-Arylation of Alkylamines
To a solution of MeCN (4 mL), alkylamine (0.25 mmol) and silylaryl triflate (0.225 mmol) was added CsF (0.4 mmol). The reaction mixture was allowed to stir at room temperature and monitored by TLC to establish completion of the reaction. The resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product.
N-Propargylaniline (Table 2, entry 3)
Purification by flash chromatography (25:1 hexane/EtOAc) afforded the indicated compound (19 mg) in a 62% yield as a light yellow oil. The 1H NMR spectrum matchs the literature data;34 13C NMR (75 MHz, CDCl3) δ 147.1, 129.4, 118.9, 113.7, 81.2, 71.5, 33.9.
General Procedure for the Diarylation of Amines and Sulfonamides
To a solution of MeCN (4 mL), the amine or sulfonamide (0.25 mmol) and silylaryl triflate (0.6 mmol) was added CsF (1.2 mmol). The reaction mixture was allowed to stir at room temperature for 3 d for N-arylated amines and 1 d for N-arylated sulfonamides. The resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product.
Triphenylamine (Table 1, entry 20)
Purification by flash chromatography (100:1 hexane/EtOAc) afforded the indicated compound (60 mg) in a 98% yield as a white solid: mp 123-124 °C (lit.35 mp 125-126 °C). The 1H and 13C NMR spectra match the literature data.36
N,N-Diphenylbenzenesulfonamide (Table 3, entry 3)
Purification by flash chromatography (100:1 hexane/EtOAc) afforded the indicated compound (77 mg) in a 99% yield as a white solid: mp 120-121 °C; 1H NMR (300 MHz, CDCl3) δ 7.73-7.70 (m, 2H), 7.62-7.45 (m, 3H), 7.34-7.23 (m, 10H); 13C NMR (75 MHz, CDCl3) δ 141.6, 140.7, 133.0, 129.5, 129.1, 128.5, 127.9, 127.7. HRMS m/z 309.0829 (calcd C18H15NO2S, 309.0823).
General Procedure for the O-Arylation of Phenols
To a solution of MeCN (4 mL), the phenol (0.25 mmol) and the silylaryl triflate (0.375 mmol) was added CsF (0.75 mmol). The reaction mixture was allowed to stir at room temperature for 1 d and the resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product.
Diphenyl ether (Table 4, entry 1)
Purification by flash chromatography (100:1 hexane/EtOAc) afforded the indicated compound (39 mg) in a 92% yield as a colorless oil. The 1H and 13C NMR spectra match the literature data.37
General Procedure for the O-Arylation of Carboxylic Acids
To a solution of MeCN (4 mL), the carboxylic acid (0.25 mmol) and the silylaryl triflate (0.5 mmol) was added CsF (1.0 mmol). The reaction mixture was allowed to stir at room temperature for 1 d and the resulting solution was washed with brine (20 mL) and extracted with diethyl ether (20 mL). The combined ether fractions were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the desired product.
Phenyl benzoate (Table 5, entry 1)
Purification by flash chromatography (100:1 hexane/EtOAc) afforded the indicated compound (40 mg) in an 81% yield as a white solid: mp 69-70 °C (lit.38 mp 68-70 °C). The 1H and 13C NMR spectra match the literature data.39
Supplementary Material
Acknowledgements
We are grateful to the National Institutes of Health (KU Chemical Methodologies and Library Development Center of Excellence, P50 GM069663) for their generous financial support.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for all previously unknown products. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Negwer M. Organic-Chemical Drugs and their Synonyms. 