

Abstract
Using microarrays made with cDNAs from subtracted placental libraries, we show increased hemoglobin production in the preeclamptic placenta. Heme and hemoglobin may cause endothelial damage and inflammation and drive pathological changes in the placenta if they are released there.
Study Objective:
To create a library enriched in cDNAs from preeclamptic placentas to print on to microarrays for placental profiling of preeclampsia (PE) and high risk pregnancies.
Design:
Prospective study.
Setting:
University women's clinic and academic research laboratory.
Patients:
Ten patients with PE, 5 with PE and bilateral notching, 5 with bilateral notching without PE, and 15 normotensive patients were recruited.
Interventions:
Placenta and placenta bed biopsies were collected after delivery.
Main Outcome Measures:
Subtracted libraries of PE transcripts were produced, and cDNAs from these libraries were used to make PE-specific cDNA arrays. Results were verified quantitatively using real-time PCR and histologically using in situ hybridization and immunohistochemistry.
Results:
30 genes were significantly altered in at least one group comparison. Differences in two candidate genes were confirmed using quantitative rt-PCR. Hemoglobin α2 and γ transcripts were significantly over-expressed in the PE placenta. Scattered cells in the placenta and placental blood vessels were shown to express genes encoding these hemoglobin-chains.
Conclusions:
We demonstrate increased hemoglobin production in the PE placenta. The hemoglobin may be released into the placenta blood vessel lumen. Free heme and hemoglobin are potent toxins which cause endothelial damage and inflammation.
Keywords: hematopoiesis, endothelial damage, hypoxia, inflammation
Introduction
Preeclampsia (PE) complicates 3 to 7% of pregnancies (1). Known for centuries, it remains a leading cause of maternal mortality and morbidity. Clinical manifestations of PE (hypertension and proteinuria) appear after the 20th week of gestation, and treatment of these problems is symptomatic and less effective than one would wish it to be. Delivery of the fetus and removal of the placenta is the only curative treatment, a fact that has lead to the generally accepted theory that placental pathology is central to the development of PE.
It is believed that inadequate placentation, resulting in reduced placental perfusion, is an early step in the development of PE (2). Reduced perfusion associated with increased vascular resistance can be detected with Doppler ultrasound (3), and women with evidence of increased resistance in the uterine arteries early in their pregnancies (“notching”) have a higher risk of developing PE than women without this finding (4). As PE progresses, the maternal vascular bed is affected, and a general endothelial inflammation is seen (5).
Poor placental perfusion appears to result in a cascade of pathological changes: decreased oxygen delivery (6-9), oxidative stress, formation of reactive oxygen species, endothelial damage (10), increased vascular permeability (11), and inflammation. The role of oxidative stress in PE has been studied by many investigators. Increases in oxidized low-density lipoprotein (12), and transferrin saturation (13) in women with PE are both thought to reflect systemic oxidative stress. Local placental oxidative stress has also been implicated by the findings of increased levels of hypoxia inducible transcription factors 1α and 2α in the preeclamptic placenta (14).
A number of attempts have been made to identify the genetic basis of susceptibility to PE in families. Linked loci have been found on chromosomes 2 (2p13 (15, 16) and 10 (10q22q22 (17)) but to date no specific genes in the linked loci have been implicated in causing the problem. However, changes in the expression of several genes in the placenta have been proposed to affect the development of PE. We have recently shown that calmodulin 2 and v-rel reticuloendotheliosis (RELA) are significantly downregulated in PE (18). Both are calcium-dependant proteins which activate the NFKB transcription factor family members. Additional array-based studies of the placenta have revealed that a number of other genes involved in intercellular signaling, metabolism, host-pathogen interactions, and apoptosis may have altered expression in PE (19, 20). Notable among these is the soluble FMS-related tyrosine kinase-1 (sFLT1), which is upregulated in preeclampsia, leading to increased systemic levels of sFLT1 that fall after delivery (21). Increased sFLT1 in preeclamptic women was associated with decreased circulating levels of free vascular endothelial growth factor (VEGF) and placental growth factor (PLGF). These appear to cause endothelial dysfunction in vitro that can be rescued by exogenous VEGF and PLGF. Administration of sFLT1 to pregnant rats induces hypertension, proteinuria, and glomerular endotheliosis, the classic lesion of preeclampsia, and there is evidence in human subjects that increased levels of sFLT1 and reduced levels of PLGF at the beginning of the second trimester of pregnancy predict the development of PE later (22).
In the present study, we printed arrays with preeclampsia-associated cDNAs obtained from subtracted libraries (23), and compared gene expression in placenta samples from women with PE, bilateral notching, PE plus bilateral notching and healthy controls. Our goal in doing this was to look for alterations in genes that might contribute to or protect the placenta from PE.
Material and Methods
Sample Collection
Placental tissue was collected at the Department of Obstetrics and Gynecology, Lund University Hospital. The sampling, performed with written consent, was approved by the Ethical Committee Review Board for studies in human subjects. Placental tissue from 10 preeclamptic, 15 normal pregnancies, 5 patients with bilateral notch and 5 patients with bilateral notch as well as preeclampsia were included in the study (Table 1). Placental bed samples (see below) from 5 of the patients with PE and 5 of the controls were also collected. Blood samples from 5 patients with preeclampsia and 5 controls were also collected before delivery. Preeclampsia was defined as blood pressure > 140/90 mm Hg and proteinuria > 0.3 g/L (24). Patients with essential hypertension or other systemic diseases were excluded. Placenta samples were collected at birth, immediately frozen and stored at −80°C. Both caesarian and vaginal deliveries were included.
