

Abstract
Activation of c-Met signaling and β-catenin mutations are frequent genetic events observed in liver cancer development. Recently, we demonstrated that activated β-catenin can cooperate with c-Met to induce liver cancer formation in a mouse model. Cyclin D1 is an important cell cycle regulator that is considered to be a downstream target of β-catenin. To determine the importance of cyclin D1 as a mediator of c-Met and β-catenin induced hepatocarcinogenesis, we investigated the genetic interactions between cyclin D1, β-catenin and c-Met in liver cancer development using mouse models. We co-expressed cyclin D1 with c-Met in mice and found cyclin D1 to cooperate with c-Met to promote liver cancer formation. Tumors induced by cyclin D1/c-Met had a longer latency period, formed at a lower frequency, and appeared to be more benign compared to those induced by β-catenin/c-Met. In addition, when activated β-catenin and c-Met were co-injected into cyclin D1 null mice, liver tumors developed despite the absence of cyclin D1. Intriguingly, we observed a moderate accelerated tumor growth and increased tumor malignancy in these cyclin D1 null mice. Molecular analysis demonstrated an up-regulation of cyclin D2 expression in cyclin D1 null tumor samples, indicating that cyclin D2 may replace cyclin D1 in hepatic tumorigenesis. Together, our results suggest that cyclin D1 functions as a mediator of β-catenin during HCC pathogenesis, although other molecules may be required to fully propagate β-catenin signaling. Moreover, our data suggest that cyclin D1 expression is not essential for liver tumor development induced by c-met and β-catenin.
Keywords: HCC, Cyclin D1, Wnt pathway, β-catenin, c-Met
INTRODUCTION
Human hepatocellular carcinoma (HCC) is the most widespread and grievous form of malignancies to be diagnosed in adults (1). HCC is becoming a concern in developed nations, particularly the U.S., where at least 17,000 people are projected to die of liver cancer in 2008 (www.cancer.org). Patients with hepatitis B or C viral infections are at a higher risk of developing HCC, especially when the infection is accompanied by liver cirrhosis (2, 3). Liver transplantation and surgical resection are considered the most effective treatments of HCC. However, surgical treatments are only appropriate for a minority of patients. Therefore, there is an urgency to identify potential therapeutic targets for treatment of this deadly malignancy.
HCC progression is known to ensue a stepwise sequence of events (4). Separate genetic or epigenetic aberrations are thought to be involved in each step during hepatic carcinogenesis. These changes involve alterations in the expression or assembly of an oncogene or a tumor suppressor gene. Over-expression of the proto-oncogene c-Met is a common perturbation known to occur in HCC (5, 6). c-Met encodes a receptor tyrosine kinase, which becomes activated upon binding to ligand hepatocyte growth factor (HGF) or scatter factor (SF). When stimulated, c-Met becomes phosphorylated, and triggers MAPK signaling through the Ras-Raf-Mek pathway (7). It has been demonstrated in mouse models that activation of c-Met can promote liver cancer development (8). Another pathway frequently mutated and activated is the wnt/β-catenin signaling pathway. Wnt signals by binding to the frizzled family of receptors, which initiates a signaling cascade involving dishevelled, GSK3, Axin, APC, and regulates the nuclear localization and activation of β-catenin (9, 10). β-catenin subsequently binds to TCF-4, a member of the TCF/LEF family of transcriptional factors, and induces downstream gene expression. Multiple targets have been identified for activated β-catenin, many of which appear to be tissue specific (9, 10). One of the well-characterized targets for activated β-catenin is cyclin D1 (11, 12).
Cyclin D1 (CCND1) belongs to the D-type cyclin family, which also includes cyclin D2 and D3. CCND1 interacts with Cdk4/6, which in turn phosphorylates the RB protein, thereby promoting the transition from the G1 to S phase of the cell cycle (13, 14). CCND1 can cooperate with other oncogenes, including Ras, Src, and E1A to transform cells (15–17). It has been shown that over-expression of CCND1 in liver can induce HCC in a transgenic mouse model (18). However, CCND1 transgenic mice develop HCC over a long period of time (17 months) and at a relatively low frequency (20 to 30%) (18, 19). This observation suggests that over-expression of CCND1 alone may not be sufficient for HCC development, and that a secondary mutation may be necessary to cooperate with CCND1 to induce HCC. In CCND1 knockout mouse models, approximately 75% of CCND1−/− mice survive past their first month (20, 21). The surviving mice though appear smaller than normal and have some developmental defects in their retinas and mammary glands, are fertile and have a similar life span as wild type mice (20, 21). Using these CCND1 null mice, it has been found that CCND1 expression is required in certain tumor types in combination with different signaling pathways. For example, CCND1 is necessary for oncogenic Ras- and HER2-, but not wnt-1- or Myc-induced breast cancer (22). In colon cancer models induced by loss of APC, CCND1 is found to function as a tumor severity modifier and is required for efficient intestinal adenoma formation (23–25). Interestingly, in activated β-catenin induced breast cancer, loss of CCND1 accelerates tumor development (26). Currently, how CCND1 contributes to and how it interacts with other oncogenic signals during liver cancer development remains unknown.
In a recent study, we conducted genetic analyses of tumors induced by human c-Met (hMet) in a mouse model (27). We found that β-catenin mutations are the second hit during malignant transformation, and these constitutively active β-catenin mutants are required to cooperate with hMet to promote hepatic carcinogenesis (27). In this report, we confirmed CCND1 expression is upregulated in liver tumor samples induced by hMet and β-catenin. We studied genetic interactions among CCND1, hMet, and β-catenin in hepatocarcinogenesis using murine models. Our results provide novel insight into how D-type cyclins function to promote liver cancer development in vivo.
MATERIALS AND METHODS
Constructs and reagents
The pT3-EF1α-hMet and pT3-EF1α-ΔN90-β-catenin, as well as pCMV/SB (the hyperactive sleeping beauty expression vector) constructs used for animal injections were previously described (27). Human cyclin D1 was cloned into pT3-EF1α via the Gateway PCR cloning strategy (Invitrogen, Carlsbad, CA). All plasmids were purified using the endotoxin free maxi prep kit (Sigma, St. Louis, MO) before being injected into the mice.
Mice breeding, genotyping and hydrodynamic injections
Wildtype FVB/N mice were obtained from Charles River (Wilmington, MA), and cyclin D1+/− mice (in FVB/N background) were obtained from the Jackson Laboratory (Bar Harbor, Maine). CCND1+/− mice were bred together to obtain CCND1−/− mice, and the genotyping procedure was as described (21). The injected mice were monitored weekly, and sacrificed when appropriate or when they showed visibly enlarged livers or became moribund. All mice were housed, fed and treated in accordance with protocols approved by the committee for animal research at the University of California, San Francisco.
