

Abstract
Short functional peptide motifs cooperate in many molecular functions including protein interactions, protein trafficking, and posttranslational modifications. Viruses exploit these motifs as a principal mechanism for hijacking cells and many motifs are necessary for the viral life-cycle. A virus can accommodate many short motifs in its small genome size providing a plethora of ways for the virus to acquire host molecular machinery. Host enzymes that act on motifs such as kinases, proteases, and lipidation enzymes, as well as protein interaction domains, are commonly mutated in human disease, suggesting that the short peptide motif targets of these enzymes may also be mutated in disease; however, this is not observed. How can we explain why viruses have evolved to be so dependent on motifs, yet these motifs, in general do not seem to be as necessary for human viability? We propose that short motifs are used at the system level. This system architecture allows viruses to exploit a motif, whereas the viability of the host is not affected by mutation of a single motif.
Keywords: Motif, Disease, Virus, Drugs, Posttranslational modification, Review
2. INTRODUCTION
Scientific inquiry over the past century has yielded a profound increase in our understanding of human disease along with advancement in its therapy. Diseases arise from inherited genetic or epigenetic disorders that are not embryonic lethal, or from environmental factors such as toxins and pathogens. Many genetic diseases are caused by mutations in coding regions of proteins imparting either a loss of normal function or gain of an abnormal function. Since domains are the fundamental folding and functional units of proteins, it is logical to expect that disruption of domains could lead to disease. Many such mutations in domains are observed and can lead to aberrant protein folding, aberrant aggregation, or have direct consequences on enzymatic, binding, or other functions of proteins. These mutations represent a major mechanistic class of disease-causing mutations.
In addition to domains, proteins also contain short motifs (generally shorter than 10 amino acids) that are the targets of protein domains. These motifs enable critical cellular functions such as binding to other targets, protein trafficking, regulation, and posttranslational modifications. Since proteins use these motifs for a wide array of functions, one might expect that these short motifs, like their functional domain counterparts, would commonly be exploited by pathogens and mutated in disease. This review represents the first overview and consolidation of the role of short functional motifs involved in viral infection, human disease, and drug design.
There are 1000s of published short functional motifs that are most often defined by consensus sequence motifs or by position specific-scoring matrices. Throughout this manuscript, consensus motifs are represented using the single letter amino acid (IUPAC) code and regular expression motif syntax used by Prosite (a domain database): “x” indicates any amino acid in this position, residues in “[]” indicate allowable residues at a position, “<” or “>” indicate location on either the N- or C-terminus, respectively (1). For example, a LxCxE consensus motif binds retinoblastoma protein. Minimotif Miner, DiliMot, Scansite, SlimDisc, and Eukaryotic Linear Motif (ELM) are web-based tools for exploring the role of short motifs in protein and proteomes (2-6).
3. VIRUSES USE SHORT FUNCTIONAL MOTIFS TO HIJACK HOST PROTEINS
Other than the few genome-encoded enzymes, viruses must depend on their host’s machinery for nearly all aspects of their life-cycle. Therefore it is not surprising that viruses use short functional motifs for binding to host proteins and recruiting host proteins to posttranslationally modify and traffic viral proteins to specific subcellular compartments. Although some reviews have summarized how a specific motif is used by viruses, the broad-based exploitation of short motifs by viruses has not been previously summarized (7-9).
Table 1 shows a collection of motifs that are commonly used by viruses to acquire host molecular machinery. In general, many types of viruses use these motifs at all stages of their life-cycles, and utilization of these motifs is not limited to specific species, with motifs used by many different species of plants and animals. Viruses exploit the ability of host enzymes to posttranslationally modify their proteins. In addition to the sources in Table 1, several previous reviews cover the myristoylation, prenylation, N-glycosylation, phosphorylation, and cleavage of viral proteins by host enzymes (9-20).
Table 1.
Motifs in viruses
| Motif | Viruses | PubMed IDs |
|---|---|---|
| SH3 binding | Influenza, Hepatitis B and C, HIV, SIV, Herpesvirus saimiri, bovine leukemia virus, Epstein-Barr, Avian sarcoma virus | 17881440, 16935477, 16684552, 16530520, 16051843, 15784897, 15767257, 14993658, 12692296, 12379843, 12466492, 11533201, 11375055, 10756028, 10837147, 10660579, 10574946, 9778343, 8681387, 7859737, 7687742 |
| SH2 binding | Influenza, Herpesvirus 8, HIV, Jaagsiekte sheep retrovirus, Epstein-Barr virus, Kaposi’s sarcoma-associated herpesvirus, Hepatitis virus, polyomavirus, SIV | 17485529, 17170431, 9705913, 18029356, 17610928, 17311000, 17485529, 16963558, 16940534, 11707404, 10439044, 10364375, 9705913, 8985339, 8794306 |
| PDZ binding | T-cell leukemia virus, adenovirus, influenza, papillomavirus, polyomavirus | 17855372, 17715223, 17314165, 17672298, 17287269, 17121805, 16263794, 16042787, 15507652, 15507623, 15286176, 15003862, 14972558, 14625386, 12569363, 11598001, 11571640, 11127819, 11077444, 948211, 9326658, 9192623 |
| 14-3-3 binding | Kaposi’s sarcoma-associated herpesvirus, Hepatitis, HIV, adeno-associated virus, polyomavirus, minute virus | 17108038, 17141194, 16895903, 16895903, 15708996, 15003870, 10644344, 9420259, 8892871 |
| Integrin binding | Epstein-Barr, White Spot Syndrome Virus, Adenovirus, herpesvirus 8, Herpes simplex virus, Cytomegalovirus, West nile virus, Coxsackievirus, Norovirus, echovirus, parechovirus, encephalitis virus, bluetongue virus, flavivirus, yellow fever virus | 17945327, 17655902, 17427949, 16943295, 16760393, 15878864, 15613334, 15604426, 15494436, 15475343, 15194773, 14610179, 12601761, 11559835, 1146204, 11264382, 10982329, 10603320 |
| Dileucine sorting | HIV, SIV, Moloney murine leukemia virus, Mason-Pfizer monkey virus, Nipah virus, bovine leukemia virus, herpes simplex virus, Pseudorabies | 10366557, 17267500, 16978406, 16873251, 16051847, 15731282, 15254202, 11222723, 10814565, 10559313, 9811611 |
| prenylation | Pseudorabies, Herpesvirus, hepatitis, Influenza virus | 14581565, 11238466, 7512154, 8116240, 14557627, 12050399, 16573201 |
| myristoylation | HIV, Herpes simplex virus, Abelson murine leukemia virus, Arenavirus, geminivirus, Lassa virus, Vaccinia virus, Lentivirus, Hepatitis virus, poliovirus, Junin virus, mouse osteosarcoma | 17881447, 17881447, 17581989, 17427808, 17083745, 16973586, 16021629, 15613341, 15452271, 12509984, 10653654, 10611737, 9162016 |
| Rb binding | simian virus 40, papillomavirus, Maize streak virus, T-cell leukemia virus, Geminivirus, cytomegalovirus, adenovirus, polyomavirus, JC virus, nanovirus, banana bunchy top virus, Bean yellow dwarf virus, rubella, plant DNA virus, potato virus X, varicella-zoster virus, Reovirus, poliovirus, foot-and-mouth disease virus, budgerigar fledgling disease virus | 17581980, 16411741, 15722542, 15580311, 14741123, 12021356, 11921304, 10958676, 10936097, 10779361, 10640570, 10191192, 10073691, 9495340, 7664747, 7734960, 8389801, 1548757, 1850017, 1848606, 2166807, 2194551, 2157900, 2535744 |
Viruses also take advantage of a number of known short binding motifs for binding to host proteins. In addition to the sources presented in Table 1, previous reviews cover the viral use of motifs to bind host SH2 domains, PDZ domains, retinoblastoma protein (Rb), and Integrin receptors (21-27). Many viral proteins are known to have immunoreceptor tyrosine based motifs (ITAMs) that are usually found in transmembrane immune receptors.
Quite often viral consensus binding or posttranslational modification motifs are critical for the viral life-cycle and viral replication. A few of the many examples are provided. A PxxP motif in HIV Nef activates Src family kinases and is important for HIV replication (28, 29). A PPPY motif in Marbug Virus VP40 protein is important for its interaction with Tsg101 and viral budding (30). The N-myristoylation motif on the East African Cassava Mosaic Cameroon virus AC4 protein is responsible for plasma membrane targeting and pathogenicity (31). The carboxy-terminal PDZ binding motif of the E6 human papillomavirus protein is important for viral proliferation and maintenance of viral copy number (32).
There are many motifs in viral proteins that are important for the viral life-cycle but for which host targets are not yet known or perhaps mediate interaction between two viral proteins. For example, a YPPL motif in F13L vaccinia virus protein is required for efficient release of extracellular enveloped viruses (33). A KKR motif in cytoplasmic tail of Nef in Nipah Virus is important for its fusogenic activity (34). A PGQM motif in Gag is required for HIV infection (35, 36). A WxxF motif found in Vif mediates interaction with Vpr (37).
So why are short functional motifs so critical for viral function? One possibility is that viruses select motifs because motifs are often involved in the regulation of host molecules; the virus needs to deregulate pathways and then re-regulate these pathways to suit its needs. Therefore, motifs provide ready access to switching on and off specific cell processes, effectively allowing reprogramming of the cell. A single consensus motif may also provide the virus access to many factors that bind this motif. For example, a viral PxxP motif may target an array of different host factors containing SH3 domains. Short motifs may also provide a means by which the virus uses its small proteome to acquire many different host functions.
4. MOTIFS MUTATED IN HUMAN DISEASE
4.1. Binding and trafficking motifs
Since short peptide motifs are so important for the viral life-cycle and many short motifs are functional in mammalian proteins, it is a reasonable conjecture that mutation of motifs might represent a considerable fraction of disease-causing mutations in humans. However, it is not yet known the degree to which the short functional motifs are mutated in disease. We reviewed the literature and identified 20 diseases that are associated, at least in part with mutations that are in, or juxtaposed to short consensus motifs (Table 2).
Table 2.