Table 1. Patient demographics.
Clinical data, at delivery, of patients included in the array study.
| PE | PE+N | Notch | Control | |
|---|---|---|---|---|
| n | 10 | 5 | 5 | 15 |
| Maternal age (years) | 28.5 ± 2.9 | 32.4 ± 4.3 | 31.8 ± 5.8 | 31 ± 3.3 |
| Gestational age (days)A | 264 ± 16 | 245 ± 29 | 251 ± 22 | 280 ± 10 |
| Systolic pressure (mmHg)B | 155.8 ± 10.9 | 153.2 ± 5.9 | 115 ± 5 | 116.1 ± 10.6 |
| Diastolic pressure (mmHg)B |
107.4 ± 6.5 | 100.6 ± 2.3 | 73.8 ± 8.2 | 73.3 ± 6 |
| Proteinuria (g/l)B | 2.6 ± 1.6 | 2.7 ± 2.1 | 0.4 ± 0.5 | ND |
| Birth weight (g)C | 2990 ± 270 | 2145 ± 925 | 2524 ± 912 | 3659 ± 424 |
All results presented as mean ± SD. PE = preeclampsia, PE+N = preeclampsia with notch, ND = not detected.
Kruskal-Wallis test showed a statistically significant distribution of p = 0.01.
Kruskal-Wallis test showed a statistically significant distribution of p < 0.0001.
Kruskal-Wallis test showed a statistically significant distribution of p = 0.004 after normalizing birth weight with gestational age.
Tissue Sampling and Handling
Placental samples were collected immediately after delivery. A 10×10×10 mm cube of villous tissue was removed from the central part of the placenta avoiding macroscopic areas of necrosis and infarction. 10×10×10 mm cubes of myometrial tissue were collected from women undergoing caesarian section. The samples were immediately frozen on dry ice, and stored at −80°C until RNA was extracted. The tissue was not thawed prior to RNA extraction or cryosectioning to ensure the highest possible RNA integrity.
Blood was sampled using the PreAnalytiX PAXgene™ blood RNA system (Qiagen). Paxgene tubes were stored according to manufacturer's instructions, first 24h at −20°C and thereafter in −80°C.
RNA Extraction
Placenta
Total RNA was extracted from frozen tissue using Trizol® (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. Proteoglycan and polysaccharide were removed by performing a high-salt precipitation with 0.8 M sodium citrate and 1.2 M sodium chloride. RNA concentration was spectrophoretically determined using a Nanodrop (Nanodrop Technologies). RNA integrity was determined by denaturing 1% agarose gel electrophoresis with 6.7% formalin and 1X MOPS buffer. Samples were stored in RNAse free water at −80°C until usage. Prior to usage samples were once more precipitated and washed with 70% ethanol to remove Trizol residues.
Blood samples
Total RNA was extracted using Paxgene Blood RNA Kit according to manufacturer's protocol (Qiagen, Valencia, USA). RNA integrity was determined by denaturing 1% agarose gel electrophoresis with 6.7% formalin and 1X MOPS buffer. Samples were stored in RNAse free water at −80°C until usage. Prior to usage samples were precipitated on ice with 0.8 M sodium citrate, 1.2 M sodium chloride and 70% ethanol. Samples were washed using RNeasy MiniKit (Qiagen, Valencia, USA) and eluted in RNAse-free H2O.
Subtracted Libraries
Tissue from five women with severe PE (two of which also had notching) and eight controls was used to prepare two subtracted libraries as described previously (25). These samples were not included in the groups used for gene expression analysis in order to avoid bias. First full-length, cap-trapped cDNA libraries were made from both normal and preeclamptic placental RNAs (26). Subsequently, the two libraries were subtracted. Drivers were made from each of the amplified libraries using the vector sequences (25). Then the driver from the preeclamptic placental library used to subtract the normal placentas library and vice versa at Cot=75 with 20 fold excess of driver in a final volume of 5 μl, in 12 mM sodium phosphate (NaPi) buffer, for 8 hours at 42°C in presence of 50% formamide. At the end, HAP chromatography allowed removal of double strand DNA. The stringent condition employed eliminated most of the cDNA clones: 5900 and 1400 colonies could be picked from the subtracted preeclamptic and the control libraries, respectively, and these were subsequently sequenced.
PCR amplification of printed probes
cDNA clones were amplified using QuantiTaq™ polymerase and M13F / M13R primers as described by the manufacturer's protocol (GeneCopoeia, Germantown, USA).
For each reaction, 5 μl plasmid templates were mixed with 1× PCR reaction buffer (Cat No. QU-02-10 GeneCopoeia), 0.2 mM dNTPs (Cat No. QT-04-250 GeneCopoeia), 1 μM of M13F / M13R primer respectively, 1.5mM MgCl2 and 1 U QuantiTaq polymerase (Cat No. QT-01-500 GeneCopoeia). PCR amplification was conducted with one cycle of 2 minutes of initial heating to 96°C, 29 cycles of denaturing at 94°C for 1 minute, annealing at 55°C for 30 seconds, and extension at 72°C for 2 minutes, and one final cycle of holding at 72°C for 5 minutes. PCR products were purified using the NucleoFast 96 PCR Clean-Up Kit (ClonTech, Mountain View, USA). Briefly, PCR product was transferred to a NucleoFast 96 PCR plate and contaminants were filtered through by means of vacuum pressure. Membranes were washed once with 100 μl of nuclease-free water after which membranes were dried. Purified PCR product was recovered by adding 80μl recovery buffer and incubating the plates on shaker for 10 minutes. PCR products were verified on 1.2% agarose gels, 1.2g agarose, 100ml 1× TAE buffer (40mM tris pH 8.3, 1mM EDTA) and 6μl etidium bromide using 1× TAE as running buffer.