Hepatocyte isolation and transfection
Primary mouse hepatocyte isolation was performed using standard collagenase perfusion method as described (28). The hepatocytes were transfected with plasmids using Targefect-Hepatocyte (Targeting Systems, El Cajon, CA) according to the manufacturer’s instructions.
Histology
Animals were euthanized and their livers removed and rinsed in PBS. Samples collected from the livers were either immediately frozen for RNA and protein extraction or fixed overnight in freshly prepared cold 4% paraformaldehyde. Fixed tissue samples were embedded in paraffin. Five micron sections were placed on slides and stained with hematoxylin and eosin to observe morphology of the cells.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry staining was performed using ABC kit (Vector Laboratories, Burlingame, CA) as previously described (27). Immunofluorescence was performed in the similar manner, except that the appropriate Alexa labeled secondary antibody (Invitrogen) was applied following incubation with the primary antibody. Antibody dilutions were as follows: anti-β-catenin, 1:200, anti-E-cadherin, 1:1000 and anti-GS, 1:500 (BD Bioscience, San Jose, CA); anti-Ki67, 1:150 and anti-CCND1 (SP4) 1:75 (Lab vision, Fremont, CA).
Real-time RT-PCR
Total RNA was extracted from frozen liver tissues or primary hepatocytes using Trizol (Invitrogen) and digested with DNase I to remove genomic DNA contamination. Sybergreen based real-time RT-PCR was carried out as described (29) and rRNA was used as an internal control. Transcript quantification was performed in triplicate for every sample and reported relative to rRNA. The primer pair sequences are as described previously (29).
Preparation of lysates and western blotting
Liver tumors were frozen on dry ice upon harvesting, and homogenates were sonicated in lysis buffer (150 mM NaCl, 1.0% IGEPAL, 0.5% DOC, 0.1% SDS, 50 mM Tris (pH 8.0)) and centrifuged at 14,000 rpm at 4°C. Supernatant were boiled in Laemlli sample buffer for western blot analysis. The antibodies used are as follows: anti-Cyclin D1 (Ab3, 1:1000, Lab Vision, or C-20, 1:500, Santa Cruz Biotech); anti-Cyclin D2 (M-20, 1:500, Santa Cruz Biotech); anti-Cyclin D3 (C-16, 1:300, Santa Cruz Biotech.); anti-Cdk4 (C-22, 1:500, Santa Cruz Biotech); anti-Cdk6 (1:300, Santa Cruz Biotech.); anti-Cdk2 (M-2, 1:500, Santa Cruz Biotech); anti-GS, 1:1000; anti-actin 1:5000 (Sigma); anti-Erk, 1:1000; anti-phospho-Erk 1:1000; anti-phospho-Met 1:1000 (Cell signaling); and anti-V5 1:5000 (Invitrogen). Western blots were quantified using the ImageJ software (http://rsbweb.nih.gov/ij/).
Array based comparative genomic hybridization (aCGH)
Mouse CGH arrays were obtained from the UCSF Cancer Center Array Core. The arrays contained 2896 BAC clones spotted in triplicate, with an average spacing between clones of approximately 1MB. Array hybridization and data analyses were the same as previously described (30).
RESULTS
Cyclin D1 is induced in hMet/β-catenin tumors
In a recent study, we characterized a unique and efficient model of HCC in the mouse, driven by the inducible expression of the human receptor tyrosine kinase c-Met (hMet). Tumors arise sporadically in livers of such mice and all show mutation and activation of β-catenin (27). Using hydrodynamic transfection, we demonstrated that while neither hMet or activated β-catenin alone is able to promote HCC development, the cooperation between aberrant β-catenin signaling and activated Met is required for genesis of HCC in the mouse model (27).
To determine the molecular mechanisms of how activated β-catenin contributes to hepatic carcinogenesis, we searched for genes that are up-regulated by activated β-catenin in the mouse HCC samples. We determined the expression levels of candidate β-catenin targets: Axin 2, glutamine synthetase (GS), cyclin D1 (CCND1), and c-myc, by quantitative real time RT-PCR in 5 paired HCC and non-tumor liver tissues from hMet transgenic mice (Fig. 1A). We chose to use the liver samples from hMet transgenic mice instead of those from hMet and β-catenin injected mice because in hMet transgenic mice, non-tumor liver tissues also overexpress hMet.
Figure 1. Cyclin D1 is induced in hMet/β-catenin tumors.

(A) Quantitative real-time RT-PCR analyses of β-catenin candidate target genes Axin2, GS, CCND1, and c-Myc in 5 paired HCC and non-tumor liver tissues from hMet transgenic mice. Values are displayed as log2 ratio of tumor versus non-tumor liver. (B) Immunohistochemical staining in normal (left), HCC from hMet transgenic (middle) or HCC from hydrodynamic co-transfection of hMet and ΔN90-β-catenin (right). First row: H&E staining; Second row: β-catenin; Third row: glutamine synthetase (GS); and Fourth row: CCND1. (C) Representative Western blots showing GS and CCND1 expression in two paired tumor and non-tumor liver samples of hMet transgenic mice. Actin was used as the loading control.
Therefore, the genes that are up-regulated in tumors versus non-tumor tissues are most likely due to β-catenin activation. As expected, both Axin2 and GS expressions were approximately 23 and 38-fold higher in HCC samples than in surrounding non-tumor liver tissue. CCND1 was also found to be expressed 7-fold higher in HCC than in non-tumor liver tissue. However, c-Myc expression levels did not show significant differences (Fig. 1A).
To further validate our observations, we assayed the protein expression of CCND1 and GS. Immunohistochemical staining of liver tumor tissues revealed nuclear staining of β-catenin in HCC lesions, indicating the activation of β-catenin (Fig. 1B). In normal liver, GS was found to be only expressed in hepatocytes immediately adjacent to the central vein. In contrast, in HCC, GS expression was detected in virtually all malignant hepatocytes (Fig. 1B). Analysis of CCND1 expression revealed there was little or weak expression of CCND1 in normal mouse liver cells. However, strong nuclear staining of CCND1 was apparent in the tumor samples (Fig. 1B). The expression patterns of these genes are similar for all tumors from hMet transgenic mice and hMet/β-catenin injected mice (Fig. 1B). The up-regulation of CCND1 and GS in tumor tissues was also confirmed by Western blotting (Fig. 1C).