7th ed. Akademie Verlag GmbH; Berlin: 1994. An International Survey. [Google Scholar]; (b) Czarnik AW. Acc. Chem. Res. 1996;29:112–113. [Google Scholar]; (c) Hong Y, Senanayake CH, Xiang T, Vandenbossche CP, Tanoury GJ, Bakale RP, Wald SA. Tetrahedron Lett. 1998;39:3121–3124. [Google Scholar]; (d) Romero DL, Morge RA, Genin MJ, Biles C, Busso M, Resnick L, Althaus IW, Reusser F, Thomas RC, Tarpley WG. J. Med. Chem. 1993;36:1505–1508. doi: 10.1021/jm00062a027. [DOI] [PubMed] [Google Scholar]; (e) Evans DA, DeVries KM. In: Glycopeptide Antibiotics, Drugs and the Pharmaceutical Sciences. Nagarajan R, editor. Vol. 63. Marcel Decker, Inc.; New York: 1994. pp. 63–104. [Google Scholar]; (f) Pettit GR, Singh SB, Niven ML. J. Am. Chem. Soc. 1988;110:8539–8540. [Google Scholar]; (g) Jung ME, Rohloff JC. J. Org. Chem. 1985;50:4909–4913. [Google Scholar]; (h) Atkinson DC, Godfrey KE, Myers PL, Philips NC, Stillings MR, Welbourn AP. J. Med. Chem. 1983;26:1361–1364. doi: 10.1021/jm00364a005. [DOI] [PubMed] [Google Scholar]; (i) Nicolaou KC, Christopher NCB. J. Am. Chem. Soc. 2002;124:10451–10455. doi: 10.1021/ja020736r. [DOI] [PubMed] [Google Scholar]
- 2.CRC . In: Handbook of Pesticides. Milne GWA, editor. CRC Press; Boca Raton: 1994. [Google Scholar]
- 3.Turner SR, Strohbach JW, Tommasi RA, Aristoff PA, Johnson PD, Skulnick HI, Dolak LA, Seest EP, Tomich PK, Bohanon MJ, Horng MM, Lynn JC, Chong K-T, Hinshaw RR, Watenpaugh KD, Janakiraman MN, Thaisrivongs S. J. Med. Chem. 1998;41:3467–3476. doi: 10.1021/jm9802158. [DOI] [PubMed] [Google Scholar]
- 4.(a) Goodson FE, Hartwig JF. Macromolecules. 1998;31:1700–1703. [Google Scholar]; (b) Goodson FE, Hauck SI, Hartwig JF. J. Am. Chem. Soc. 1999;121:7527–7530. [Google Scholar]; (c) Singer RA, Sadighi JP, Buchwald SL. J. Am. Chem. Soc. 1998;120:213–214. [Google Scholar]
- 5.(a) Ullmann F. Ber. Dtsch. Chem. Ges. 1903;36:2382–2384. [Google Scholar]; (b) Goodbrand HB, Hu N-X. J. Org. Chem. 1999;64:670–674. [Google Scholar]; (c) Ullmann F. Chem. Ber. 1904;37:853–854. [Google Scholar]
- 6.a) Lindley J. Tetrahedron. 1984;40:1433–1456. [Google Scholar]; (b) Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359–1370. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 7.Kwong FY, Buchwald SL. Org. Lett. 2003;5:793–796. doi: 10.1021/ol0273396.Kwong FY, Klapars A, Buchwald SL. Org. Lett. 2003;5:581–584. doi: 10.1021/ol0171867.He H, Wu Y-J. Tetrahedron Lett. 2003;44:3385–3386.For a recent review, see: Sawyer JS. Tetrahedron. 2000;56:5045–5065.Theil F. Angew. Chem., Int. Ed. Engl. 1999;38:2345–2347. doi: 10.1002/(sici)1521-3773(19990816)38:16<2345::aid-anie2345>3.0.co;2-5.
- 8.(a) Wolfe JP, Marcoux J-F, Buchwald SL. Acc. Chem. Res. 1998;31:805–818. [Google Scholar]; (b) Zim D, Buchwald SL. Org. Lett. 2003;5:2413–2415. doi: 10.1021/ol034561h. [DOI] [PubMed] [Google Scholar]; (c) Harris MC, Huang X, Buchwald SL. Org. Lett. 2002;4:2885–2888. doi: 10.1021/ol0262688. [DOI] [PubMed] [Google Scholar]; (d) Wolfe JP, Tomori H, Sadighi JP, Yin J, Buchwald SL. J. Org. Chem. 2000;65:1158–1174. doi: 10.1021/jo991699y. [DOI] [PubMed] [Google Scholar]; (e) Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buchwald SL. J. Am. Chem. Soc. 1999;121:4369–4378. [Google Scholar]; (f) Marcoux J, Doye S, Buchwald SL. J. Am. Chem. Soc. 1997;119:10539–10540. [Google Scholar]; (g) Kuwabe S-I, Torraca KE, Buchwald SL. J. Am. Chem. Soc. 2001;123:12202–12206. doi: 10.1021/ja012046d. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hartwig JF. Acc. Chem. Res. 1998;31:853–860. [Google Scholar]; (b) Hartwig JF. Angew. Chem., Int. Ed. Engl. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (c) Mann G, Incarvito C, Rheingold AL, Hartwig JF. J. Am. Chem. Soc. 1999;121:3224–3225. [Google Scholar]
- 10.(a) Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937–2940. [Google Scholar]; (b) Decicco CP, Song S, Evans DA. Org. Lett. 2001;3:1029–1032. doi: 10.1021/ol015572i. [DOI] [PubMed] [Google Scholar]
- 11.Chan DMT, Monaco KL, Wang R, Winters MP. Tetrahedron Lett. 1998;39:2933–2936. [Google Scholar]
- 12.(a) Combs AP, Rafalski M. J. Comb. Chem. 2000;2:29–32. doi: 10.1021/cc9900394. [DOI] [PubMed] [Google Scholar]; (b) Yin J, Buchwald SL. J. Am. Chem. Soc. 2002;124:6043–6048. doi: 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]
- 13.Deng B-L, Lepoivre JA, Lemiere G. Eur. J. Org. Chem. 1999:2683–2689. [Google Scholar]