Arrays
Microarrays printed with our 800 cDNA-clones as well as an oligoset containing 27,000 oligos (OPERON v 2.1 human 70 mer oligo set, Operon, Huntsville, USA) were produced at the Swegene DNA Microarray Resource Center, Department of Oncology, Lund University, Sweden (http://swegene.onk.lu.se). Our 800 clones were printed in three replicates on each slide as a mean of quality control, allowing us to remove spots affected by local artifacts. Probes were dissolved in Corning Universal Spotting solution (Corning, Acton, USA) and printed on aminosilane coated glass slides (UltraGAPS, Cat. No. C40017, Corning) using a MicroGrid2 robot (BioRobotics, Cambridgeshire, UK) equipped with MicroSpot 10K pins (BioRobotics). Following printing, arrays were placed in a desiccator to dry for 48 hours, rehydrated for 1 second over steaming water, snap dried on a hot plate (98°C), and UV cross-linked (800mJ/cm2).
Fluorescence labeled cDNA target was prepared using the Corning ChipShot™ labeling system according to manufacturers' instructions (Cat. No. C40056, Corning). Briefly, Cy3-labeled cDNA was prepared using 5 μg of total RNA from each individual experimental sample (Table 1), and Cy5-labeled cDNA was prepared using 5 μg of normal placental RNA to serve as a reference. Cy3-dCTP (Cat. No. PA53021) and Cy5-dCTP (Cat. No. PA55021) were obtained from Amersham (Amersham Biosciences, Buckinghamshire, UK). Pretreatment of arrays, hybridization, and post-hybridization wash was carried out according to manufacturer's instructions (Corning ChipShot™ labeling system). Obtained reference cDNA from the labeling reactions was pooled in order to have homogenous reference cDNA.
Image Analysis
Hybridized microarrays were imaged using an Agilent G2565AA microarray scanner (Agilent Technologies, Palo Alto, CA). Fluorescence intensities were extracted using Gene Pix Pro 4.0 software (Axion Instruments Inc., Foster City, CA). Spots affected by veils, grains of dust or other contaminants were removed from further analysis.
Data Analysis
Data files obtained from Gene Pix Pro were analyzed using BASE (27). Spots that were filtered for low channel intensity, with a cut off at 100, and signal to noise ratio, removing spots with intensity less than two times stronger than the background. The data were then normalized using Lowess normalization. The oligo-set co-printed with our cDNA-clones were used in the normalization procedure to improve its reliability. Replicate ratios were quality controlled as previously described (28). Briefly, the mean ratio, standard deviation (SD), and the coefficient of variation (CV) were calculated for the replicates. CV was compared with average intensity from the nearest 200 neighbors. Replicates differing more than 1 SD from average intensity were iterated, removing the most outlying spot within the replicate, and then re-compared CV with average intensity. At least two spots had to remain in each replicate to be allowed for further analysis. Mean ratios were calculated from remaining replicates, missing values were imputed using KNNimpute (29) and group comparison analysis were performed using Significance Analysis of Microarray (SAM) test (30).
Bioinformatics analysis
The Gene Ontology (GO) database was used to classify genes according to their biological process or molecular function. GoMiner (31) was used to obtain GO annotations. GO terms for the genes identified as significantly increased or decreased in the microarray were retrieved.
Real-Time PCR amplification
cDNA synthesis
cDNA was synthesized with reverse transcriptase according to manufacturer's protocols (Applied Biosystems, Foster City, USA). A 50 μl reaction containing 0.5 μg total RNA, 1X TaqMan RT buffer, 5.5mM MgCl2, 500μM dNTPs, 2.5μM random hexamers, 0.4 U/μl RNase inhibitor and 1.25 U/μl MultiScribe Reverse Transcriptase was used. The reactions were incubated at 25°C for 10 minutes, at 48°C for 30 minutes and finally 5 minutes at 95°C. Samples were stored at −20°C until analysis.
Analysis
Gene transcripts were assayed by means of real-time PCR using an ABI PRISM® 7000 sequence detection system (Applied Biosystems). Primers and probes were designed using the Primer Express® software program or ordered from Assays on-Design/Demand™ (Applied Biosystems). The primers targeted different exons of the genes of interest to avoid amplifying contaminating genomic DNA. Reactions were carried out in a 25 μl final volume containing: 1× Universal PCR Master Mix (Applied Biosystems), 0.25 μmol/l probe, 0.9 μmol/l of forward and reverse primers respectively, and 1 μl of 10 ng/μl of cDNA. The thermal cycling conditions were initiated by UNG activation at 50°C for 2 minutes and an initial denaturation at 95°C for 10 minutes. Then 40 cycles were run: 95°C for 15 seconds, 60°C for 1 minute. Two negative controls with no template were included in every set of amplifications. β-actin was used as a reference to normalize the signal from the sample. Quantitation was achieved by making a calibration curve using serial 4-fold dilutions of the template DNA (0.08-80ng). Results are expressed as ratios with β-actin as the denominator.
Statistics
A Mann-Whitney U-test was used to evaluate the significance of differences between groups. P<0.05 was considered statistically significant. Results are presented as scatter–plots. A Kruskal-Wallis test was performed to include multi variable testing.