Our data suggest that CCND1 expression is induced by β-catenin during hepatic carcinogenesis. We next examined if this occurrence also takes place in normal hepatocytes. Our data suggest that CCND1 expression is induced by β-catenin during hepatic carcinogenesis. We next examined if this occurrence also takes place in normal hepatocytes. To access the induction of CCND1 by β-catenin, we expressed an activated form of β-catenin, ΔN90-β-catenin, into mice using hydrodynamic transfection, and their expression levels were analyzed using immunofluorescent staining. We found that while sporadic activated β-catenin staining could be visualized in normal hepatocytes, no CCND1 expression was detected in the same cells (Supplemental Fig. 1A). Consistent with our previous results, we observed co-localization of β-catenin and CCND1 in tumor cells induced by c-Met/ΔN90-β-catenin (Sup. Fig. 1A). To confirm our findings, we transfected primary mouse hepatocytes with ΔN90-β-catenin (Sup. Fig. 1B). While we can detect strong expression of the β-catenin target gene Axin2, no up-regulation of CCND1 was observed (Sup. Fig. 1C). These experiments suggest that CCND1 is not a direct target for β-catenin in normal hepatocytes, but rather CCND1 expression is induced during hepatic carcinogenesis.
In conclusion, we found that CCND1 expression is up-regulated in mouse liver tumors induced by hMet and β-catenin. Consistent with our observation, over-expression of CCND1, but not c-Myc, has been reported in two HCC mouse models involving the activation of β-catenin: conditional APC knockout mice and RasV12 and β-catenin double conditional transgenic mice (31, 32). Since CCND1 is an important factor in the regulation of cell cycle progression, we hypothesized that CCND1 may be a key mediator of β-catenin in promoting hepatic carcinogenesis.
Cyclin D1 cooperates with hMet to induce HCC in mice
To test the hypothesis that cyclin D1 is a mediator of activated β-catenin signaling during hepatic carcinogenesis, we investigated whether overexpression of CCND1 can substitute for activated β-catenin and cooperate with hMet to induce HCC in mice. We used hydrodynamic transfection to co-express CCND1 and hMet in mouse liver. Histological examination revealed that none of the nine mice that were injected with hMet/CCND1 showed any signs of tumor development between 11 to 22 weeks post injection (Fig. 2). Tumor formation was first observed in these mice at 25 weeks post injection. In total, 6 of the 19 mice injected with CCND1 and hMet developed liver tumors within 29 weeks post injection (Fig 2). Gross examination of livers of these mice also showed multiple small lesions scattered throughout the surface of the liver (Sup. Fig. 2). In contrast, mice expressing only hMet (n=9) or CCND1 alone (n=10) failed to develop liver cancer during this time period (Fig. 2). The fact that tumors were only observed when CCND1 and hMet are co-expressed suggests that over-expression of CCND1 cooperates with hMet to promote liver cancer formation.
Figure 2. Cyclin D1 cooperates with hMet to induce HCC in mice.
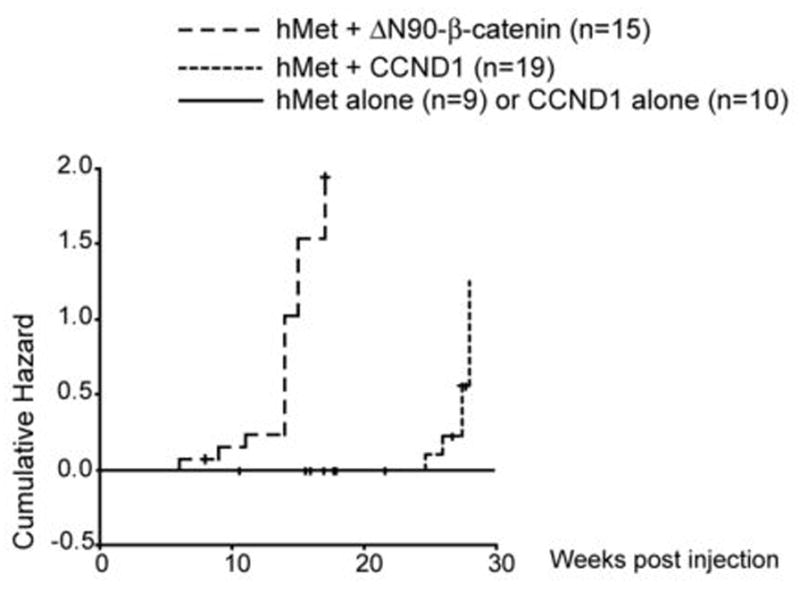
Cumulative hazard curve comparing the latency and frequency of tumor development in hMet alone, CCND1 alone, hMet/CCND1, and hMet/ΔN90-β-catenin injected mice. The cumulative hazard represents the relative probability of tumor development in each condition.
We found that co-expression of hMet and ΔN90-β-catenin induces HCC in 12 out of 15 mice within 17 weeks post injection (Fig. 2), much earlier than mice injected with CCND1 and hMet. We also observed that the frequency of tumor development in hMet and ΔN90-β-catenin (80%) was higher than that of hMet and CCND1 injected mice (32%) (Fig. 2). Together the data suggest that tumor initiation is likely to be much later in hMet/CCND1 injected mice compared with hMet/β-catenin injected mice.
Molecular features of liver tumors induced by CCND1/hMet
To gain further insight into the molecular features of liver tumors induced by hMet/CCND1, we examined tumor samples using histological analysis and quantitative RT-PCR. We found upregulation of liver tumor specific marker AFP in hMet/CCND1 tumor samples (Fig. 3C), confirming the neoplastic nature of these cells. Histological examination of liver tissue from hMet/CCND1 injected mice revealed that the majority of the tumors appeared to be adenoma that compressed the surrounding non-tumorous liver parenchyma (Fig. 3A). Increased plate thickness and trabecular disorganization were rarely observed in these tumors (Fig. 3A). This is distinct from tumors found in hMet transgenic mice or mice injected with hMet/ΔN90-β-catenin, where a majority of the tumors display features consistent with malignant HCC (Fig. 1B). Nuclear staining of CCND1 was observed, revealing the presence of CCND1 in neoplastic hepatocytes, but rarely in non-tumor liver tissues (Fig. 3A). The level of CCND1 expression in hMet/CCND1 tumor cells is similar to what we observed in tumors from hMet/ΔN90-β-catenin injected mice. Over-expression of hMet (with c-terminal V5 tag) was confirmed by Western blotting using the V5 antibody (Fig. 3B); and the activation of hMet as illustrated by the high levels of phospho-Met and phospho-Erk in hMet/CCND1 tumor cells (Fig. 3B). To rule out the possibility that the tumors induced by hMet/CCND1 were due to endogenous mutations of β-catenin, we analyzed β-catenin and its target gene expression in tumor samples. We found no evidence of activation of β-catenin signaling in tumor cells, as only membrane β-catenin staining was observed (Fig. 3A), and no up-regulation of β-catenin target genes Axin2 and GS was detected by real-time RT-PCR (Fig 3C and data not shown). Altogether, these analyses support our hypothesis that hMet and cyclin D1 together promote liver adenoma formation in mice.