Binding and trafficking motifs mutated in disease
| Disease | Protein | Motif Mutation | Motif function | Loss or gain of function | References |
|---|---|---|---|---|---|
| Usher Syndrome | SANS | DTxL>→VTxL> | PDZ binding motif | LOF | (41) |
| Idiopathic Hypercalciuria | Claudin 15 | TRV>→RRV> | PDZ binding motif for ZO-1 | LOF | (39) |
| Congenital Adrenal Hypoplasia | DAX-1 | LLxLxL→ LLxPxL LPxLxL |
Nuclear Receptor Binding motif | LOF | (130, 131) |
| Autoimmune Polyendocrinopathy-candidiasis-ectodermal Dystrophy | autoimmune regulator | LxxLL→ deletion | Nuclear Receptor Binding motif | LOF | (63) |
| Systemic Lupus Erythematosus | Ltk |
GxxYxxM→ ExxYxxM |
PI3-kinase SH2 binding motif | GOF | (46) |
| Diabetes mellitus | Irs-1 |
TxxxYxxM→ RxxxYxxM |
PI3-kinase SH2 binding motif | GOF | (45) |
| Pleiotropic Malformation Syndrome | STRA6 | YTLL→YMLL | Stat5 SH2 binding motif | LOF | (48) |
| Lesch-Nyhan Syndrome | Hypoxanthine-guanine phosphoribosyl transferase |
VLIVEDIIDTG → DLIVEDIIDTG VSIVEDIIDTG VLSIVEDIIDTG VLTVEDIIDTG VLMVEDIIDTG VLIMEDIIDTG VLIVEVIIDTG VLIVEGIIDTG VLIVEDKIDTG VLIVEDITDTG VLIVEDIIDTD |
phosphoribosyl-pyrophosphate binding motif | LOF | (65, 66) |
| Idiopathic Dilated Cardiomyopathy1 | Nebulette | SNxxYK or SKxxYK | Actin binding motif | LOF | (59) |
| Retinitis Pigmentosa | Rhodopsin | xVxPx> → xMxPx> xVxSx> deletion |
Arf4 bindng motif | LOF | (60, 61) |
| Noonan Syndrome | Raf-1 | RSxSTP→ RLxSTP SSxSTP RSxFTP RSxPTP RSxSRP RSxSIP RSxSTS RSxSTL |
14-3-3 binding motif | GOF | (43) |
| Nonsyndromic Hearing Loss | Myo7A | [VI]QxxxRGxxxR → [VI]QxxxRGxxxC |
IQ calmodulin binding motif | LOF | (56) |
| LEOPARD Syndrome | Raf-1 | RSxSTP → RLxSTP |
14-3-3 binding motif | GOF | (43) |
| Ovarian Cancer | Raf-2 | RSxSxP → RSxAxP |
14-3-3 binding motif | GOF | (44) |
| Myofbrillar Myopathy | ZASP | 26 residue motif | α-actinin binding motif | LOF | (42) |
| Colorectal carcinogenesis, Melanomas | β-catenin | DSGHDS →DSGHDF |
β-TrCP ubiquitin ligase adaptor recognition domain | LOF | (53, 54) |
| Liddle’s Syndrome | ENaC β subunit | PPPxY→PSPxY, PPLxY, PPSxY, PPRxY, PPPxH, PPHxY | Nedd4 WW domain binding motif | LOF | (49, 50) |
| Brugada Syndrome | Nav 1.5 Channel | VPIAxxESD→ VPIAxxKSD |
Ankyrin G binding motif | LOF | (55) |
| Rheumatoid Arthritis | PTP-N22 | PPLPxRTxxxxIV → PPLPxWTxxxxIV |
Csk SH3 domain binding motif | LOF | (57) |
| Tricho-rhino-phalangeal Syndromes | TRPS1 transcription factor | RRRTRKR→ RRRTRKC RRRTRKH |
Nuclear localization motif | LOF | (67) |
Polymorphisms are risk factors for this motif.
4.1.1. PDZ binding motifs
PDZ domains bind to different classes of short consensus motifs at the C-termini of proteins (38). Ushers Syndrome and Hypercalciuria are known to be caused by mutations of PDZ binding motifs that bind to proteins with PDZ domains. Mutations in the tight junction protein Claudin 16 cause Familial Hypercalciuria which progressively leads to renal failure. One mutation identified in Claudin 16 (T233RV235> → RRV>) causes a loss of function in this PDZ binding motif, blocks its association with ZO-1, and mistargets Claudin 16 to lysozomes (39).
Ushers syndrome is a heterogeneous disease with seven loci associated with deafness and blindness. The PDZ domain of Harmonin associates with a PDZ binding motif in SANS, two of the proteins associated with this disease (40). In a Turkish kindred, a mutation of D458TxL461 → VTxL> (where “>” indicates the C-terminus) was identified (41). Although the mutation is juxtaposed to the class I PDZ consensus motif ([ST]x[ILV]>), the mutation is predicted to disrupt this interaction based on structure models generated from other PDZ/motif complexes. Another mutation in a consensus PDZ binding motif was identified in the ZASP 2s isoform, a protein associated with muscular dystrophy, but the role of this motif in binding proteins with PDZ domains has not yet been determined (42).
4.1.2. 14-3-3 binding motifs
Both Noonan and LEAOPARD Syndromes result from mutations in Raf-1 (43). These syndromes present with common features including cardiac abnormalities, Facial Dysmorphia and short stature. All residues in Raf-1 that are important for binding 14-3-3 (R256SxSTP261) are mutated in Noonan Syndrome, and a S257L mutation has been observed in a patient with LEAOPARD Syndrome. Some of these mutations were shown to block binding to 14-3-3. Since 14-3-3 binding is a negative regulator of Raf-1 kinase activity, these mutations produce a gain of function in Raf-1 kinase. Ser259, a key residue in this motif that is also phosphorylated and mutated to Ala in ovarian cancer, but the effect of 14-3-3 binding to Raf-1 in ovarian cancer has not been explored (44).
4.1.3. SH2 binding motifs
SH2 domains bind phosphotyrosine residues. Mutation of an SH2 binding motif has not been shown to cause disease, however there are several reports that suggest SH2 motifs play a role in human disease. Diabetes mellitus can be caused by rare mutations in IRS-1 that lead to insulin resistance. Mutation of T608xxxYxxM615→ RxxxYxxM in IRS-1 alters a residue near the consensus YxxM motif for binding to the SH2 domain in the p85 subunit of phosphatidylinositol 3-kinase (PI3K) (45). This mutation effects association of IRS-1 with PI3K and insulin-induced PI3K activity, suggesting a role for a residue that is located four residues N-terminal to the critical tyrosine.
Likewise, a similarly important role for residues N-terminal to the tyrosine is suggested in Lupus, an autoimmune disease where the immune system attacks healthy cells (46). In systemic lupus erythematosus, two polymorphisms in Leukocyte Tyrosine Kinase (Ltk) are more prevalent in patients with disease than in control subjects (46). These mutations surround a Y753xxM756 motif for binding the SH2 domain of the p85 subunit of PI3K. One polymorphism results in a gain of function mutation of G750xxYxxM756→ E750xxYxxM756 that enhances binding to PI3K, suggesting that residue 750 is important for SH2 domain binding. In support of this observation, examination of the structure of the complex between PI3 kinase SH2 domain and a cKit peptide that contains the sequence TxxYxxM shows contacts between the Thr residue of cKit and Asn56 of the SH2 domain (47).
Pleiotropic Malformation Syndrome results from mutations in STRA6 a protein identified based on its retinoic acid-induced expression (48). A Y643TLL646→YMLL mutation in STRA6 has been identified in a consensus binding motif for the Stat5 SH2 domain, however the effect of this mutation of Stat5 and Stat5 signaling needs to be validated.
4.1.4. Ubiquitin ligase binding motifs
Liddle’s Syndrome presents with salt-sensitive hypertension and is associated with mutations in the β and γ subunits of the epithelial sodium channel (ENaC). The majority of missense mutations are concentrated in a cytoplasmic P614PPxY618 motif near the C-terminus of the β subunit (49, 50). Mutation of this motif increases the channel activity several fold and leads to retention of the activated channel at the cell surface. This short motif binds to the ubiquitin ligase Nedd4 through its WW domain and plays a role in stability of the channel (51, 52). Ubiquitin ligation is also effected in melanoma cell lines, where a D32SGHDS37 →DSGHDF mutation has been observed in β-catenin. The mutated SxxxS consensus motif in this sequence is normally phosphorylated and targeted for degradation by β-TrCP, an adaptor for a ubiquitin ligase (53, 54).
4.1.5. Ankyrin G binding motif
Brugada Syndrome is a fatal cardiac arrhythmia where an E1053K mutation has been identified in the Nav 1.5 sodium channel (55). This mutation alters a V1047PIAxxESD1055 motif in the cytosolic loop that binds to an unknown target on the surface of cardiomyocytes.
4.1.6. IQ Calmodulin binding motif
Mutations in Myosin 7A have been identified in patients with Nonsyndromic Hearing Loss (56). A R853C mutation disrupts an IQ motif ([VI843]QxxxRGxxxR853) known for binding to calmodulin. Experiments examining Calmodulin dependent vasoconstriction support a functional role for this motif in Calmodulin binding and a loss of function in hearing loss.
4.1.7. SH3 binding motif
Mutations in Protein Tyrosine Phosphatase (PTPN22) have been identified in patients with Rheumatoid Arthritis (57). The R620W mutation alters the P615PLPxRTxxxxIV627 consensus motif for binding the Csk SH3 domain and reduces its affinity for Csk (58).
4.1.8. Actin binding motif
Polymorphisms in the Nebulette protein are associated with an increased risk for Idiopathic Dilated Cardiomyopathy (59). Two of these polymorphisms (Asn654 or Lys654 are located in a actin binding motif (S653[DEQNS]xxYK658) and may affect binding to actin (59).
4.1.9. Arf4 binding motif
Mutations in the C-terminus of Rhodopsin cause Retinitis Pigmentosa (60, 61). Missense mutations (V345M and P347S) within, and a truncation mutation that eliminates a five residue xVxPx> motif from the C-terminus, blocks binding of Rhodopsin to the Arf4 GTPase, an interaction important for trafficking of Rhodopsin to the rod outer segment.
4.1.10. Nuclear hormone receptor binding motif
Mutations in two similar hormone receptor binding motifs have been identified. L295P and L297P mutations in the L294LxLxL299 motif in DAX-1 were identified in Congenital Adrenal Hypoplasia. This motif is thought to be involved in recruitment of transcription factor co-regulators. The more conventional motif for nuclear hormone receptor binding is a LxxLL (62). While not conclusive, a frameshift mutation at position Ala505 near the C-terminus of autoimmune regulator protein deletes a L516xxLL520 motif which may be involved in Autoimmune Polyendocrinopathy-candidiasis-ectodermal Dystrophy (63).