In situ hybridization (ISHH)
The hybridizations were conducted as previously described (18). Cryostat sections were thaw mounted onto sialinized slides, which were stored at −80°C until they were used. Fresh frozen tissue was employed to maximize mRNA detection. Sections were fixed, dehydrated, dilipidated, and hybridized as previously described (32). Hybridizations were carried out for 20-24 hours in 55°C with 2×106 cpm of denatured 35S-cRNA probe per 80 μl hybridization buffer (20 mM Tris-HCl (pH 7.4), 1 mM EDTA (pH 8.0), 300mM NaCl, 50% formamide, 10% dextran sulphate, 1× Denhardt's 25 mg/ml yeast tRNA, 100 μg/ml salmon sperm DNA, 250 μg/ml total yeast RNA (fraction XI, Sigma, St. Louis, USA), 150 mM dithiothreitol (DTT), 0.15% sodium thiosulfate (NTS) and 0.15% sodium dodecyl sulphate (SDS). Following washes, slides were apposed to Kodak Hyperfilm Biomax MR for 2 days, after which they were coated with nuclear track emulsion (NTB-3, Kodak, Rochester, USA). Slides were exposed for 3 (Hbα2, Hbγ2) respectively 4 (Hbβ) weeks at 4°C, after which they were developed in Dektol (Kodak), fixed and counterstained with a Giemsa stain.
Immunohistochemistry
Immunohistochemistry was performed on 5 samples from the PE group and 5 from the control group respectively. Fresh frozen sections, 12 μm thick, of the placenta samples were fixed by immersion in 4% buffered formaldehyde for 10 min at room temperature. Sections were then incubated in a blocking solution (Powerblock; Zymed, Carlsbad, USA) for 30 minutes at RT. The fetal Hb antibody (Bethyl Laboratories, Montgomery, USA), raised in sheep, was diluted 1:500 in the diluent containing 1×PBS with 0.05% Triton X and 2% NGS. Following an hour RT incubation the sections were rinsed and transferred into a 1:1000 dilution of an anti-sheep CY3 antibody raised in donkey (Jackson laboratories, Bar Harbor, USA) for an hour at RT. The sections were then rinsed, coverslipped with 0.1M Tris and viewed under a Leica DMA 6000 inverted fluorescent microscope. Pictures were taken using Volocity software.
Results
Eight hundred unique clones were identified in the subtracted libraries that we made and sequenced. To determine whether the expression levels of the transcripts corresponding to these cDNAs were indeed altered in preeclamptic versus control placentas, we used them to print microarrays that allowed us to study the four groups of samples described earlier.
The gene expression profiles for all groups were compared using a false discovery rate based analysis method, with a q-value <0.05 as the cutoff. Thirty genes were significantly altered in at least one between-group comparison (Figure 1).
Figure 1. Heatmap of significant genes.
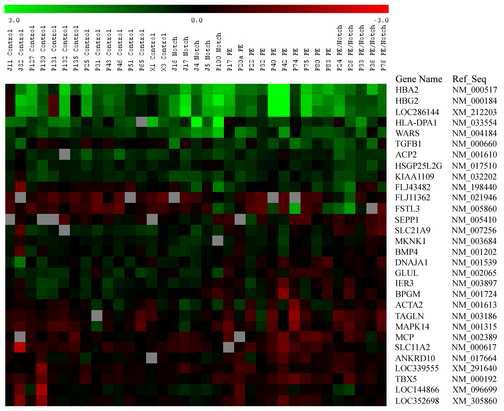
Expression profile of genes found significantly changed in at least one group comparison. After filtering genes for signal to noise ratio above 2.0 and fold change above 1.4, a total of 30 unique genes were obtained. In the figure genes are sorted on mean expression ratio from high to low in the PE group. A cut off q-value at 0.005 was selected.
GO terms associated with each gene in Figure 1 were tabulated. The annotations included hemoglobin complex (GO 5883), oxygen transport (GO 15671), regulation of actin polymerization (GO 8064), and regulation of actin filament length (GO 30832).
The most markedly altered genes were reassayed by means of real-time PCR. The levels of Hbα2 (p=0.004), Hbγ (p=0.003) and Hbβ (p=0.02) mRNAs were found to be the most significantly increased in PE samples vs. controls (Figure 2a,b,c) and also in samples from PE with notching compared to controls (Hbα2 p=0.02, Hbγ p=0.03 and Hbβ p=0.04). (Hbβ was not represented on the array, but was examined because of changes detected in Hbα2 and Hbγ.) Major histocompatibility complex, class II, DP alpha 1 (HLA-DPA1) was significantly upregulated in the group with notching compared to all other groups (p=0.01 against PE without notching, p=0.02 against PE with notching, and p=0.01 against control) (Figure 2d). Tryptophanyl-tRNA synthetase (WARS) was significantly over-expressed in notch without PE compared to controls (p=0.01). Glutamate-ammonia ligase (GLUL) was significantly over-expressed in samples from PE with notch compared to controls (p=0.009). Finally, solute carrier family 11, member 2 (SLC11A2), a facilitated glucose transporter, was significantly increased in PE with notch vs. PE without notch, and vs. controls (p=0.04 and p=0.01 respectively). β-actin levels did not differ among the groups (data not shown).
Figure 2. Real-time PCR quantification of Hbα2, Hbγ, Hbβ and HLA-DPA1 in placenta and blood samples.