Figure 3. Molecular features of liver tumors induced by hMet/CCND1.

(A) Immunohistochemical staining of liver tumor tissues from hMet/CCND1 mice: H&E, CCND1, β-catenin and Ki67 staining (left to right). (B) Western blot analyses showing the expression of V5 tagged hMet, activation of hMet (phospho-Met) and elevated MAPK signaling (phospho-Erk) in hMet/CCND1 tumor cells; (C) Quantitative real time-PCR analyses of liver tumor markers in normal liver (NT), hMet/CCND1 tumors and hMet/ΔN90-β-catenin tumor samples. In all cases, the expression in normal liver was set to 1 and used to normalize all the other samples.
Next, we examined the genes involved in cell cycle, apoptosis and cell adhesion in CCND1/hMet tumor samples. We observed frequent cell proliferation in tumor cells as indicated by positive staining for the proliferative marker, Ki67 (Fig. 3A). This observation is also confirmed by the high expression of cell cycle regulatory genes, cyclins B1 and E1, as well as Cdk inhibitor p21Cip1 in the tumors (Fig. 3C). In addition, tumor cells expressed high levels of the anti-apoptotic protein, survivin, as well as the angiogenic gene, Ang2 (Fig. 3C). Immunostaining revealed that tumor cells are positive for the cell-cell adhesion molecule, E-cadherin (data not shown). This observation is confirmed by up-regulation of E-cadherin as indicated by real-time PCR analysis (Fig. 3C). While there are striking differences in tumor latency and incidence between the hMet/ΔN90-β-catenin and hMet/CCND1 injected mice, molecular features of both tumors are similar to a certain extent. For example, elevated expressions of cyclins, survivin, Ang2, E-cadherin, and p21Cip1 were observed in all tumor samples examined (Fig. 3C).
In conclusion, our study demonstrates that over-expression of CCND1 can cooperate with hMet to promote liver cancer formation, supporting the hypothesis that CCND1 is a critical downstream signaling molecule of activated β-catenin. However, the differences between tumors induced by hMet/CCND1 and hMet/ΔN90-β-catenin indicate that other factors, in addition to CCND1, may be required to fully transduce the signaling generated by aberrant β-catenin.
CCND1 expression is not required for tumor development induced by activated β-catenin plus hMet
Since CCND1 expression is induced by hMet and β-catenin in liver tumors, we next determined whether the expression of CCND1 is required for activated β-catenin and hMet to promote HCC development in vivo. Towards this aim, we generated CCND1 knockout mice in the FVB/N background. These CCND1 null mice show similar phenotypes as described in the C57/BL6 background (20, 21). Liver tissues appeared to be normal in these mice. ΔN90-β-catenin and hMet were co-injected into CCND1+/− and CCND1−/− mice, and tumor development was monitored.
We found that hMet and ΔN90-β-catenin could induce liver cancer development independent of the CCND1 genotype. In particular, we observed 7 out of 8 CCND1−/− mice developed tumors within 11 weeks post injection; 8 out of 10 CCND1+/− mice developed tumors 14 weeks post injection, and 12 out of 15 wild type mice developed tumors 17 weeks post injection (Fig. 4). Intriguingly, we noticed that the CCND1 gene dosage affected the onset and progress of hepatocarcinogenesis: tumors progressed earlier with lower CCND1 gene doses, i.e., CCND1−/− mice demonstrated the fastest tumor progression, whereas wild type mice showed the longest latency time for tumor development (Fig. 4). Tumors from CCND1−/− or CCND1+/− mice are multi-focal and scattered around the liver, similar to what has been observed in wild type FVB/N mice (Sup. Fig. 2).
Figure 4. Accelerated tumor development induced by hMet/ƒΔN90-β-catenin in cyclin D1 knockout mice.
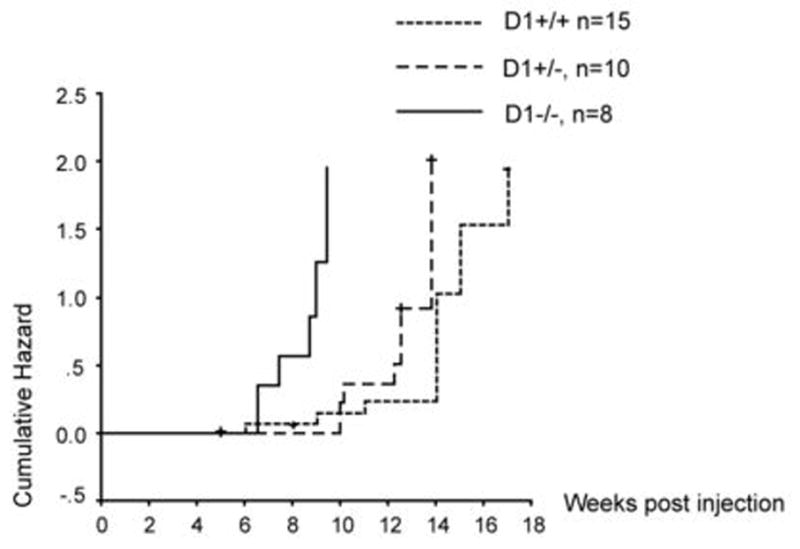
Cumulative hazard curve comparing the latency and frequency of tumor development induced by hMet/ΔN90-β-catenin in wildtype, CCND1+/− or CCND1−/− mice. The cumulative hazard represents the relative probability of tumor development in each condition.