4.1.11. α-actinin binding motif
A 26 amino acid region of ZASP contains a ZM motif with the consensus definition of Q[YF]NxPxx[ML]YSxxx[IL] (64). Mutations A147T and A165V surrounding this motif are mutated in ZASP patients with Myofibrillar Myopathy (42). While the ZM motif binds to α-actinin, it is not known whether these mutations impair α-actinin binding.
4.1.12. 5-phosphoribosyl-1-pyrophosphate binding motif
Hypoxanthine-guanine Phosphoribosyltransferase is an enzyme that binds 5-phosphoribosyl-1-pyrophosphate through a V130LIVEDIIDTGK141 motif in its active site (65, 66). In Lesch-Nyhan Syndrome, 11 different mutations spanning the majority of these residues abrogate or impair the enzymatic activity.
4.1.13. Trafficking motifs
Several protein trafficking motifs are known to be mutated in disease. TRPS1 is a transcription factor that is deleted or mutated in Tricho-rhino-phalangeal Syndromes (TRPS) (67). The last Arg of known RRRTRKR nuclear localization motif is mutated. R952H or R952C mutations in these patients blocks nuclear translocation of TRPS1 in a transfected cell line. In HCN2 and Ether-a-go-go Related Gene (HERG), a C-terminal RxR motif is involved in endoplasmic reticulum retention (68, 69). In several patients with long QT syndrome the C-terminus is truncated, and mutation of these residues reduces channel conductance in transfected cells, thus the effects of the deletions are thought to be due to defective trafficking of the mutant protein through this ER retention motif. Although not directly mutated, a C-terminal dileucine motif involved in trafficking is lost in C-terminal deletions of ATP7A in Menkes Disease and a C-terminal peroxisomal SKL> targeting motif is lost in Maolonyl-CoA Decarboxylase, the gene mutated in Malonyl-CoA Decarboxylase Deficiency (70, 71).
4.2. Posttranslational modification motifs
Several mutations in phosphorylation sites, protease cleavage sites, and N-glycosylation sites are known to play a role in disease.
4.2.1. Phosphorylation sites
Kinases are one of the largest gene families in mammalian proteomes, and it is not surprising that deregulation of kinase activity is a common theme in disease. One treatment strategy has been to develop inhibitors of kinases. This has been particularly effective in cancer where drugs inhibiting a number of different kinases are effective and in current practice. Most well-studied kinases are known to phosphorylate multiple substrates, thus it is not known whether the efficacy of these drugs is due to broad inhibition of phosphorylation of many substrates or through selective inhibition of one or several key regulatory phosphorylation events.
Given that many phosphorylation consensus sequences can be abolished or created by introduction of a missense mutation and the fact that kinase inhibitors are effective for diseases, we expected that there would be many examples of disease causing mutations in consensus sequences of kinase substrates that effect substrate phosphorylation. While this is a very broad subject and it is difficult to be comprehensive, a thorough literature search revealed only seven known diseases in which mutation of phosphorylation sites are associated with disease (Table 3).
Table 3.
Motifs for posttranslational modifications mutated in disease
| Phosphorylation sites | |||||
| Liver Glycogenosis Type II | Phosphorylase kinase | RxxT→ RxxI | Phosphorylation site | LOF | (76) |
| Pleiotropic Malformation Syndrome | STRA6 | RxT→ CxT | PKA phosphorylation site | LOF | (48) |
| Maturity-onset Diabetes of the Young | HNF4α | RxG→RxS | PKA phosphorylation site | GOF | (73) |
| Schizophrenia | Synapsin III | SP→NP | MAPK phosphorylation site | LOF | (75) |
| Bukitt’s Lymphoma | cMyc | PTxxxS→ several | GSK3 phosphoylation site | LOF | (74) |
| Familial Advanced Sleep Phase Syndrome | Per2 | S662G within binding site | CKIε binding site | LOF | (72) |
| Familial Dysautonomia | IKAP | RIVT→PIVT | CamKII phosphorylation Site | LOF | (77) |
| Protease Cleavage sites | |||||
| Rickets | FGF-23 | RxxR→QxxR, RxxQ, RxxW | Protease cleavage site | LOF | (79) |
| Hypohidrotic Ectodermal Dysplasia | ectodysplasin-A | RxKR→ RxNR, CxKR, HxKR | Furin cleavage site | LOF | (81) |
| Intrauterine Growth Retardation | IGF1 Receptor | RKRR→RKQR | Protease cleavage site | LOF | (80) |
| Nonsyndromic Deafness | Low density-lipoprotein receptor A | RIVGG→LIVGG | Protease cleavage site | LOF | (86) |
| Haemophilia | Factor VIII | R372C, R1689C | Protein C, Thrombin cleavage site | LOF | (84, 85) |
| N-linked glycosylation | |||||
| (Many diseases) | ----- | ----- | N-glycosylation site | GOF | (89) |
| Retinitis Pigmentosa | Rhodopsin | NxS→NxN | N-glycosylation site | LOF | (90) |
| Metachromatic Leukodystrophy | Sphingolipid Activator protein B | NxT→HxT | N-glycosylation site | LOF | (92) |
| Creutzfeld-Jacob disease | Prion | NxT→NxA | N-glycosylation site | LOF | (91) |
Patients with familial advanced sleep phase syndrome contain a S662G mutation in the Per2 gene which is within a domain that binds to Casein Kinase Iε (72). This mutation blocks CK1ε mediated phosphorylation of Per2. Maturity-onset Diabetes of the Young can be caused by a gain of function G115S mutation in the hepatocyte nuclear factor 4α transcription factor (73). This mutation introduces a new consensus site for phosphorylation by PKA and PKA mediated phosphorylation of this site reduces transcriptional activity mediated by this transcription factor. Mutation of T58 in cMyc is a mutational hotspot in many lymphomas. This site is phosphorylated and mediates proteosome degradation of cMyc (74). A S470N polymorphism in Synapsin III is observed in schizophrenic patients more often than unaffected individuals (75). The polymorphism eliminates a MAPK phosphorylation site and the polymorphism is also associated with reduced phosphorylation of Synapsin III.
Several phosphorylation sites are implicated in disease, but phosphorylation by a specific kinase has not yet been demonstrated. X-linked Liver Glycogenosis Type II have a deficiency in Phosphorylase Kinase activity. Several of these mutations cluster around a R1111EMT1114 sequence with a T→I mutation or an insertion after R1111; These mutations eliminate a consensus phosphorylation site for several kinases (76). In patients with Familial Dysautonomia, the IκB kinase complex-associated protein (IKAP) has a R696→P mutation in a RxxT consensus sequence for Calmodulin Kinase II phosphorylation (77). This mutation leads to reduced phosphorylation of IKAP in biosynthetic labeling experiments. In Plieotrophic Malformation Syndrome, a homozygous mutation in STRA6 is observed (48). This mutation causes a R655C residue change that eliminates a consensus phosphorylation site (RxT) for Protein Kinase A.
We were surprised at how few phosphorylation sites were known to be mutated in disease. This result supports the hypothesis that deregulation of a kinase results in phosphorylation of multiple downstream targets and that more than one target is necessary for causing disease. We can therefore generalize that activating a kinase likely causes phosphorylation of multiple downstream targets and can cause disease, but phosphomimetic mutations of a single phosphorylation site that is a target of one of these kinases does not lead to disease with the exception of the gain of function site in Hepatocyte nuclear factor 4α transcription factor as noted above. This suggests that drugs that target kinases act at a systemic level by blocking multiple important phosphorylation events.
4.2.2. Protease cleavage sites
As discussed above for kinases, another general class of therapeutic compounds that block enzyme activity is inhibitors of proteases. For example, inhibitors targeting the HIV protease are one of the currently used strategies to control HIV infection (78). Most proteases cleave many substrates. For example, all prohormones are processed at dibasic cleavage sites by a small number of Prohormone Convertases.
We wanted to know if there were any diseases that resulted from incorrect protease processing of a specific site. Five examples were identified and are shown in Table 3. FGF23 is processed from a prohormone at a R176xxR179 subtilisin-like proprotein convertase cleavage site. In autosomal dominant Hypophosphatemic Rickets, R176Q, R179Q, and R179W mutations are observed in FGF23 (79). These mutations block proteolytic processing of FGF23. Insulin like Growth Factor 1 Receptor (IGF-1R) is processed from a pro-receptor by proteolytic cleavage. In Intrauterine Growth Retardation, a heterozygous mutation in IGF-1R mutates the proteolysis consensus sequence from 707RKRR710 to RKQR (80). This mutation leads to increased amounts of proreceptor and decreased IGF binding to cells. Furin cleaves Ectodysplasin-A at a RVRRNKR159 consensus cleavage site. 20% of patients with X-linked Hypohidrotic Ectodermal Dysplasia have a mutation in one of the five basic residues, with three of these mutations located within a consensus furin cleavage site suggesting that defective proteolytic processing is one mechanisms of this disease (81). von Willebrand disease is caused by mutations in von Willebrand Factor. One proband has a mutation that codes for R760C change in amino acid sequence that eliminates a protease consensus site for Furin (82). This mutation leads to the presence of unprocessed von Willebrand factor in the plasma. In venous thrombosis patients a R506Q mutation has been identified in Factor V protease. These mutations eliminate a cleavage site for activated Protein C responsible for Factor V degradation and result in increased thrombin generation and hypercoagulation (83).
In several cases, the literature search revealed proteases that had mutations that blocked the processing of the protease from its pro- (inhibitory/zymogen) form. Thrombin cleaves Factor VIII, a protease in the proteolytic blood clotting cascade. In Haemophilia A patients with R372C or R1689C mutations, consensus sequences for Thrombin cleavage is altered and leads to increased blood levels of Factor VIII. These results suggest that cleavage and activation of Factor VIII protease is blocked by these mutations (84, 85). Mutations in Non-syndromic Deafness map to a serine protease (TMPRSS3) which is cleaved at a 216RIVGG220 sequence to activate the zymogen. A R216L mutation was found in this motif which impairs proteolytic activation, results in completely inactive enzyme, and is likely responsible for disease presentation in this patient (86).