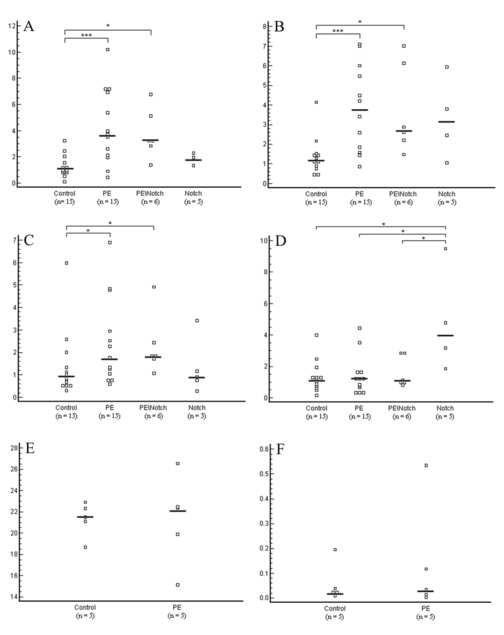
All values are normalized against the amount of β-actin and presented as scatter plots. (A) Hbα mRNA expression in the placenta. Significant changes were found between PE vs. controls (p=0.004) and between PE\Notch (PE with notching) vs. controls (p=0.03). (B) Hbγ relative mRNA values showing significant changes between PE vs. controls (p=0.003) and between PE\Notch vs. controls (p=0.03). (C) Hbβ showed significant overexpression in PE vs. controls (p=0.02) and in PE\Notch vs. controls (p=0.04). (D) HLA-DPA1 mRNA was overexpressed in the notch group vs. all other groups: vs. controls (p=0.01), vs. PE (p=0.01), and vs. PE with notching (p=0.02). (E) Hbα mRNA expression in blood samples. No changes were seen between the groups. (F) Hbγ mRNA expression was very low in blood samples. No changes were seen between PE and controls.
In situ hybridization revealed nucleated Hbα- and Hbγ-expressing cells scattered throughout the villous section in both PE and control samples (Figure 3). Placentas from PE patients seemed to have more Hb containing cells than control samples, and the signals per cell appeared to be more intense than in controls. In several of the samples studied, Hb-positive cells were associated with the walls of blood vessels, with several cells free in the lumen. Many single cells were found in the villous space. Based on their morphology, location, and distribution, they are not likely to be trophoblasts. Placental bed samples from both PE and control subjects showed a weaker Hb mRNA expression compared to samples from the villous section. Positive cells were detected in blood vessels, and few positive cells were seen in the surrounding myometrial tissue.
Figure 3. In situ hybridizations from placenta and placenta bed samples.
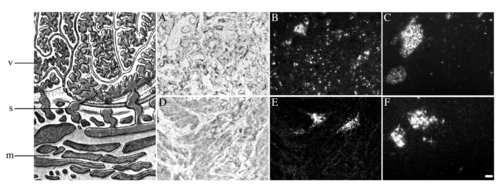
Schematic image of the human placenta showing the villous section (V) of the placenta and below a placenta bed section (M) with spiral arteries (S) in between. (A) A light field image of Hbα mRNA-expression in a representative preeclamptic placenta sample. Hbα expression was especially seen in and around blood vessels. However, several scattered cells in the villous section are also seen. (B) Darkfield image of the same section. (C) A darkfield image of Hbα mRNA expression from a representative control placenta. Compared with PE placentas, the control placentas show fewer Hbα-expressing cells in the villous section. (D) Light field image from a representative myometrial sample from a PE patient. Hbα expression is only seen in the spiral arteries, no expression is seen in the myometrial tissue. (E) The same myometrial section in dark field. (F) A myometrial sample from a control placenta. Hbα mRNA expression is similar to the expression seen in PE myometrium. mRNA expression for Hbγ and Hbβ were similar to that for Hbα (data not shown). Scale-bar is 100μm.
To correlate protein- with RNA-expression, immunohistochemistry was performed. Hbγ-expression was especially relatively abundant in the lumens of placental blood vessels in PE samples and also near the endothelial cells in the vascular walls as well as in the extra-vascular space of the villous stroma. The control placenta samples had some Hbγ-expression in the vascular endothelium, but no expression in the vascular lumen (Figure 4).
Figure 4. A representative image of Hbγ protein expression in the placenta.
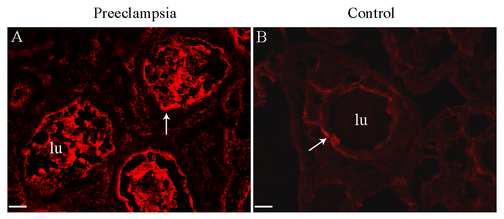
Protein expression is shown with a red fluorescent marker. In the PE placenta there is a strong expression of Hbγ in the vascular lumen (lu), but Hbγ is also expressed in the vascular endothelium (arrow) as well as in the extra-vascular section of the villous stroma (A). The placenta from normotensive controls however, showed no expression of Hbγ in the vascular lumen (B) but Hbγ is expressed in the vascular endothelium (arrow). Scale bar is 25 µm.
Discussion
Making and sequencing subtracted cDNA libraries allows one to detect possible changes in mRNA-expression between two defined groups (25, 33). In our libraries we found 800 unique cDNA-clones that appeared to differ between placental samples from women with PE and samples from healthy pregnant women. Several of the transcripts represented by these cDNAs were subsequently shown with microarray and quantitative RT-PCR to be significantly altered between the PE and the control groups. The changes seen in the hemoglobin genes were among the most interesting, and our discussion will focus on these.