Histological analyses revealed tumor lesions from CCND1+/− and CCND1−/− mice emerge as hepatocellular carcinoma with cytological atypia and frequent trabecular disorganization (Fig. 5A and data not shown). This is further verified by high expression levels of AFP in these tumor samples (Fig. 5D). Ectopic expression of β-catenin is validated by nuclear and cytoplasmic staining in tumor cells (Fig. 5A). Expression of hMet in tumor samples is indicated by the presence of the V5 marker. The activation of hMet signaling is confirmed by an increase in the expression of phospho-Met and phospho-Erk (Fig. 5C). CCND1 expression was not detected in tumors from CCND1−/− mice by immunohistochemical staining or Western blotting (Fig. 5A and Fig. 6A).
Figure 5. Molecular features of liver tumors induced by hMet/ƒΔN90-β-catenin in different Cyclin D1 genetic backgrounds.
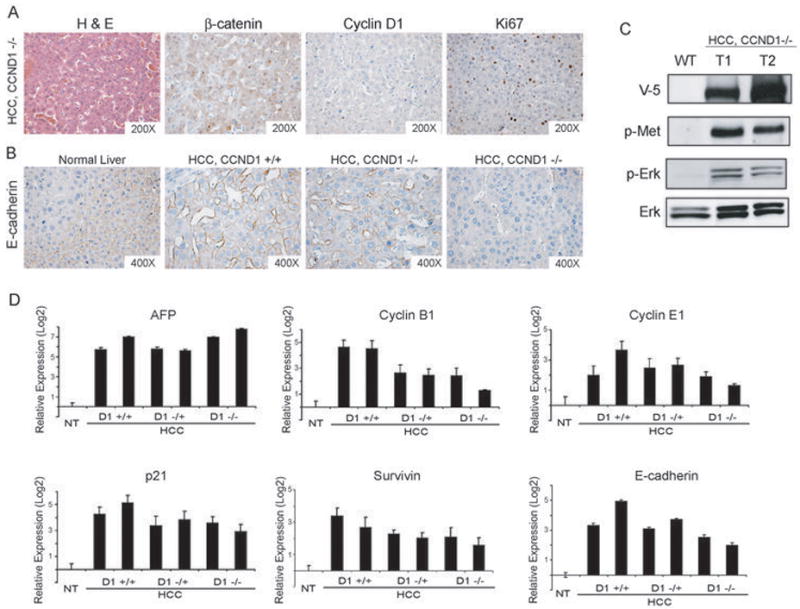
(A) Immunohistochemical staining of liver tumor tissues from hMet/ΔN90-β-catenin injected CCND1−/− mice: H&E, β-catenin, CCND1, and Ki67 staining (left to right). (B) Immunohistochemical staining of E-cadherin expression in normal liver, liver tumors from wildtype or CCND1−/− mice; (C) Western blot analyses showing the expression of V5 tagged hMet, activation of hMet (phospho-Met), and elevated MAPK signaling (phospho-Erk) in hMet/ΔN90-β-catenin;CCND1−/− tumor cells (D) Quantitative real time-PCR analyses of tumor markers in normal liver, liver tumors from wildtype, CCND1+/− or CCND1−/− mice. In all cases, the expression in normal liver was set to 1 and used to normalize all the other samples.
Figure 6. Expression in D-type cyclins and Cdks in mouse liver tissues.
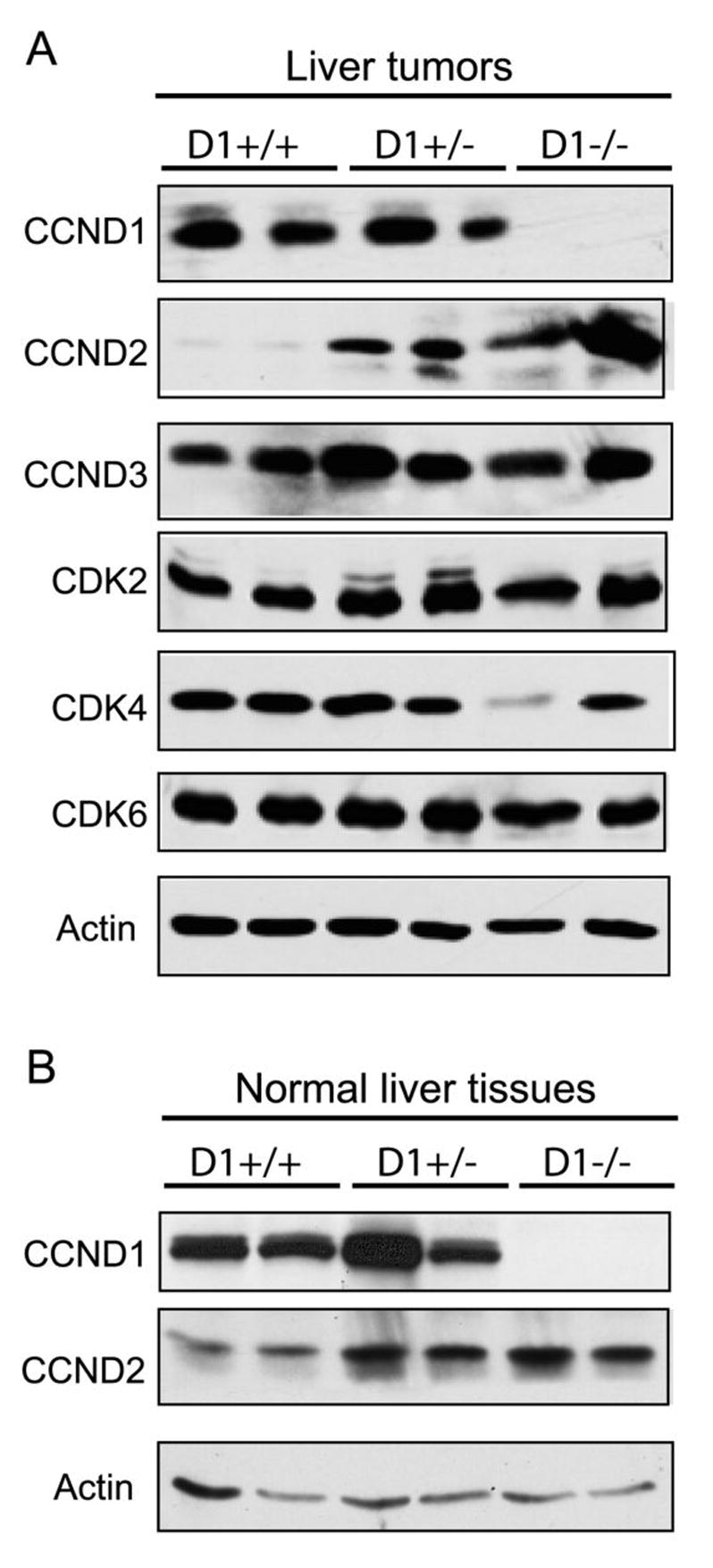
(A) Western blotting analysis of the expression of CCND1, CCND2, CCND3, Cdk2, Cdk4 and Cdk6 in liver tumor samples with different CCND1 genetic backgrounds. (B) Expression of CCND1 and CCND2 in normal liver tissues from mice with different CCND1 genetic backgrounds. Actin was used as the loading control.