All disease caused by mutation of a protease substrate were loss of function, we did not identify a case where disease was caused by introduction of a new proteolytic site that lead to aberrant processing of a protein. Although not in Table 3, there are a number of mutations in Amyloid Precursor Protein (APP) (including French, Iranian, Austrian, and German mutations) that surround the γ-secretase cleavage site and are involved in Alzheimer’s disease (87). Thus, altered processing of APP identifies another motif targeted in disease. While the Swedish mutant of APP has a KM→NL mutation in the β-secretase cleavage site, these changes do not alter the proteolytic activity toward this site (88).
4.2.3. N-glycosylation sites
Aberrant N-glycosylation seems to be common in disease. There are 32 known gain of function N-glycosylation mutations that span a wide array of disease and have previously been reviewed (89). There are a few loss of function glycosylation mutations. In Retinitis Pigmentosa a Ser/Asn substitution in the NxS N-glycosylation consensus sequence eliminates an N-glycosylation site (90). In Creutzfeld-Jacob disease a T183A substitution is observed in at least one patient. This mutation is in the N-glycosylation consensus sequence (NxT) of Prion protein that eliminates glycosylation at this site (91). In Metachromatic Leukodystrophya a N215H mutation in Sphingolipid Activator Protein B is observed. This mutation eliminates a NxT N-glycosylation site, caused loss of glycosylation, and inactivated the protein (92).
4.2.4. Discussion of motifs mutated in disease
Our review of the literature identified 20 binding motifs and 15 motifs for posttranslational modifications that are mutated in human disease. There were no unifying themes regarding type of motif, disease, organ, etc. Since many motifs are known to cause disease, the 35 binding and posttranslational modification motifs summarized in this review suggest that a disproportionately small number of short functional motifs are mutated in disease. However, more than 32 N-glycosylation motifs are associated with disease (89). Since N-glycosylation motifs play an important role in folding of proteins that transit through the secretory pathway, this motif is unique among short consensus motifs because it plays a role in protein folding and misfolding is a major disease mechanism. There are a number of possible explanations as to why so few motifs are mutated in disease which are further discussed in section 6.
5. MOTIF MIMETIC DRUGS
5.1. Receptor agonists
The majority of drugs approved by the Food and Drug Administration (FDA) target receptors and enzymes. A few peptide hormone receptors bind to short peptide hormones that can be considered motifs. Examples include Enkephalins, Vasopressin, Oxytocin, Thyroid Releasing Hormone, Cholecystokinin, and Somatostatin-14 peptides. In addition, evolutionary conservation of sequences for peptide hormones often shows a conserved cluster of ∼5 residues, suggesting that a short motif core of peptide hormone residues may be responsible for interaction with their receptors. Consistent with this hypothesis mapping of important contact residues for growth hormone binding to the Growth Hormone Receptor shows a similar size core binding motif (93, 94). In several cases, peptide hormones are therapeutically used: Somatuline Depot® (Lanreotide) is a Somatostatin octapeptide analog recently approved for treating Acromegalic patients and Byetta® (Exenatide) is similar to Glucagon-like Peptide 1 (GLP-1) and is used for treating Type II Diabetes. Several recombinant or synthetic peptide hormones such as Insulin, Somatostatin and Angiotensin are approved for various endocrinological disorders.
Non-peptide agonists have been identified that bind to the Insulin, Thrombopoietin, Wassopressin, Somatostatin, Bradykinin, Cholecystokinin, Angiotensin, Melatonin, Growth Hormone, Opioid, and Glucagon-like Peptide 1 receptors. Thus, in the case of receptors small molecule agonists that mimic core regions of peptide hormones seems to be a viable strategy for drug design. Several examples are provided. Supprelin LA® (Histrelin) is a Gonadotropin Releasing Hormone (GnRH) agonist used for treating the early onset of puberty. Demerol® (Meperidine) is an Opiate agonist for the κ-opioid receptor; Morphine, Fentanyl, Methadone, Oxycodone, and Codeine are agonists for the μ-opioid receptor. A comparison of Morphine as a mimetic of Enkaphalin peptides is shown in Figure 1.
Figure 1.
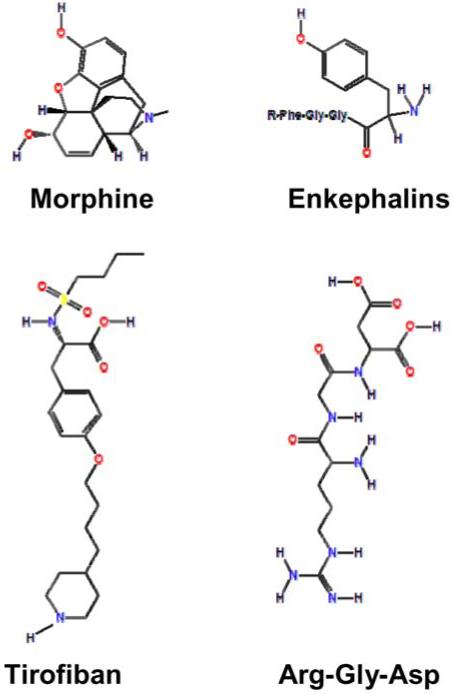
Chemical structures of FDA-approved motif mimetic drugs. Comparisons of the chemical structures of small compounds (Morphine and Aggrastat® (Tirofiban)) to the peptide motifs they mimic (Enkephalins and RGD Integrin ligands), respectively. The R group in Enkephalins can be Met for Met-enkaphalin, Leu for Leu-enkephalin, or other peptide for longer Enkephalin peptides such as β-endorphin.
5.2. POSTTRANSLATIONAL MODIFICATION INHIBITORS
5.2.1. Kinase inhibitors
Despite the many important functions of protein phosphorylation in cells, there are no known drugs that directly target protein phosphorylation sites in proteins. However, over the past decade a number of kinase inhibitors have emerged for treating various cancers and immunosupression. Stutnet® (Sunitinib) is a receptor tyrosine kinase (RTK) inhibitor for treating gastrointestinal tumors. Similarly, Sprycel® (Dasatinib) is a broad-based tyrosine kinase inhibitor that inhibits Bcr-Abl, Src, and other protein tyrosine kinases for treating chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL). Gleevec® (Imatinib Mesylate) is a general protein tyrosine kinase inhibitor approved for use in CML and Iressa® (Gefitinib) is a tyrosine kinase inhibitor for lung cancer (95). Another broad-based Ser/Thr and Tyr kinase inhibitor is Nexavar® (Sorafenib) which is used for advanced Renal Cell Carcinoma. Tarceva® (Erlotinib) is an Epidermal Growth Factor Receptor inhibitor used for treating Non-small Cell Lung Cancers (96). The majority of these small compound inhibitors of kinase activity are relatively non-specific, yet therapeutically effective. A number of neutralizing antibodies to specific RTKs are also in clinical use for various types of cancer. These have advantages in specificity, but therapeutic antibodies are limited by delivery when compared to small molecule drugs. Beyond cancer, Siromilus® (Rapamycin) is used to block organ rejection by inhibiting mTOR, a kinase involved in cytokine-stimulated T-cell proliferation (97).
Protein kinases, including the targets of most of the aforementioned inhibitors, generally phosphorylate many cellular proteins. Since many of these inhibitors have specificity limited to a precise subset of kinases in the kinome, it is likely that these drugs inhibit multiple phosphorylation events in cells. It is not yet clear whether the benefit of these drugs is from a single phosphorylation site or inhibition of multiple sites. Since very few known phosphorylation site mutations lead to disease (previous section) and no known drugs target phosphorylation sites, the actions of these drugs are likely mediated through perturbation of a number of different phosphorylation sites and act at a systemic level.
5.2.2. Protease inhibitors
Proteases cleave recognition motifs in protein substrates. Several mutations in protease motifs lead to disease (previous section), suggesting that at lease some proteases are specific and might make good targets for drugs. There are several FDA approved protease inhibitors. Tritace ® (Ramipril), Vasotec® (Enalapril), Accupril® (Quinapril), Monopril® (Fosinopril), and Lotensin® (Benzapril) are a substrate mimetic Angiotensin-converting Enzyme inhibitors (98). Novastan® (Argatroban) is a Thrombin inhibitor. Januvia® (Sitagliptin) is a DPP-4 dipeptidyl peptidase enzyme inhibitor for treating Type II Diabetes. There are numerous inhibitors of the HIV proteases and this approach has been one of the principal means for treating Autoimmune Immunodeficiency Syndrome (AIDS) (78).
5.2.3. Other posttranslational modifications
Many proteins are modified by lipidation of specific motifs in the amino and carboxy-termini of proteins. Two drugs in this class are approved by the FDA. Zovirax® (Acyclovir) is an antiviral myristoylation inhibitor that blocks N-terminal myristoylation of viral proteins, again highlighting the critical importance of motifs in viral infection. Valtrex® (Valacyclovir) is rapidly converted to Acylovir and is for treating herpes simplex virus. Another lipidation motif is a CAAx> box (A = aliphatic residue) which is on the C-terminus of proteins that are modified by covalent attachment of farnysyl or geranylgernayl group on the Cys residue. This modification is catalyzed by Farnesyl Transferase I and Gernanylgeranyl Transferases I and II, respectively. Inhibitors for these enzymes are being explored for treatment of cancer (99, 100). Farnysyl Transferase inhibitors reached Phase III trials, but, by themselves produced modest effects and are now being explored for treatment in combination with other drugs and are currently in clinical trials. Inhibitors of the Geranylgeranyl Transferases are also being explored (12. 100).
5.3. Inhibitors of protein-protein interactions in treating disease
Despite an enormous research effort toward understanding and inhibiting protein-protein interactions, no drugs that disrupt intracellular protein-protein interactions are currently approved by the FDA for treatment of disease. This is likely due, in part, to the focus of big pharmaceutical companies on receptors, ion channels, and enzymes. Notwithstanding, this is an active area of research.
Potential extracellular targets are the Integrin Receptors. Proteins with an RGD motif bind to approximately ½ of the 20 known Integrin Receptors; these receptors mediate focal adhesion attachment to the extracellular matrix. Investigators have attempted to exploit this motif in various ways, most of which have been extensively reviewed elsewhere, but are summarized below.
Two RGD mimetic drugs are antagonists of the Integrin αIIbβ3 (Glycoprotein IIb/IIIa Receptor), which is involved in platelet thrombosis. Eptifibatide® (Integrilin) is a cyclic hexapeptide containing the RGD sequence that binds to the Glycoprotein IIb/IIIa Receptor (101-105). Co-administered with aspirin or Clopidogrel (low molecular weight heparin), this cocktail is used to treat Unstable Angina or a Non-ST-segment-elevation Myocardial Infarction. Similarly, Aggrastat® (Tirofiban) is a low molecular weight synthetic RGD mimetic (Figure 1) used to treat Unstable Angina and Non-Q-wave Myocardial Infarction (101).