Both microarray and qPCR experiments showed increased expression of Hbα and Hbγ mRNA in PE vs. controls. In situ hybridization showed an increased number of cells expressing Hb in placental samples from PE vs. control subjects. The fact that Hb expressing cells were associated with vessel walls, may either indicate that the cells are migrating into or out of the vessels, or that there are binding sites on vessel walls for these cells. Myometrial vessels lack Hb+ nucleated cells while placental vessels in PE subjects are rich in them. Furthermore, Hb mRNA levels are equivalent in PE and normal patients' blood samples. These findings suggest that the Hb+ cells in PE placental vessels are not maternal, but rather fetal in origin. Neonatals from pregnancies complicated with intrauterine growth restriction or intrauterine hypoxia have a higher number of nucleated red blood cells in their periphery circulation as well as in the cord blood vessels (34, 35). Taken together with the in situ results, the cells described may therefore be fetal erythroblasts migrating from the fetal into the maternal side of the placenta.
Hb expression is normally increased in response to low oxygen levels and it has been argued that the placenta in PE suffers from poor perfusion and hypoxia. Moreover, the placenta can produce the hematopoietic factor activin A (36), and levels of activin A are increased in PE (37). This factor may act locally; the murine (38, 39) and human (40, 41) placenta have been suggested to function as hematopoietic organs. Thus, increased hematopoietic activity in the preeclamptic placenta could explain the elevated levels of hemoglobin mRNA that we observed. However, it is not certain whether gestational age contributes to hemoglobin expression, but it may certainly affect the production.
If these Hb-producing cells turn over quickly, they might release high levels of heme into the extra-villous space and the placental blood vessels. Indeed, our immunohistochemistry shows high levels of hemoglobin in the PE placenta blood vessel lumen. The control placenta on the other hand showed no release of hemoglobin into the blood vessels. To make matters worse, hemolysis in necrotic and thrombotic areas of the PE placenta may add to the amount of free heme there.
Free heme is a potent redox agent that can cause severe tissue damage through the creation of reactive oxygen species (ROS). Heme oxidizes several lipids including low-density lipoproteins (LDL) (42), converting them into cytotoxic peroxides which cause endothelial damage. Furthermore, heme can directly damage cell membranes by disrupting them and oxidizing membrane proteins leading to increased membrane permeability and cytolysis.
Free heme is usually degraded by heme oxygenase (HO). In preeclampsia there is both decreased expression of HO-2 (43, 44) and reduced activity of HO (45). This may lead to impaired heme degradation and contribute to the build-up of heme in the placenta. Furthermore, reduced production of carbon monoxide from heme by HO may contribute to increased vascular tone and reduced placental blood flow (46).
Free heme together with reduced placental blood flow induces an inflammatory response, which may result in further endothelial cell damage. The generation of ROS by heme leads to the activation of NF-κB transcription factors (47), which stimulate the production of adhesion molecules (such as ICAM-1 and VCAM-1) and the recruitment of leukocytes. In fact, heme has recently been shown to stimulate neutrophil migration via chemotactic receptors (48). In addition, heme induces neutrophil activation and the expression of several cytokines such as interleukin-8 (49). However, heme can also exert an immune response by activating the toll like receptor 4, and inducing secretion of tumor necrosis factor-a (TNFa) (50).
Thus, infiltration of the placenta by large numbers of Hb positive cells is a worrisome sign. Heme released from these cells could be quite harmful and may be responsible for much of the placental pathology associated with PE. Whether heme toxicity drives sFLT1 synthesis and secretion remains to be determined.
In conclusion, our findings suggest that Hb genes are over expressed in a subpopulation of cells in the preeclamptic placenta. Whether these cells are intrinsic to the placenta or migrants from the fetus is unknown. The production of agents that stimulate hematopoiesis by placental cells in response to reduced perfusion and possibly local hypoxia may contribute to the formation, recruitment and distribution of the cells. While they seem to be present in the placenta of subjects who had normal pregnancies, their increase in placentas from PE patients is a matter of concern. If they turn over rapidly and release their Hb (and heme) excessively, they may damage adjacent structures including the vascular endothelium.
Acknowledgements
We like to thank Jason Inman (The Institute for Genomic Research, MD, USA) for helping with cDNA identification, Irene Larsson for helping with the laboratory work, Marcus Ringnér (PhD, Assistant Professor, Department of Theoretical Physics), and Patrik Medstrand, (PhD, Associate professor, Department of Genomics and Bioinformatics) for helping us with the statistical and bioinformatic analysis.
This work was funded by grants from the Swedish Research Council: 5775, Anna Lisa & Sven Erik Lundgrens foundation for Medical Research, Crawford foundation, Magnus Bergvalls foundation, Swedish Society for Medical Research and the Intramural Research Program of NIDCR, NIH.