We further assayed the molecular signatures of tumors from different CCND1 genetic backgrounds. Increased cell proliferation was detected in tumors from CCND1−/− mice, as indicated by positive Ki67 staining (Fig.5A). The over-expression of cyclin B1, E1, and Cdk inhibitor p21Cip1 in tumors from CCND1−/− as well as CCND1+/− and wildtype mice further verify this observation (Fig.5D). The anti-apoptotic gene survivin is also found to be over-expressed in all tumor samples (Fig. 5D).
We next examined the expression of the cell-cell adhesion molecule, E-cadherin. In the normal liver, hepatocytes show weak staining of E-cadherin around the periportal area (Fig. 5B) (33). Tumors induced by hMet and β-catenin in wild type mice displayed ubiquitous expression of E-cadherin in all tumor cells (Fig. 5B). In contrast, there appeared to be a heterogeneous pattern of E-cadherin expression in tumors from CCND1−/− mice: while some tumor nodules retained E-cadherin staining, others showed no expression (Fig. 5B). Because loss of E-cadherin has been linked with malignant phenotype, our data indicates that tumors from cyclin D1 null mice show moderate accelerated tumor growth and increased malignancy.
A study by Calvisi et. al. demonstrated that mouse liver tumors with β-catenin activation have a stable genome (34). Consistent with this observation, we found that tumors induced by hMet and ΔN90-β-catenin also have a stable genome, with no abnormal chromosomal gains or losses (Sup. Fig. 4). We examined genomic instability in tumor samples from CCND1−/− mice using array based comparative genomic hybridization. Similar to what we observed in wildtype mice, liver tumors induced by hMet and ΔN90-β-catenin in CCND1−/− mice have no genomic instabilities (Sup. Fig. 4). The study therefore suggests that the accelerated tumor growth in CCND1−/− mice is not due to the increased genomic instability of these tumor samples.
Up-regulation of CCND2 in CCND1 null liver and liver tumor samples
Our preliminary studies demonstrate that mRNA of all three members of the D-type cyclin family is expressed in the mouse liver (data not shown). To determine whether the loss of cyclin D1 was compensated by the other two D-type cyclins, we assayed for the protein expression of cyclin D2 and D3 in tumor tissues from wildtype, CCND1+/− and CCND1−/− mice. Strikingly we observed that while CCND2 is expressed at very low levels in wildtype tumor samples, its expression is significantly up-regulated in tumors from CCND1+/− and CCND1−/− mice (Fig. 6A). The expression of CCND3 remains the same in all tumor samples. We further examined the expression of the D-type cyclin partners, Cdk4 and Cdk6, as well as Cdk2, which binds to cyclin E and A. We found the expression of Cdk2 and Cdk6 to be independent of tumor genotypes. However, the expression of Cdk4, a major signaling partner of CCND1, in CCND1−/− mice tumors is significantly decreased by 60% in comparison to tumors from wildtype mice (Fig. 6A and Sup. Fig.3).
We next determined whether the up-regulation of CCND2 also compensates for the loss of CCND1 during normal liver development, or occurs only during tumorigenesis. We assayed the expression of CCND1 and CCND2 in normal wildtype liver and in the liver of CCND1 null mice. We found that CCND2 expression is increased in normal liver samples compared to CCND1+/− and CCND1−/− mice (Fig. 6B). The results suggest that CCND2 replaces CCND1 function when the CCND1 gene is deleted in the liver and the CCND2/Cdk6 complex replaces the CCND1/Cdk4 complex during hepatic carcinogenesis.
DISCUSSION
In this manuscript, we showed that CCND1 expression is up-regulated in liver tumors induced by hMet/β-catenin. Consistent with the studies by Cadoret A. and colleagues (35), we found that CCND1 is not a direct target of activated β-catenin in normal mouse hepatocytes. On the other hand, over-expression of CCND1 has been found to be correlated with β-catenin activation in multiple mouse liver tumor models (31, 32, 36). Recently, Zeng G. et. al. reported that RNA-mediated β-catenin knockdown in human HCC cell lines with activated β-catenin mutations leads to decreased expression of CCND1 (37). Together, all the data suggest that CCND1 is likely to be induced by activated β-catenin during hepatic carcinogenesis. Our experiments demonstrate that CCND1 can partially substitute activated β-catenin and cooperates with hMet to induce liver cancer formation in vivo, thus providing additional evidence of CCND1 as a target of β-catenin during malignant transformation.
While co-expression of CCND1 and hMet can induce liver cancer formation in mice, we found that the tumors induced by CCND1/hMet require longer latency, form at a lower frequency, and appear to be more benign compared with tumors induced by β-catenin/hMet. These observations indicate that CCND1 is only part of the signaling triggered by β-catenin activation, and other molecules may be required to fully transduce the aberrant β-catenin signaling. Other targets of β-catenin include Tbx3 (38) and Gpr49 (39), both found to be up-regulated in liver tumors induced by hMet/β-catenin (Patil M.A. et al., unpublished results). Tbx3, a member of the T-box transcriptional repressor family, has been found be over-expressed in melanoma, breast cancer, and ovarian cancer (40–42). In addition, Tbx3 has been shown to be a potent inhibitor of p19Arf and hence, a regulator of the p19Arf-MDM2-p53 pathway (43). Thus, Tbx3 may provide a novel link between activated β-catenin and p19Arf tumor suppressor pathways. Gpr49, also known as Lgr5, is an orphan G protein-coupled receptor. Gpr49 has been found to be over-expressed in human colon and ovarian tumors and is proven to be a marker for intestinal stem cells (44, 45). Altogether, it would be of great importance to elucidate how Tbx3, Grp49, and CCND1 function together and mimic the activity of β-catenin in cooperation with c-Met to induce liver cancer.