In addition to its approved medical uses, the RGD motif is also being explored in several ways for treatment of cancer. RGD peptides can also be used to treat tumors by targeting toxins, radioactive chemicals, antimitotic drugs, and DNA replication inhibitors to tumors (106-113). Similarly, tumors and tissues can be visualized using RGD peptide conjugates (109, 108, 114). Cilengitide is a RGD cyclic pentapeptide in Phase II trial that inhibits angiogenesis (115-117). Other uses of RGD peptides are in increasing attachment to solid supports for increasing adhesion of medical implants and grafts, and in treatment of Acute Renal Injury, and Osteoporosis (118-122).
Several other motifs are being explored as drug targets. Aminopeptidase N is a protease expressed on the cells surface of tumor blood vessels. A hexapeptide/Tumor Necrosis Factor conjugate containing a NGR motif acts as a tumor-homing conjugate (123). Some motifs that bind to intracellular domains are being explored as drug targets. PDZ motifs have well defined binding clefts and proteins containing PDZ domains are involved in pain perception, Epilepsy, Cancer, Schizophrenia, and Parkinson’s disease (124). Drugs that target PDZ-mediated interactions are under investigation. High affinity peptides that bind the SH3 domain of the Crk adaptor are under investigation as anticancer drugs (125). Peptides targeting the Grb2 and Crk SH2/SH3 adaptors are under investigation for treating several diseases (126). NK109 is an antitumor drug that binds to a PNxxxxP motif present in the C2 domain of Protein Kinase Cα (127). Nutlin-3 is a small molecule inhibitor that blocks the motif-driven interaction of MDM2 and MDMX with p53 (128). Treatment of mouse models of Retinoblastoma with Nutlin-3 and a Topoisomerase inhibitor Hycamantin® (topotecan hydrochloride) drastically reduced tumor burden.
6. PERSPECTIVE ON SHORT FUNCTIONAL MOTIFS IN HUMAN HEALTH
6.1. Molecular vulnerability of the human body to short motifs
Investigation of disease causing mutations, drugs, pathogens, and disease models teach us about the vulnerability of the human body. Disease causing mutations and FDA approved drugs suggest that enzymes, hormones, and receptors are particularly vulnerable sites. We questioned whether short functional motifs are also a point of human vulnerability. With the exception of N-glycosylation motifs which effects protein folding in the endoplasmic reticulum, our literature analysis revealed that few short functional motifs are known to be mutated in human disease. We must consider several possible explanations: 1. mutation of many short motifs are embryonic lethal and most individuals harboring such mutations die 2. short motifs have unimportant functions 3. short motifs are important at the systemic level, but any single motif rarely effects viability 4. the majority of functional motifs have not been identified so that we are unaware of their role in disease, or 5. the majority of diseases have not been identified so we are not aware of the role of motifs in the yet-to-be-discovered disease.
To gain a brute-force understanding at the genome level and address these possible explanations, we examined some statistics of mouse models using the Mouse Genome Database (129). Of the 4,289 genes thus far deleted in targeted knock-out mice, 1897 are lethal, suggesting that ∼44% of mouse genes are necessary for survival. Of the 1,897 genes required for mouse viability, 1,742 were lethal at embryonic, perinatal, or postnatal stage and another 155 were lethal at a later stage in life. These 155 genes produce viable organisms with defects that cause death later in life and can be considered an equivalent of disease in humans. By extrapolation, the disease causing genes represent only a small percentage of mouse genes.
While we cannot rule out that a large proportion of human motif mutations result in embryonic lethality, this seems unlikely in consideration of the small proportion of genes that cause disease in mice. Although mutation of ∼44 % of mice genes results in lethality, motifs often only diminish function of a protein and we did not identify any cases where mutation of a motif is similar to a null phenotype for the gene. We are in the early stages of short motif discovery and it is possible that many more mutated motifs will be identified as more motifs and more specific motif definitions are accumulated. In our literature analysis, a common theme was an observation that residues juxtaposed to a consensus sequence, or within a portion of a motif not thought to impart specificity, were mutated. This suggests that current motif definitions may not be exact and need improvement. However, to this point it seems like mutation of motifs that result in disease is a rare occurrence.
We favor the hypothesis that most motifs are important at the systemic level, but mutation of a single motif does not perturb the system enough to affect viability. Since few intracellular motifs are mutated in disease (sections 3 and 4), it appears that human cellular physiology is not critically dependent on any one motif for function. For example, if a kinase phosphorylates 50 targets and five are critical for viability then knocking out the kinase will cause death, but knocking out any one phosphorylation site will not. In this scenario, all five critical phosphorylation sites would need to be mutated to cause death. Effectively, this system architecture reduces vulnerability of the organism. However, opportunistic pathogens such as viruses could readily exploit cellular systems that are driven through short motif recognition and this seems to be the case (see section 2). There are a couple of specific cases, where this does not hold true. Asparagine-linked glycosylation motifs are involved in the protein folding quality control pathway of the endoplasmic reticulum and both gain and loss of function mutations for this motif are found in disease (89). We think this is a point of vulnerability because of its role in effecting folding, as misfolding is a primary disease mechanism. The other case is that for extracellular peptide hormones that have core motifs that bind to receptors. Many of the cell-to-cell signaling systems are not redundant, vulnerable to mutation in disease, and may be one reason why such a larger proportion of drugs target receptors and peptide hormones.
One consideration regarding our current knowledge of motifs is that motifs may need to be much more specific than currently motif definitions describe. Many motifs have highly redundant motif definitions and are found at greater than 100,000 copies in the human proteome; clearly not all of these putative sites are used. This over-prediction of short motifs may be due to the fact that motifs must work within a specific context such as being on the protein surface, be localized to a specific compartment, or being expressed with its target in the same cell or during a specific point of development. This may hinder our ability to recognize motifs that are mutated in disease.
6.2. Insights into drugs design strategies
The majority of FDA-approved drugs target enzymes, receptors, and channels. Many of these drugs are screened based on a specific target. Most of these targets are likely to have non-redundant functions in cells. Only a few drugs that mimic short motifs exist (Figure 1) and in general, drugs that block protein-protein interactions are not routinely used in treatment of disease. This could be because pharmaceutical companies do not explore protein-protein interactions as potential drug targets. However, as covered in Section 5, there has been reasonable interest in exploring a number of motifs as druggable targets. Since viruses have evolved to use motifs for essential functions by hijacking host proteins, one potential area where motif mimetic drugs may be useful is for treating viral infection and infection by other pathogens. Another way that motif mimetic drugs may be useful is as a cocktail targeting multiple targets. For example, an inhibitor of a tyrosine kinase can disrupt phosphorylation of many proteins and processes. If the few critical phosphorylation sites were known, a cocktail of motif mimetic drugs that that block phosphorylation of the key sites might be just as effective as the tyrosine kinase inhibitor, but present less problems with toxicity through non-desirable effects of the drug on other phosphorylation sites and cell processes. Such drug cocktails may be more effective by perturbing the systems in different ways. For example, in controlling the spread of HIV, a protease inhibitor cocktail that targets the HIV protease and reverse transcriptase is more effective than a single protease inhibitor. Much remains to be learned about motifs, but given their widespread role in cell function and physiology, they are like to be part of the future of drug design.
7. ACKNOWLEDGEMENTS
We would like to thank Dr. Michael Gryk for critically reading this manuscript and want to acknowledge members of the Minimotif Miner team for intellectual discussions related to topics of this review. We thank the NIH (GM079689 to M.R.S.) and the University of Connecticut Partnership for Excellence in Structural Biology (K.K.) for funding. Krishna Kadaveru and Jay Vyas contributed equally to this work.
Footnotes
Publisher's Disclaimer: This is an, un-copyedited, author manuscript that has been accepted for publication in the Frontiers inBioscience”. Cite this article as appearing in the Journal of Frontiers in Bioscience. Full citation can be found by searching the Frontiersin Bioscience (http://bioscience.org/search/authors/htm/search.htm) following publication and at PubMed(http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed) following indexing. This article may not be duplicated orreproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code)without permission of the copyright holder, the Frontiers in Bioscience. From the time of acceptance following peer review, the full finalcopy edited article of this manuscript will be made available at http://www.bioscience.org/. The Frontiers in Bioscience disclaims anyresponsibility or liability for errors or omissions in this version of the un-copyedited manuscript or in any version derived from it by the National Institutes of Health or other parties.