Microarrays and protocols were obtained from the Swegene DNA Microarray Resource Center in Lund, supported by the Knut and Alice Wallenberg foundation through the Swegene consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roberts JM, Cooper DW. Pathogenesis and genetics of pre-eclampsia. Lancet. 2001;357:53–6. doi: 10.1016/s0140-6736(00)03577-7. [DOI] [PubMed] [Google Scholar]
- 2.Kaar K, Jouppila P, Kuikka J, Luotola H, Toivanen J, Rekonen A. Intervillous blood flow in normal and complicated late pregnancy measured by means of an intravenous 133Xe method. Acta obstetricia et gynecologica Scandinavica. 1980;59:7–10. doi: 10.3109/00016348009160077. [DOI] [PubMed] [Google Scholar]
- 3.Papageorghiou AT, Yu CK, Cicero S, Bower S, Nicolaides KH. Second-trimester uterine artery Doppler screening in unselected populations: a review. J Matern Fetal Neonatal Med. 2002;12:78–88. doi: 10.1080/jmf.12.2.78.88. [DOI] [PubMed] [Google Scholar]
- 4.Thaler I, Weiner Z, Itskovitz J. Systolic or diastolic notch in uterine artery blood flow velocity waveforms in hypertensive pregnant patients: relationship to outcome. Obstet Gynecol. 1992;80:277–82. [PubMed] [Google Scholar]
- 5.Redman CW, Sargent IL. Preeclampsia and the systemic inflammatory response. Semin Nephrol. 2004;24:565–70. doi: 10.1016/s0270-9295(04)00127-5. [DOI] [PubMed] [Google Scholar]
- 6.Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, et al. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab. 2005;90:4299–308. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajakumar A, Brandon HM, Daftary A, Ness R, Conrad KP. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta. 2004;25:763–9. doi: 10.1016/j.placenta.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Bowen RS, Gu Y, Zhang Y, Lewis DF, Wang Y. Hypoxia promotes interleukin-6 and -8 but reduces interleukin-10 production by placental trophoblast cells from preeclamptic pregnancies. J Soc Gynecol Investig. 2005;12:428–32. doi: 10.1016/j.jsgi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Vaiman D, Mondon F, Garces-Duran A, Mignot TM, Robert B, Rebourcet R, et al. Hypoxia-activated genes from early placenta are elevated in preeclampsia, but not in Intra-Uterine Growth Retardation. BMC Genomics. 2005;6:111. doi: 10.1186/1471-2164-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts JM, Lain KY. Recent Insights into the pathogenesis of pre-eclampsia. Placenta. 2002;23:359–72. doi: 10.1053/plac.2002.0819. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Gu Y, Zhang Y, Lewis DF. Evidence of endothelial dysfunction in preeclampsia: decreased endothelial nitric oxide synthase expression is associated with increased cell permeability in endothelial cells from preeclampsia. American journal of obstetrics and gynecology. 2004;190:817–24. doi: 10.1016/j.ajog.2003.09.049. [DOI] [PubMed] [Google Scholar]
- 12.Qiu C, T TTP, Vadachkoria S, Muy-Rivera M, Sanchez S, Williams M. Oxidized low-density lipoprotein (Oxidized LDL) and the risk of preeclampsia. Physiol Res. 2005 doi: 10.33549/physiolres.930813. [DOI] [PubMed] [Google Scholar]
- 13.Hubel CA, Kozlov AV, Kagan VE, Evans RW, Davidge ST, McLaughlin MK, et al. Decreased transferrin and increased transferrin saturation in sera of women with preeclampsia: implications for oxidative stress. American journal of obstetrics and gynecology. 1996;175:692–700. doi: 10.1053/ob.1996.v175.a74252. [DOI] [PubMed] [Google Scholar]
- 14.Rajakumar A, Doty K, Daftary A, Harger G, Conrad KP. Impaired oxygen-dependent reduction of HIF-1alpha and -2alpha proteins in pre-eclamptic placentae. Placenta. 2003;24:199–208. doi: 10.1053/plac.2002.0893. [DOI] [PubMed] [Google Scholar]
- 15.Arngrimsson R, Sigurard ttir S, Frigge ML, Bjarnad ttir RI, Jonsson T, Stefansson H, et al. A genome-wide scan reveals a maternal susceptibility locus for pre-eclampsia on chromosome 2p13. Hum Mol Genet. 1999;8:1799–805. doi: 10.1093/hmg/8.9.1799. [DOI] [PubMed] [Google Scholar]
- 16.Moses EK, Lade JA, Guo G, Wilton AN, Grehan M, Freed K, et al. A genome scan in families from Australia and New Zealand confirms the presence of a maternal susceptibility locus for pre-eclampsia, on chromosome 2. Am J Hum Genet. 2000;67:1581–5. doi: 10.1086/316888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dijk M, Mulders J, Poutsma A, Konst AA, Lachmeijer AM, Dekker GA, et al. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet. 2005;37:514–9. doi: 10.1038/ng1541. [DOI] [PubMed] [Google Scholar]
- 18.Hansson SR, Chen Y, Brodszki J, Chen M, Hernandez-Andrade E, Inman JM, et al. Gene expression profiling of human placentas from preeclamptic and normotensive pregnancies. Mol Hum Reprod. 2006 doi: 10.1093/molehr/gal011. [DOI] [PubMed] [Google Scholar]
- 19.Han JY, Kim YS, Cho GJ, Roh GS, Kim HJ, Choi WJ, et al. Altered gene expression of caspase-10, death receptor-3 and IGFBP-3 in preeclamptic placentas. Molecules and cells. 2006;22:168–74. [PubMed] [Google Scholar]
- 20.Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, Udagawa Y. Microarray Analysis of Differentially Expressed Fetal Genes in Placental Tissue Derived from Early and Late Onset Severe Pre-eclampsia. Placenta. 2006 doi: 10.1016/j.placenta.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–58. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 23.Carninci P, Shibata Y, Hayatsu N, Sugahara Y, Shibata K, Itoh M, et al. Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res. 2000;10:1617–30. doi: 10.1101/gr.145100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milne F, Redman C, Walker J, Baker P, Bradley J, Cooper C, et al. The pre-eclampsia community guideline (PRECOG): how to screen for and detect onset of pre-eclampsia in the community. BMJ (Clinical research ed. 2005;330:576–80. doi: 10.1136/bmj.330.7491.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirozane-Kishikawa T, Shiraki T, Waki K, Nakamura M, Arakawa T, Kawai J, et al. Subtraction of cap-trapped full-length cDNA libraries to select rare transcripts. Biotechniques. 2003;35:510–6, 8. doi: 10.2144/03353st04. [DOI] [PubMed] [Google Scholar]