In our study, we have shown that expression of CCND1 is not required in murine HCC pathogenesis induced by activated β-catenin and hMet. Therefore, while CCND1 expression is upregulated in liver cancer cells, cyclin D1 expression appears to be dispensable for c-Met and β-catenin’s activity in liver tumor development. Unexpectedly, we observed that hMet and ΔN90-β-catenin induced tumor development was accelerated by the loss of cyclin D1. Tumor cells appeared to be more aggressive, with frequent E-cadherin negative tumors present in the HCCs of CCND1−/− mice. Interestingly, increased breast tumorigenesis has also been observed in CCND1−/− mice when they were crossed with mice expressing activated β-catenin targeted to the mammary gland (26). Our study provides additional evidence that cyclin D1 plays divergent roles under different oncogenic signals and in diverse cell types. For each specific oncogenic signal and each cell type, one has to assay for the tumorigenic activity of the oncogene in a CCND1 null background in order to elucidate the requirements of cyclin D1 in the specific circumstance.
What are the molecular mechanisms for the accelerated and more aggressive phenotype observed in CCND1 knockout mice? One possible clue comes from our investigation of the expression of other D-type cyclins and Cdks in the CCND1−/− tumor samples. Increased expression of CCND2 in CCND1+/− and CCND1−/− tumors strongly suggests that this member of D-type cyclin family can replace cyclin D1. In addition, lack of cyclin D1 seems to decrease the Cdk4 protein level, likely through reduced stability of free Cdk4. Whether CCND2/Cdk6 is more efficient in the phosphorylation of Rb than CCND1/Cdk4 during liver tumor development warrants further investigation, and may provide functional roles for CCND2 in hepatic carcinogenesis. A recent study supports a positive role for cyclin D2 in tumorigenesis, where cyclin D2 transgenic mice are more susceptible to developing skin tumors, a characteristic that is not shared by cyclin D1 and D3 transgenic mice generated under the same promoter (46, 47). In addition, D-type cyclins play Cdk-independent roles in certain cell types. For example, D-type cyclins bind to nuclear receptors such as androgen, estrogen, and vitamin D receptors to regulate the expression of several genes in prostate, mammary gland and skin keratinocytes (48, 49). While the interaction of D-type cyclins with nuclear receptors has not been described in liver, whether cyclin D1 acts through this pathway during liver tumorigenesis clearly needs to be evaluated. A third possibility is that D-type cyclins may modulate tumorigenesis via regulating cell types other than hepatocytes. For example, D type cyclins may regulate tumor immunity or angiogenesis, and the more rapid tumor growth in CCND1 null mice may be due to the reduced immune response or more robust angiogenesis since CCND1 is deleted in all cell types in mice. This hypothesis can be tested by generating hepatocyte specific deleted CCND1 mice using the albumin Cre system. If we fail to observe this accelerated tumor growth phenotype in these mice, the result will support an additional non-hepatocyte role of D type cyclins during HCC pathogenesis.
It has been speculated that small molecules targeted against CCND1/Cdk4 may be useful as therapeutic reagents against human tumors. However, our study and the study by Rowlands et al suggest that we need to be cautious about such treatments, since it could lead to unfavorable consequences under certain conditions. For example, CCND1/Cdk4 inhibitors may not be suitable for patients who have chronic HBV or HCV infection, as these patients are at a greater risk of developing HCC, and loss of CCND1/CDK4 activity may accelerate the progression of this malignancy.
Supplementary Material
Acknowledgments
We would like to thank Sandra Huling of the UCSF Liver Center Morphology Core for histology support; Ali Naqvi of the Cell and Tissue biology Core for isolating mouse hepatocytes; and Greg Hamilton of the UCSF Cancer Center Array Core for array CGH analysis. We would also like to thank Aaron Tward, Holger Willenbring and Andrei Goga for helpful discussion. This work is supported by NIH K01CA096774 and R21CA131625 to X.C, R01CA116328 to M.R-P, as well as P30DK026743 to the liver center.
Abbreviations
- HCC
hepatocellular carcinoma
- hMet
human c-Met
- CCND1
Cyclin D1
- AFP
alpha-fetoprotein
- IHC
Immunohistochemistry
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag H. Epidemiology of hepatocellular carcinoma. Clinics in Liver Disease. 2001;5:87–107. doi: 10.1016/s1089-3261(05)70155-0. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB. Hepatocellular carcinoma: an epidemiologic view. J Clin Gastroenterol. 2002;35:S72–8. doi: 10.1097/00004836-200211002-00002. [DOI] [PubMed] [Google Scholar]
- 4.Feitelson MA, Sun B, Satiroglu Tufan NL, Liu J, Pan J, Lian Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene. 2002;21:2593–604. doi: 10.1038/sj.onc.1205434. [DOI] [PubMed] [Google Scholar]
- 5.Ueki T, Fujimoto J, Suzuki T, Yamamoto H, Okamoto E. Expression of hepatocyte growth factor and its receptor, the c-met proto-oncogene, in hepatocellular carcinoma. Hepatology. 1997;25:619–23. doi: 10.1002/hep.510250321. [DOI] [PubMed] [Google Scholar]
- 6.Kaposi-Novak P, Lee JS, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006;116:1582–95. doi: 10.1172/JCI27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 8.Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. 2001;153:1023–34. doi: 10.1083/jcb.153.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cadigan KM. Wnt signaling--20 years and counting. Trends Genet. 2002;18:340–2. doi: 10.1016/s0168-9525(02)02707-5. [DOI] [PubMed] [Google Scholar]
- 10.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 12.Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–47. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 14.Ewen ME, Lamb J. The activities of cyclin D1 that drive tumorigenesis. Trends Mol Med. 2004;10:158–62. doi: 10.1016/j.molmed.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Lee RJ, Albanese C, Stenger RJ, et al. pp60(v-src) induction of cyclin D1 requires collaborative interactions between the extracellular signal-regulated kinase, p38, and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp60(v-src) signaling in breast cancer cells. J Biol Chem. 1999;274:7341–50. doi: 10.1074/jbc.274.11.7341. [DOI] [PubMed] [Google Scholar]