- 14.Sekar G, Singh VK. J. Org. Chem. 1999;64:287–289. doi: 10.1021/jo981196c. [DOI] [PubMed] [Google Scholar]
- 15.(a) Yamamoto T. Synth. Commun. 1979;9:219–222. [Google Scholar]; (b) Ramesh C, Kubota Y, Miwa M, Sugi Y. Synthesis. 2002:2171–2173. [Google Scholar]
- 16.Beyer H, Walter W. Handbook of Organic Chemistry. Prentice Hall Europe; London-Munich: 1996. p. 497. [Google Scholar]
- 17.(a) Fischer E. Ber. 1895;28:3254–3257. [Google Scholar]; (b) Butts J. J. Am. Chem. Soc. 1931;53:3560–3561. [Google Scholar]
- 18.Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211–1214. [Google Scholar]
- 19.(a) Yoshida H, Shirakawa E, Honda Y, Hiyama T. Angew. Chem., Int. Ed. Engl. 2002;41:3247–3249. doi: 10.1002/1521-3773(20020902)41:17<3247::AID-ANIE3247>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Yoshida H, Fukushima H, Ohshita J, Kunai A. Angew. Chem., Int. Ed. 2004;43:3935–3938. doi: 10.1002/anie.200460009. [DOI] [PubMed] [Google Scholar]; (c) Yoshida H, Sugiura S, Kunai A. Org. Lett. 2002;4:2767–2769. doi: 10.1021/ol0262845. [DOI] [PubMed] [Google Scholar]
- 20.(a) Peña D, Escudero S, Pérez D, Guitián E, Castedo L. Angew. Chem., Int. Ed. Engl. 1998;37:2659–2661. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2659::AID-ANIE2659>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Yoshida H, Watanabe M, Fukushima H, Ohshita J, Kunai A. Org. Lett. 2004;6:4049–4051. doi: 10.1021/ol048298b. [DOI] [PubMed] [Google Scholar]
- 21.Beller M, Breindl C, Riermeier TH, Tillack A. J. Org. Chem. 2001;66:1403–1412. doi: 10.1021/jo001544m. [DOI] [PubMed] [Google Scholar]
- 22.(a) Liu Z, Larock RC. Org. Lett. 2003;5:4673–4675. doi: 10.1021/ol0358612. [DOI] [PubMed] [Google Scholar]; (b) Liu Z, Larock RC. Org. Lett. 2004;6:99–102. doi: 10.1021/ol0361406. [DOI] [PubMed] [Google Scholar]
- 23.Peña D, Pérez D, Guitián E, Castedo L. J. Am. Chem. Soc. 1999;121:5827–5828. [Google Scholar]
- 24.Yoshida H, Ikadai J, Shudo M, Ohshita J, Kunai A. J. Am. Chem. Soc. 2003;125:6638–6639. doi: 10.1021/ja034571d. [DOI] [PubMed] [Google Scholar]
- 25.Silylaryl triflate 1e was prepared from 2-bromo-4,6-dimethylphenol in a manner similar to the preparation of silylaryl triflate 1c.
- 26.4-Aminopyridine and 2-aminopyridine react with the silylaryl triflate 1a to afford an unknown salt; no N-arylated products were isolated under our usual reaction conditions.
- 27.Liu Z, Zhang X, Larock RC. J. Am. Chem. Soc. 2005;127:15716–15717. doi: 10.1021/ja055781o. [DOI] [PubMed] [Google Scholar]
- 28.Wittig G, Reichel B. Ber. 1963;96:2851–2858. [Google Scholar]
- 29.No simple N-arylation product from indole is isolated, when indole is allowed to react with silylaryl triflate 1a under our usual reaction conditions.
- 30.Kessar SV. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Pergamon Press; Oxford, England: 1991. pp. 483–515. [Google Scholar]
- 31.(a) He H, Wu YJ. Tetrahedron Lett. 2003;44:3385–3386. [Google Scholar]; (b) Yin J, Buchwald SL. Org. Lett. 2002;4:1101–1104. doi: 10.1021/ol005654r. [DOI] [PubMed] [Google Scholar]
- 32.Desmarets C, Schneider R, Fort Y. J. Org. Chem. 2002;67:3029–3036. doi: 10.1021/jo016352l. [DOI] [PubMed] [Google Scholar]
- 33.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J. Org. Chem. 2002;67:5553–5566. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida Y, Tanabe Y. Synthesis. 1997:533–535. [Google Scholar]
- 35.Haga K, Oohashi M, Kaneko R. Bull. Chem. Soc. Jpn. 1984;57:1586–1590. [Google Scholar]
- 36.Shi L, Wang M, Fan Ch.-A., Zhang F-M, Tu Y-Q. Org. Lett. 2003;5:3515–3517. doi: 10.1021/ol0353868. [DOI] [PubMed] [Google Scholar]
- 37.Ma D, Cai Q. Org. Lett. 2003;5:3799–3802. doi: 10.1021/ol0350947. [DOI] [PubMed] [Google Scholar]
- 38.Glatzhofer DT, Roy RR, Cossy KN. Org. Lett. 2002;4:2349–2352. doi: 10.1021/ol026051d. [DOI] [PubMed] [Google Scholar]
- 39.Lee CK, Yu JS, Kim SH. J. Heterocycl. Chem. 1998;35:835–841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.