8. REFERENCES
- 1.Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A. The PROSITE database, its status in 2002. Nuc Acids Res. 2002;30:235–238. doi: 10.1093/nar/30.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balla S, Thapar V, Luong T, Faghri T, Huang CH, Rajasekaran S, del Campo JJ, Shin JH, Mohler WA, Maciejewski MW, Gryk M, Piccirillo B, Schiller SR, Schiller MR. Minimotif Miner, a tool for investigating protein function. Nat Methods. 2006;3:175–177. doi: 10.1038/nmeth856. [DOI] [PubMed] [Google Scholar]
- 3.Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DMA, Ausiello G, Brannetti B, Costantini A, Ferre F, Maselli V, Via A, Cesareni G, Diella F, Superti-Furga G, Wyrwicz L, Ramu C, McGuigan C, Gudavalli R, Letunic I, Bork P, Rychlewski L, Kuster B, Helmer-Citterich M, Hunter WN, Aasland R, Gibson TJ. ELM server: a new resource for investigating short functional sites in modular eukaryotic proteins. Nuc Acids Res. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neduva V, Russell RB. DILIMOT: discovery of linear motifs in proteins. Nuc Acids Res. 2006;34:W350–W355. doi: 10.1093/nar/gkl159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nuc Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davey NE, Shields DC, Edwards RJ. SLiMDisc: short, linear motif discovery, correcting for common evolutionary descent. Nuc Acids Res. 2006;34:3546–3554. doi: 10.1093/nar/gkl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grande SM, Ross SR, Monroe JG. Viral immunoreceptor-associated tyrosine-based activation motifs: potential players in oncogenesis. Future Oncol. 2006;2:301–310. doi: 10.2217/14796694.2.2.301. [DOI] [PubMed] [Google Scholar]
- 8.Grande SM, Bannish G, Fuentes-Panana EM, Katz E, Monroe JG. Tonic B-cell and viral ITAM signaling: Context is everything. Immunol Rev. 2007;218:214–234. doi: 10.1111/j.1600-065X.2007.00535.x. [DOI] [PubMed] [Google Scholar]
- 9.Boutin JA. Myristoylation. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- 10.Maurer-Stroh S, Eisenhaber F. Myristoylation of viral and bacterial proteins. Trends Microbiol. 2004;12:178–185. doi: 10.1016/j.tim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Harper DR, Gilbert RL. Viral Lipoproteins - the Role of Myristoylation. Biochem Soc Trans. 1995;23:553–557. doi: 10.1042/bst0230553. [DOI] [PubMed] [Google Scholar]
- 12.Einav S, Glenn JS. Prenylation inhibitors: a novel class of antiviral agents. J. Antimicrob Chemother. 2003;52:883–886. doi: 10.1093/jac/dkg490. [DOI] [PubMed] [Google Scholar]
- 13.Glenn JS. Prenylation of HDAg and antiviral drug development. Curr Top Microbiol Immunol. 2006;307:133–149. doi: 10.1007/3-540-29802-9_7. [DOI] [PubMed] [Google Scholar]
- 14.Grose C. Glycoproteins encoded by varicella-zoster virus: biosynthesis, phosphorylation, and intracellular trafficking. Annu Rev Microbiol. 1990;44:59–80. doi: 10.1146/annurev.mi.44.100190.000423. [DOI] [PubMed] [Google Scholar]
- 15.Sugrue RJ. Viruses and glycosylation: an overview. Methods Mol Biol. 2007;379:1–13. doi: 10.1007/978-1-59745-393-6_1. [DOI] [PubMed] [Google Scholar]
- 16.Vigerust DJ, Shepherd VL. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 2007;15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.zur Wiesch JS, Schmitz H, Borowski E, Borowski P. The proteins of the Hepatitis C virus: Their features and interactions with intracellular protein phosphorylation. Arch Virol. 2003;148:1247–1267. doi: 10.1007/s00705-003-0115-8. [DOI] [PubMed] [Google Scholar]
- 18.Jakubiec A, Jupin I. Regulation of positive-strand RNA virus replication: the emerging role of phosphorylation. Virus Res. 2007;129:73–79. doi: 10.1016/j.virusres.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrd CM, Hruby DE. Vaccinia virus proteolysis--a review. Rev Med Virol. 2006;16:187–202. doi: 10.1002/rmv.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kido H, Okumura Y, Yamada H, Le TQ, Yano M. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharmacol Des. 2007;13:405–414. doi: 10.2174/138161207780162971. [DOI] [PubMed] [Google Scholar]
- 21.Bliska J. How pathogens exploit interactions mediated by SH3 domains. Chem Biol. 1996;3:7–11. doi: 10.1016/s1074-5521(96)90076-9. [DOI] [PubMed] [Google Scholar]
- 22.Hiraiwa A, Ishibashi M. Binding of viral oncogenic proteins and PDZ domains of cellular proteins. Tanpakushitsu Kakusan Koso. 43:237–243. 198. [PubMed] [Google Scholar]
- 23.Lee C, Cho Y. Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev Med Virol. 2002;12:81–92. doi: 10.1002/rmv.340. [DOI] [PubMed] [Google Scholar]
- 24.Oldak M, Smola-Hess S, Maksym R. Integrin beta4, keratinocytes and papillomavirus infection. Int J Mol Med. 2006;17:195–202. [PubMed] [Google Scholar]
- 25.Switala-Jelen K, Dabrowska K, Opolski A, Lipinska L, Nowaczyk M, Gorski A. The biological functions of beta 3 integrins. Folia Biol. 2004;50:143–152. doi: 10.14712/fb2004050050143. [DOI] [PubMed] [Google Scholar]
- 26.Triantafilou K, Takada Y, Triantafilou M. Mechanisms of integrin-mediated virus attachment and internalization process. Crit Rev Immunol. 2001;21:311–322. [PubMed] [Google Scholar]
- 27.Nemerow GR, Stewart PL. Role of alpha(v) integrins in adenovirus cell entry and gene delivery. Microbiol Mol Biol Rev. 1999;63:725–734. doi: 10.1128/mmbr.63.3.725-734.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trible RP, Emert-Sedlak L, Smithgall TE. HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J Biol Chem. 2006;281:27029–27038. doi: 10.1074/jbc.M601128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fackler OT, Wolf D, Weber HO, Laffert B, D’Aloja P, Schuler-Thurner B, Geffin R, Saksela K, Geyer M, Peterlin BM, Schuler G, Baur AS. A natural variability in the proline-rich motif of Nef modulates HIV-1 replication in primary T cells. Curr Biol. 2001;11:1294–1299. doi: 10.1016/s0960-9822(01)00373-6. [DOI] [PubMed] [Google Scholar]
- 30.Urata S, Noda T, Kawaoka Y, Morikawa S, Yokosawa H, Yasuda J. Interaction of Tsg101 with Marburg Virus VP40 Depends on the PPPY Motif, but Not the PT/SAP Motif as in the Case of Ebola Virus, and Tsg101 Plays a Critical Role in the Budding of Marburg Virus-Like Particles Induced by VP40, NP, and GP. J Virol. 2007;81:4895–4899. doi: 10.1128/JVI.02829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fondong VN, Reddy RVC, Lu C, Hankoua B, Felton C, Czymmek K, Achenjang F. The consensus N-myristoylation motif of a geminivirus AC4 protein is required for membrane binding and pathogenicity. Mol Plant Microbe Interact. 2007;20:380–391. doi: 10.1094/MPMI-20-4-0380. [DOI] [PubMed] [Google Scholar]
- 32.Lee CH, Laimins LA. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J Virol. 2004;78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honeychurch KM, Yang G, Jordan R, Hruby DE. The vaccinia virus F13L YPPL motif is required for efficient release of extracellular enveloped virus. J Virol. 2007;81:7310–7315. doi: 10.1128/JVI.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aguilar HC, Matreyek KA, Choi DY, Filone CM, Young S, Lee B. Polybasic KKR Motif in the Cytoplasmic Tail of Nipah Virus Fusion Protein Modulates Membrane Fusion by Inside-Out Signaling. J Virol. 2007;81:4520–4532. doi: 10.1128/JVI.02205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubota S, Adachi Y, Copeland TD, Oroszlan S. Binding of Human Prothymosin-Alpha to the Leucine-Motif/Activation Domains of Htlv-I Rex and Hiv-1 Rev. Eur J Biochem. 1995;233:48–54. doi: 10.1111/j.1432-1033.1995.048_1.x. [DOI] [PubMed] [Google Scholar]
- 36.Srivastava M, Cartas M, Rizvi TA, Singh SP, Serio D, Kalyanaraman VS, Pollard HB, Srinivasan A. HIV-1 Gag shares a signature motif with annexin (Anx7), which is required for virus replication. Proc Nat Acad Sci U S A. 1999;96:2704–2709. doi: 10.1073/pnas.96.6.2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouhamdan M, Xue YN, Baudat Y, Hu BC, Sire J, Pomerantz RJ, Duan LX. Diversity of HIV-1 Vpr interactions involves usage of the WXXF motif of host cell proteins. J Biol Chem. 1998;273:8009–8016. doi: 10.1074/jbc.273.14.8009. [DOI] [PubMed] [Google Scholar]
- 38.Hung AY, Sheng M. PDZ domains: Structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- 39.Muller D, Kausalya PJ, Claverie-Martin F, Meij IC, Eggert P, Garcia-Nieto V, Hunziker W. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Amer J Hum Genet. 2003;73:1293–1301. doi: 10.1086/380418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weil D, El Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- 41.Kalay E, de Brouwer APM, Caylan R, Nabuurs SB, Wollnik B, Karaguzel A, Heister JGAM, Erdol H, Cremers FPM, Cremers CWRJ, Brunner HG, Kremer H. A novel D458V mutation in the SANS PDZ binding motif causes atypical Usher syndrome. J Mol Med-Jmm. 2005;83:1025–1032. doi: 10.1007/s00109-005-0719-4. [DOI] [PubMed] [Google Scholar]
- 42.Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005;57:269–276. doi: 10.1002/ana.20376. [DOI] [PubMed] [Google Scholar]
- 43.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, Lopez Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 44.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, Defazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esposito DL, Li YH, Vanni C, Mammarella S, Veschi S, Della Loggia F, Mariani-Costantini R, Battista P, Quon MJ, Cama A. Clinical case seminar - A novel T608R missense mutation in insulin receptor substrate-1 identified in a subject with type 2 diabetes impairs metabolic insulin signaling. J Clin Endocrin Metab. 2003;88:1468–1475. doi: 10.1210/jc.2002-020933. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Nakamura K, Jiang Y, Tsurui H, Matsuoka S, Abe M, Ohtsuji M, Nishimura H, Kato K, Kawai T, Atsumi T, Koike T, Shirai T, Ueno H, Hirose S. Gain-of-function polymorphism in mouse and human Ltk: implications for the pathogenesis of systemic lupus erythematosus. Hum Mol Genet. 2004;13:171–179. doi: 10.1093/hmg/ddh020. [DOI] [PubMed] [Google Scholar]