- 16.Robles AI, Rodriguez-Puebla ML, Glick AB, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–74. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lovec H, Sewing A, Lucibello FC, Muller R, Moroy T. Oncogenic activity of cyclin D1 revealed through cooperation with Ha-ras: link between cell cycle control and malignant transformation. Oncogene. 1994;9:323–6. [PubMed] [Google Scholar]
- 18.Deane NG, Parker MA, Aramandla R, et al. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001;61:5389–95. [PubMed] [Google Scholar]
- 19.Deane NG, Lee H, Hamaamen J, et al. Enhanced tumor formation in cyclin D1 x transforming growth factor beta1 double transgenic mice with characterization by magnetic resonance imaging. Cancer Res. 2004;64:1315–22. doi: 10.1158/0008-5472.can-03-1772. [DOI] [PubMed] [Google Scholar]
- 20.Sicinski P, Donaher JL, Parker SB, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–30. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 21.Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995;9:2364–72. doi: 10.1101/gad.9.19.2364. [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 23.Wilding J, Straub J, Bee J, et al. Cyclin D1 is not an essential target of beta-catenin signaling during intestinal tumorigenesis, but it may act as a modifier of disease severity in multiple intestinal neoplasia (Min) mice. Cancer Res. 2002;62:4562–5. [PubMed] [Google Scholar]
- 24.Hulit J, Wang C, Li Z, et al. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sansom OJ, Reed KR, van de Wetering M, et al. Cyclin D1 is not an immediate target of beta-catenin following Apc loss in the intestine. J Biol Chem. 2005;280:28463–7. doi: 10.1074/jbc.M500191200. [DOI] [PubMed] [Google Scholar]
- 26.Rowlands TM, Pechenkina IV, Hatsell SJ, Pestell RG, Cowin P. Dissecting the roles of beta-catenin and cyclin D1 during mammary development and neoplasia. Proc Natl Acad Sci U S A. 2003;100:11400–5. doi: 10.1073/pnas.1534601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tward AD, Jones KD, Yant S, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelsen CJ, Rickheim DG, Tucker MM, Hansen LK, Albrecht JH. Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J Biol Chem. 2003;278:3656–63. doi: 10.1074/jbc.M209374200. [DOI] [PubMed] [Google Scholar]
- 29.Lee SA, Ho C, Roy R, et al. Integration of genomic analysis and in vivo transfection to identify sprouty 2 as a candidate tumor suppressor in liver cancer. Hepatology. 2007 doi: 10.1002/hep.22169. [DOI] [PubMed] [Google Scholar]
- 30.Patil MA, Chua MS, Pan KH, et al. An integrated data analysis approach to characterize genes highly expressed in hepatocellular carcinoma. Oncogene. 2005;24:3737–47. doi: 10.1038/sj.onc.1208479. [DOI] [PubMed] [Google Scholar]
- 31.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 32.Colnot S, Decaens T, Niwa-Kawakita M, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–21. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hailfinger S, Jaworski M, Braeuning A, Buchmann A, Schwarz M. Zonal gene expression in murine liver: lessons from tumors. Hepatology. 2006;43:407–14. doi: 10.1002/hep.21082. [DOI] [PubMed] [Google Scholar]
- 34.Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004;126:1374–86. doi: 10.1053/j.gastro.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Cadoret A, Ovejero C, Saadi-Kheddouci S, et al. Hepatomegaly in transgenic mice expressing an oncogenic form of beta-catenin. Cancer Res. 2001;61:3245–9. [PubMed] [Google Scholar]
- 36.Gotoh J, Obata M, Yoshie M, Kasai S, Ogawa K. Cyclin D1 over-expression correlates with beta-catenin activation, but not with H-ras mutations, and phosphorylation of Akt, GSK3 beta and ERK1/2 in mouse hepatic carcinogenesis. Carcinogenesis. 2003;24:435–42. doi: 10.1093/carcin/24.3.435. [DOI] [PubMed] [Google Scholar]
- 37.Zeng G, Apte U, Cieply B, Singh S, Monga SP. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9:951–9. doi: 10.1593/neo.07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renard CA, Labalette C, Armengol C, et al. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 2007;67:901–10. doi: 10.1158/0008-5472.CAN-06-2344. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto Y, Sakamoto M, Fujii G, et al. Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology. 2003;37:528–33. doi: 10.1053/jhep.2003.50029. [DOI] [PubMed] [Google Scholar]
- 40.Fan W, Huang X, Chen C, Gray J, Huang T. TBX3 and its isoform TBX3+2a are functionally distinctive in inhibition of senescence and are overexpressed in a subset of breast cancer cell lines. Cancer Res. 2004;64:5132–9. doi: 10.1158/0008-5472.CAN-04-0615. [DOI] [PubMed] [Google Scholar]
- 41.Vance KW, Carreira S, Brosch G, Goding CR. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65:2260–8. doi: 10.1158/0008-5472.CAN-04-3045. [DOI] [PubMed] [Google Scholar]
- 42.Lomnytska M, Dubrovska A, Hellman U, Volodko N, Souchelnytskyi S. Increased expression of cSHMT, Tbx3 and utrophin in plasma of ovarian and breast cancer patients. Int J Cancer. 2006;118:412–21. doi: 10.1002/ijc.21332. [DOI] [PubMed] [Google Scholar]
- 43.Lingbeek ME, Jacobs JJ, van Lohuizen M. The T-box repressors TBX2 and TBX3 specifically regulate the tumor suppressor gene p14ARF via a variant T-site in the initiator. J Biol Chem. 2002;277:26120–7. doi: 10.1074/jbc.M200403200. [DOI] [PubMed] [Google Scholar]
- 44.McClanahan T, Koseoglu S, Smith K, et al. Identification of overexpression of orphan G protein-coupled receptor GPR49 in human colon and ovarian primary tumors. Cancer Biol Ther. 2006;5:419–26. doi: 10.4161/cbt.5.4.2521. [DOI] [PubMed] [Google Scholar]
- 45.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 46.Rojas P, Cadenas MB, Lin PC, Benavides F, Conti CJ, Rodriguez-Puebla ML. Cyclin D2 and cyclin D3 play opposite roles in mouse skin carcinogenesis. Oncogene. 2007;26:1723–30. doi: 10.1038/sj.onc.1209970. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez-Puebla ML, LaCava M, Conti CJ. Cyclin D1 overexpression in mouse epidermis increases cyclin-dependent kinase activity and cell proliferation in vivo but does not affect skin tumor development. Cell Growth Differ. 1999;10:467–72. [PubMed] [Google Scholar]
- 48.Jian Y, Yan J, Wang H, et al. Cyclin D3 interacts with vitamin D receptor and regulates its transcription activity. Biochem Biophys Res Commun. 2005;335:739–48. doi: 10.1016/j.bbrc.2005.07.141. [DOI] [PubMed] [Google Scholar]
- 49.Weigel NL, Moore NL. Cyclins, cyclin dependent kinases, and regulation of steroid receptor action. Mol Cell Endocrinol. 2007;265–266:157–61. doi: 10.1016/j.mce.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.