- 26.Carninci P, Hayashizaki Y. High-efficiency full-length cDNA cloning. Methods Enzymol. 1999;303:19–44. doi: 10.1016/s0076-6879(99)03004-9. [DOI] [PubMed] [Google Scholar]
- 27.Saal LH, Troein C, Vallon-Christersson J, Gruvberger S, Borg A, Peterson C. BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-8-software0003. SOFTWARE0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2549–57. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–5. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 30.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradley DJ, Towle HC, Young WS., 3rd Spatial and temporal expression of alpha- and beta-thyroid hormone receptor mRNAs, including the beta 2-subtype, in the developing mammalian nervous system. J Neurosci. 1992;12:2288–302. doi: 10.1523/JNEUROSCI.12-06-02288.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carninci P, Waki K, Shiraki T, Konno H, Shibata K, Itoh M, et al. Targeting a complex transcriptome: the construction of the mouse full-length cDNA encyclopedia. Genome Res. 2003;13:1273–89. doi: 10.1101/gr.1119703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baschat AA, Gembruch U, Reiss I, Gortner L, Harman CR, Weiner CP. Neonatal nucleated red blood cell counts in growth-restricted fetuses: relationship to arterial and venous Doppler studies. American journal of obstetrics and gynecology. 1999;181:190–5. doi: 10.1016/s0002-9378(99)70458-8. [DOI] [PubMed] [Google Scholar]
- 35.Bernstein PS, Minior VK, Divon MY. Neonatal nucleated red blood cell counts in small-for-gestational age fetuses with abnormal umbilical artery Doppler studies. American journal of obstetrics and gynecology. 1997;177:1079–84. doi: 10.1016/s0002-9378(97)70018-8. [DOI] [PubMed] [Google Scholar]
- 36.Florio P, Luisi S, Ciarmela P, Severi FM, Bocchi C, Petraglia F. Inhibins and activins in pregnancy. Mol Cell Endocrinol. 2004;225:93–100. doi: 10.1016/j.mce.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 37.Troeger C, Holzgreve W, Ladewig A, Zhong XY, Hahn S. Examination of maternal plasma erythropoietin and activin A concentrations with regard to circulatory erythroblast levels in normal and preeclamptic pregnancies. Fetal Diagn Ther. 2006;21:156–60. doi: 10.1159/000089068. [DOI] [PubMed] [Google Scholar]
- 38.Mikkola HK, Gekas C, Orkin SH, Dieterlen-Lievre F. Placenta as a site for hematopoietic stem cell development. Exp Hematol. 2005;33:1048–54. doi: 10.1016/j.exphem.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HK. The placenta is a niche for hematopoietic stem cells. Dev Cell. 2005;8:365–75. doi: 10.1016/j.devcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Challier JC, Galtier M, Cortez A, Bintein T, Rabreau M, Uzan S. Immunocytological evidence for hematopoiesis in the early human placenta. Placenta. 2005;26:282–8. doi: 10.1016/j.placenta.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Demir R, Kaufmann P, Castellucci M, Erbengi T, Kotowski A. Fetal vasculogenesis and angiogenesis in human placental villi. Acta Anat (Basel) 1989;136:190–203. doi: 10.1159/000146886. [DOI] [PubMed] [Google Scholar]
- 42.Camejo G, Halberg C, Manschik-Lundin A, Hurt-Camejo E, Rosengren B, Olsson H, et al. Hemin binding and oxidation of lipoproteins in serum: mechanisms and effect on the interaction of LDL with human macrophages. J Lipid Res. 1998;39:755–66. [PubMed] [Google Scholar]
- 43.Barber A, Robson SC, Myatt L, Bulmer JN, Lyall F. Heme oxygenase expression in human placenta and placental bed: reduced expression of placenta endothelial HO-2 in preeclampsia and fetal growth restriction. Faseb J. 2001;15:1158–68. doi: 10.1096/fj.00-0376com. [DOI] [PubMed] [Google Scholar]
- 44.Lash GE, McLaughlin BE, MacDonald-Goodfellow SK, Smith GN, Brien JF, Marks GS, et al. Relationship between tissue damage and heme oxygenase expression in chorionic villi of term human placenta. Am J Physiol Heart Circ Physiol. 2003;284:H160–7. doi: 10.1152/ajpheart.00738.2002. [DOI] [PubMed] [Google Scholar]
- 45.Appleton SD, Marks GS, Nakatsu K, Brien JF, Smith GN, Graham CH, et al. Effects of hypoxia on heme oxygenase expression in human chorionic villi explants and immortalized trophoblast cells. Am J Physiol Heart Circ Physiol. 2003;284:H853–8. doi: 10.1152/ajpheart.00655.2002. [DOI] [PubMed] [Google Scholar]
- 46.Kim HP, Ryter SW, Choi AM. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol. 2006;46:411–49. doi: 10.1146/annurev.pharmtox.46.120604.141053. [DOI] [PubMed] [Google Scholar]
- 47.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci U S A. 1994;91:5987–91. doi: 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca-Souza AV, et al. Heme Induces Neutrophil Migration and Reactive Oxygen Species Generation through Signaling Pathways Characteristic of Chemotactic Receptors. The Journal of biological chemistry. 2007;282:24430–6. doi: 10.1074/jbc.M703570200. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett. 2005;157:175–88. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of Heme as Activator of Toll-like Receptor 4. The Journal of biological chemistry. 2007;282:20221–9. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]