- 47.Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, Harrison SC. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat Struct Biol. 1996;3:364–374. doi: 10.1038/nsb0496-364. [DOI] [PubMed] [Google Scholar]
- 48.Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nurnberg G, Schirmer-Zimmermann H, Tolmie JL, Chitayat D, Houge G, Fernandez-Martinez L, Keating S, Mortier G, Hennnekam RC, von der Wense A, Slavotinek A, Meinecke P, Bitoun P, Becker C, Nurnberg P, Reis A, Rauch A. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furuhashi M, Kitamura K, Adachi M, Miyoshi T, Wakida N, Ura N, Shikano Y, Shinshi Y, Sakamoto K, Hayashi M, Satoh N, Nishitani T, Tomita K, Shimamoto K. Liddle’s syndrome caused by a novel mutation in the proline-rich PY motif of the epithelial sodium channel beta-subunit. J Clin Endocrinol Metab. 2005;90:340–344. doi: 10.1210/jc.2004-1027. [DOI] [PubMed] [Google Scholar]
- 50.Wang WQ, Zhou WW, Jiang L, Cui B, Ye L, Su TW, Wang JG, Li XY, Ning G. Mutation analysis of SCNN1B in a family with Liddle’s syndrome. Endocrine. 2006;29:385–390. doi: 10.1385/ENDO:29:3:385. [DOI] [PubMed] [Google Scholar]
- 51.Staub O, Yeger H, Plant PJ, Kim H, Ernst SA, Rotin D. Immunolocalization of the ubiquitin-protein ligase Nedd4 in tissues expressing the epithelial Na+ channel (ENaC) Amer J Physiol. 1997;41:C1871–C1880. doi: 10.1152/ajpcell.1997.272.6.C1871. [DOI] [PubMed] [Google Scholar]
- 52.Staub O, Dho S, Henry PC, Correa J, Ishikawa T, Mcglade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. Embo J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 53.Wu G, He X. Threonine 41 in beta-catenin serves as a key phosphorylation relay residue in beta-catenin degradation. Biochemistry. 2006;45:5319–5323. doi: 10.1021/bi0601149. [DOI] [PubMed] [Google Scholar]
- 54.Rubinfeld B, Robbins P, ElGamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 55.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolz H, Schade G, Kothe C, Mohrmann G, Hess M, Gal A. Impaired calmodulin binding of myosin-7A causes autosomal dominant hearing loss (DFNA11) Hum Mutat. 2004;24:274–275. doi: 10.1002/humu.9272. [DOI] [PubMed] [Google Scholar]
- 57.Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang QQ, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SYP, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Amer J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gregorieff A, Cloutier JF, Veillette A. Sequence requirements for association of protein-tyrosine phosphatase PEP with the Src homology 3 domain of inhibitory tyrosine protein kinase p50(csk) J Biol Chem. 1998;273:13217–13222. doi: 10.1074/jbc.273.21.13217. [DOI] [PubMed] [Google Scholar]
- 59.Arimura T, Nakamura T, Hiroi S, Satoh M, Takahashi M, Ohbuchi N, Ueda K, Nouchi T, Yamaguchi N, Akai J, Matsumori A, Sasayama S, Kimura A. Characterization of the human nebulette gene: a polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum Genet. 2000;107:440–451. doi: 10.1007/s004390000389. [DOI] [PubMed] [Google Scholar]
- 60.Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Nat Acad Sci U S A. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li TS, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: Evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Nat Acad Sci U S A. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptor. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 63.Ishii T, Suzuki Y, Ando N, Matsuo N, Ogata T. Novel mutations of the autoimmune regulator gene in two siblings with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2000;85:2922–2926. doi: 10.1210/jcem.85.8.6726. [DOI] [PubMed] [Google Scholar]
- 64.Klaavuniemi T, Kelloniemi A, Ylanne J. The ZASP-like motif in actinin-associated LIM protein is required for interaction with the alpha-actinin rod and for targeting to the muscle Z-line. J Biol Chem. 2004;279:26402–26410. doi: 10.1074/jbc.M401871200. [DOI] [PubMed] [Google Scholar]
- 65.Zoref-Shani E, Bromberg Y, Hirsch J, Feinstein S, Frishberg Y, Sperling O. A novel point mutation (I137T) in the conserved 5-phosphoribosyl-1-pyrophosphate binding motif of hypoxanthine-guanine phosphoribosyltransferase (HPRTJerusalem) in a variant of Lesch-Nyhan syndrome. Mol Genet Metab. 2003;78:158–161. doi: 10.1016/s1096-7192(03)00002-7. [DOI] [PubMed] [Google Scholar]
- 66.Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O’Neill JP. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mut Res. 2000;463:309–326. doi: 10.1016/s1383-5742(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 67.Kaiser FJ, Brega P, Raff ML, Byers PH, Gallati S, Kay TT, de Almeida S, Horsthemke B, Ludecke HJ. Novel missense mutations in the TRPS1 transcription factor define the nuclear localization signal. Eur J Hum Genet. 2004;12:121–126. doi: 10.1038/sj.ejhg.5201094. [DOI] [PubMed] [Google Scholar]
- 68.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 69.Gong QM, Keeney DR, Robinson JC, Zhou ZF. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 70.Dagenais SL, Adam AN, Innis JW, Glover TW. A novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes disease. Am J Hum Genet. 2001;69:420–427. doi: 10.1086/321290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fitzpatrick DR, Hill A, Tolmie JL, Thorburn DR, Christodoulou L. The molecular basis of malonyl-CoA decarboxylase deficiency. Amer J Hum Genet. 1999;65:318–326. doi: 10.1086/302492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Toh K, Jones C, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- 73.Oxombre B, Kouach M, Moerman E, Formstecher P, Laine B. The G115S mutation associated with maturity-onset diabetes of the young impairs hepatocyte nuclear factor 4alpha activities and introduces a PKA phosphorylation site in its DNA-binding domain. Biochem J. 2004;383:573–580. doi: 10.1042/BJ20040473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bahram F, der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–2110. [PubMed] [Google Scholar]
- 75.Porton B, Ferreira A, Delisi LE, Kao HT. A rare polymorphism affects a mitogen-activated protein kinase site in synapsin III: Possible relationship to schizophrenia. Biol Psychiatry. 2004;55:118–125. doi: 10.1016/j.biopsych.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 76.Hendrickx J, Dams E, Coucke P, Lee P, Fernandes J, Willems PJ. X-linked liver glycogenosis type II (XLG II) is caused by mutations in PHKA2, the gene encoding the liver a subunit of phosphorylase kinase. Hum Mol Genet. 1996;5:649–652. doi: 10.1093/hmg/5.5.649. [DOI] [PubMed] [Google Scholar]
- 77.Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, Ekstein J, Rubin BY. Familial dysautonomia is caused by mutations of the IKAP gene. Amer J Hum Genet. 2001;68:753–758. doi: 10.1086/318808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reeves JD, Piefier AJ. Emerging drug targets for antiretroviral therapy. Drugs. 2005;65:1747–1766. doi: 10.2165/00003495-200565130-00002. [DOI] [PubMed] [Google Scholar]
- 79.Bai XY, Miao DS, Goltzman D, Karaplis AC. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem. 2003;278:9843–9849. doi: 10.1074/jbc.M210490200. [DOI] [PubMed] [Google Scholar]
- 80.Kawashima Y, Kanzaki S, Yang F, Kinoshita T, Hanaki K, Nagaishi J, Ohtsuka Y, Hisatome I, Ninomoya H, Nanba E, Fukushima T, Takahashi S. Mutation at cleavage site of insulin-like growth factor receptor in a short-stature child born with intrauterine growth retardation. J Clinl Endocrinol Metab. 2005;90:4679–4687. doi: 10.1210/jc.2004-1947. [DOI] [PubMed] [Google Scholar]
- 81.Chen YW, Molloy SS, Thomas L, Gambee J, Bachinger HP, Ferguson B, Zonana J, Thomas G, Morris NP. Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc Nat Acad Sci U S A. 2001;98:7218–7223. doi: 10.1073/pnas.131076098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Casonato A, Sartorello F, Cattini MG, Pontara E, Soldera C, Bertomoro A, Girolami A. An Arg760Cys mutation in the consensus sequence of the von Willebrand factor propeptide cleavage site is responsible for a new von Willebrand disease variant. Blood. 2003;101:151–156. doi: 10.1182/blood-2002-04-1046. [DOI] [PubMed] [Google Scholar]
- 83.Dahlback B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost. 1995;74:139–148. [PubMed] [Google Scholar]
- 84.Pattinson JK, Millar DS, McVey JH, Grundy CB, Wieland K, Mibashan RS, Martinowitz U, Tan-Un K, Vidaud M, Goossens M. The molecular genetic analysis of hemophilia A: a directed search strategy for the detection of point mutations in the human factor VIII gene. Blood. 1990;76:2242–2248. [PubMed] [Google Scholar]
- 85.Arai M, Higuchi M, Antonarakis SE, Kazazian HH, Jr., Phillips JA, III, Janco RL, Hoyer LW. Characterization of a thrombin cleavage site mutation (Arg 1689 to Cys) in the factor VIII gene of two unrelated patients with cross-reacting material-positive hemophilia A. Blood. 1990;75:384–389. [PubMed] [Google Scholar]
- 86.Wattenhofer M, Sahin-Calapoglu N, Andreasen D, Kalay E, Caylan R, Braillard B, Fowler-Jaeger N, Reymond A, Rossier BC, Karaguzel A, Antonarakis SE. A novel TMPRSS3 missense mutation in a DFNB8/10 family prevents proteolytic activation of the protein. Human Genetics. 2005;117:528–535. doi: 10.1007/s00439-005-1332-x. [DOI] [PubMed] [Google Scholar]
- 87.Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K. Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Nat Acad Sci U S A. 1999;96:3053–3058. doi: 10.1073/pnas.96.6.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gruninger-Leitch F, Schlatter D, Kung E, Nelbock P, Dobeli H. Substrate and inhibitor profile of BACE (beta-secretase) and comparison with other mammalian aspartic proteases. J Biol Chem. 2002;277:4687–4693. doi: 10.1074/jbc.M109266200. [DOI] [PubMed] [Google Scholar]
- 89.Vogt G, Vogt B, Chuzhanova N, Julenius K, Cooper DN, Casanova JL. Gain-of-glycosylation mutations. Current Opinion in Genetics & Development. 2007;17:245–251. doi: 10.1016/j.gde.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 90.Sullivan LJ, Makris GS, Dickinson P, Mulhall LE, Forrest S, Cotton RG, Loughnan MS. A new codon 15 rhodopsin gene mutation in autosomal dominant retinitis pigmentosa is associated with sectorial disease. Arch Ophthalmol. 1993;111:1512–1517. doi: 10.1001/archopht.1993.01090110078029. [DOI] [PubMed] [Google Scholar]
- 91.Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathologica. 2004;108:476–484. doi: 10.1007/s00401-004-0913-4. [DOI] [PubMed] [Google Scholar]
- 92.Wrobe D, Henseler M, Huettler S, Pascual SIP, Chabas A, Sandhoff K. A non-glycosylated and functionally deficient mutant (N215H) of the sphingolipid activator protein B (SAP-B) in a novel case of metachromatic leukodystrophy (MLD) J Inher Metab Dis. 2000;23:63–76. doi: 10.1023/a:1005603014401. [DOI] [PubMed] [Google Scholar]
- 93.Schiffer C, Ultsch M, Walsh S, Somers W, de Vos AM, Kossiakoff A. Structure of a phage display-derived variant of human growth hormone complexed to two copies of the extracellular domain of its receptor: Evidence for strong structural coupling between receptor binding sites. J Mol Bio. 2002;316:277–289. doi: 10.1006/jmbi.2001.5348. [DOI] [PubMed] [Google Scholar]
- 94.Bernat B, Sun M, Dwyer M, Feldkamp M, Kossiakoff AA. Dissecting the binding energy epitope of a high-affinity variant of human growth hormone: Cooperative and additive effects from combining mutations from independently selected phage display mutagenesis libraries. Biochemistry. 2004;43:6076–6084. doi: 10.1021/bi036069b. [DOI] [PubMed] [Google Scholar]
- 95.Choong NW, Cohen EEW. Forthcoming receptor tyrosine kinase inhibitors. Expert Opin Ther Targets. 2006;10:793–797. doi: 10.1517/14728222.10.6.793. [DOI] [PubMed] [Google Scholar]
- 96.Larkin JMG, Eisen T. Kinase inhibitors in the treatment of renal cell carcinoma. Crit Rev Oncol Hematol. 2006;60:216–226. doi: 10.1016/j.critrevonc.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 97.Dumont FJ, Su QX. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1995;58:373–395. doi: 10.1016/0024-3205(95)02233-3. [DOI] [PubMed] [Google Scholar]
- 98.Raizada MK, Ferreira AJ. ACE2: A new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol. 2007;50:112–119. doi: 10.1097/FJC.0b013e3180986219. [DOI] [PubMed] [Google Scholar]
- 99.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 100.Philips MR, Cox AD. Geranylgeranyltransferase I as a target for anti-cancer drugs. J Clin Invest. 2007;117:1223–1225. doi: 10.1172/JCI32108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coller BS. Anti-GPIIb/IIIa drugs: Current strategies and future directions. Thromb Haemost. 2001;86:427–443. [PubMed] [Google Scholar]
- 102.Plow EF, Cierniewski CS, Xiao Z, Haas TA, Byzova TV. AlphaIIbbeta3 and its antagonism at the new millennium. Thromb Haemost. 2001;86:34–40. [PubMed] [Google Scholar]
- 103.Bennett JS. Novel platelet inhibitors. Annual Review of Medicine. 2001;52:161–184. doi: 10.1146/annurev.med.52.1.161. [DOI] [PubMed] [Google Scholar]
- 104.Schror K. Anti-integrins--new platelet function inhibitors for therapy and prevention of acute coronary syndrome. Wien Klin Wochenschr. 1999;111:90–97. [PubMed] [Google Scholar]
- 105.Theroux P. Oral inhibitors of platelet membrane receptor glycoprotein IIb/IIIa in clinical cardiology: Issues and opportunities. Amer Heart J. 1998;135:S107–S112. doi: 10.1016/s0002-8703(98)70238-8. [DOI] [PubMed] [Google Scholar]
- 106.Hallahan DE, Qu SM, Geng L, Cmelak A, Chakravarthy A, Martin W, Scarfone C, Giorgio T. Radiation-mediated control of drug delivery. Amer J Clinl Oncol. 2001;24:473–480. doi: 10.1097/00000421-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 107.Lu X, Lu D, Scully MF, Kakkar VV. Integrins in drug targeting-RGD templates in toxins. Curr Pharmaceut Des. 2006;12:2749–2769. doi: 10.2174/138161206777947713. [DOI] [PubMed] [Google Scholar]
- 108.Meyer A, Auemheimer J, Modlinger A, Kessler H. Targeting RGD recognizing integrins: Drug development, biomaterial research, tumor imaging and targeting. Curr Pharmaceutl De. 2006;12:2723–2747. doi: 10.2174/138161206777947740. [DOI] [PubMed] [Google Scholar]
- 109.Dijkgraaf I, Rijnders AY, Soede A, Dechesne AC, van Esse GW, Brouwer AJ, Corstens FHM, Boerman OC, Rijkers DTS, Liskamp RMJ. Synthesis of DOTA-conjugated multivalent cyclic-RGD peptide dendrimers via 1,3-dipolar cycloaddition and their biological evaluation: implications for tumor targeting and tumor imaging purposes. Org Biomol Chem. 2007;5:935–944. doi: 10.1039/b615940k. [DOI] [PubMed] [Google Scholar]
- 110.Shen SI, Kotamraj PR, Bhattacharya S, Li XL, Jasti BR. Synthesis and characterization of RGD-fatty acid amphiphilic micelles as targeted delivery carriers for anticancer agents. J Drug Target. 2007;15:51–58. doi: 10.1080/10611860601035212. [DOI] [PubMed] [Google Scholar]
- 111.Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T, Zhang Q. Intracellular delivery of doxorubicin with RGD-modified sterically stabilized liposomes for an improved antitumor efficacy: In vitro and in vivo. J Pharmaceut Sci. 2005;94:1782–1793. doi: 10.1002/jps.20397. [DOI] [PubMed] [Google Scholar]
- 112.Holig P, Bach M, Volkel T, Nahde T, Hoffmann S, Muller R, Kontermann RE. Novel RGD lipopeptides for the targeting of liposomes to integrin-expressing endothelial and melanoma cells. Prot Eng Des Sel. 2004;17:433–441. doi: 10.1093/protein/gzh055. [DOI] [PubMed] [Google Scholar]
- 113.Contino-Pepin C, Maurizis MC, Pucci B. Amphiphilic oligomers: a new kind of macromolecular carrier of antimitotic drugs. Curr Med Chem Anticancer Agents. 2002;2:645–665. doi: 10.2174/1568011023353732. [DOI] [PubMed] [Google Scholar]
- 114.Dunehoo AL, Anderson M, Majumdar S, Kobayashi N, Berkland C, Siahaan TJ. Cell adhesion molecules for targeted drug delivery. J Pharmaceut Sci. 2006;95:1856–1872. doi: 10.1002/jps.20676. [DOI] [PubMed] [Google Scholar]
- 115.Garanger E, Boturyn D, Dumy P. Tumor targeting with RGD peptide ligands-design of new molecular conjugates for imaging and therapy of cancers. Anticancer Agents Med Chem. 2007;7:552–558. doi: 10.2174/187152007781668706. [DOI] [PubMed] [Google Scholar]
- 116.Westlin WF. Integrins as targets of angiogenesis inhibition. Cancer J. 2001;7:S139–S143. [PubMed] [Google Scholar]
- 117.Nam JO, Kim JE, Jeong HW, Lee SJ, Lee BH, Choi JY, Park RW, Park JY, im IS. Identification of the alphavbeta3 integrin-interacting motif of betaig-h3 and its anti-angiogenic effect. J Biol Chem. 2003;278:25902–25909. doi: 10.1074/jbc.M300358200. [DOI] [PubMed] [Google Scholar]
- 118.Hersel U, Dahmen C, Kessler H. RGD modified polymers: biomaterials for stimulated cell adhesion and beyond. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 119.Healy KE, Rezania A, Stile RA. Designing biomaterials to direct biological responses. Ann N Y Acad Sci. 1999;875:24–35. doi: 10.1111/j.1749-6632.1999.tb08491.x. [DOI] [PubMed] [Google Scholar]
- 120.Goligorsky MS, Noiri E, Kessler H, Romanov V. Therapeutic effect of arginine-glycine-aspartic acid peptides in acute renal injury. Clin Exp Pharmacol Physiol. 1998;25:276–279. doi: 10.1111/j.1440-1681.1998.t01-2-.x. [DOI] [PubMed] [Google Scholar]
- 121.Goligorsky MS, Noiri E, Kessler H, Romanov V. Therapeutic potential of RGD peptides in acute renal injury. Kidney Intl. 1997;51:1487–1492. doi: 10.1038/ki.1997.204. [DOI] [PubMed] [Google Scholar]
- 122.Chorev M, Dresnerpollak R, Eshel Y, Rosenblatt M. Approach to Discovering Novel Therapeutic Agents for Osteoporosis Based on Integrin Receptor Blockade. Biopolymers. 1995;37:367–375. doi: 10.1002/bip.360370603. [DOI] [PubMed] [Google Scholar]
- 123.Curnis F, Arrigoni G, Sacchi A, Fischetti L, Arap W, Pasqualini R, Corti A. Differential binding of drugs containing the NGR motif to CD13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res. 2002;62:867–874. [PubMed] [Google Scholar]
- 124.Dev KK. Making protein interactions druggable: Targeting PDZ domains. Nat Rev Drug Discov. 2004;3:1047–1056. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 125.Kardinal C, Posern G, Zheng J, Knudsen BS, Moarefi I, Feller SM. Rational development of cell-penetrating high affinity SH3 domain binding peptides that selectively disrupt the signal transduction of Crk family adapters. Ann N Y Acad Sci. 1999;886:289–292. doi: 10.1111/j.1749-6632.1999.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 126.Feller SM, Lewitzky M. Potential disease targets for drugs that disrupt protein-protein interactions of Grb2 and Crk family adaptors. Curt Pharmacol Des. 2006;12:529–548. doi: 10.2174/138161206775474369. [DOI] [PubMed] [Google Scholar]
- 127.Morohashi K, Yoshino A, Yoshimori A, Saito S, Tanuma S, Sakaguchi K, Sugawara F. Identification of a drug target motif: An anti-tumor drug NK109 interacts with a PNxxxxP. Biochem Pharmacol. 2005;70:37–46. doi: 10.1016/j.bcp.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 128.Laurie NA, Donovan SL, Shih CS, Zhang JK, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 129.Bult CJ, Blake JA, Richardson JE, Kadin JA, Eppig JT. The Mouse Genome Database (MGD): integrating biology with the genome. Nuc Acids Res. 2004;32:D476–D481. doi: 10.1093/nar/gkh125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brown P, Scobie GA, Townsend J, Bayne RAL, Seckl JR, Saunders PTK, Anderson RA. Identification of a novel missense mutation that is as damaging to DAX-1 repressor function as a nonsense mutation. J Clin Endocrinol Metab. 2003;88:1341–1349. doi: 10.1210/jc.2002-021560. [DOI] [PubMed] [Google Scholar]
- 131.Zhang H, Thomsen JS, Johansson L, Gustafsson JA, Treuter E. DAX-1 functions as an LXXLL-containing corepressor for activated estrogen receptors. J Biol Chem. 2000;275:39855–39859. doi: 10.1074/jbc.C000567200. [DOI] [PubMed] [Google Scholar]